")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Spotlight on opicapone as an adjunct to levodopa in Parkinson’s disease: design, development and potential place in therapy
Received 28 October 2016
Accepted for publication 7 December 2016
Published 9 January 2017 Volume 2017:11 Pages 143—151
DOI https://doi.org/10.2147/DDDT.S104227
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Ádám Annus,1 László Vécsei1,2
1Department of Neurology, Faculty of Medicine, Albert Szent-Györgyi Clinical Center, University of Szeged, 2MTA-SZTE Neuroscience Research Group, Szeged, Hungary
Abstract: Parkinson’s disease (PD) is a progressive, chronic, neurodegenerative disease characterized by rigidity, tremor, bradykinesia and postural instability secondary to dopaminergic deficit in the nigrostriatal system. Currently, disease-modifying therapies are not available, and levodopa (LD) treatment remains the gold standard for controlling motor and nonmotor symptoms of the disease. LD is extensively and rapidly metabolized by peripheral enzymes, namely, aromatic amino acid decarboxylase and catechol-O-methyltransferase (COMT). To increase the bioavailability of LD, COMT inhibitors are frequently used in clinical settings. Opicapone is a novel COMT inhibitor that has been recently approved by the European Medicines Agency as an adjunctive therapy to combinations of LD and aromatic amino acid decarboxylase inhibitor in adult PD patients with end-of-dose motor fluctuations. We aimed to review the biochemical properties of opicapone, summarize its preclinical and clinical trials and discuss its future potential role in the treatment of PD.
Keywords: Parkinson’s disease, COMT inhibitors, opicapone
Introduction
Parkinson’s disease (PD) is a progressive, chronic, neurodegenerative disease characterized by rigidity, tremor, bradykinesia and postural instability secondary to dopaminergic deficit in the nigrostriatal system.1–3 Currently, disease-modifying therapies are not available, and levodopa (LD) treatment remains the gold standard for controlling motor symptoms of the disease.4–7 Nonmotor symptoms (eg, cognitive decline, psychiatric symptoms, autonomic and sleep disturbance, etc) also cause a marked decrease in the quality of life, but currently there is limited evidence that LD treatment can alleviate these symptoms. Furthermore, within 5 years of treatment, ~50% of patients develop motor fluctuations and dyskinesia.1,2,8 The pathophysiology behind motor complications is that instead of physiologic, tonic stimulation, there is a reduced and pulsatile dopaminergic stimulation of striatal neurons.9 This is due to fluctuations in the plasma concentration of LD and progressive neuronal cell death in the nigrostriatal system.10 Therefore, the aim of improving treatment is to provide a steadier and more sustained plasma concentration of LD and consequent continuous dopaminergic transmission in the basal ganglia.
LD is the precursor of dopamine (DA), and unlike DA, it can penetrate through the blood–brain barrier (BBB). However, after oral intake, LD is extensively and rapidly metabolized by peripheral enzymes, namely, aromatic amino acid decarboxylase (AADC) and catechol-O-methyltransferase (COMT). Figure 1 shows the peripheral and intraneural metabolism of LD. COMT catalyzes the transfer of a methyl group from S-adenosyl methionine (SAM) to catechol estrogens and endogenous catecholamines.11 After the transfer, the co-substrate becomes S-adenosyl homocysteine (SAHcy). The catalytic cycle is complete when SAHcy is exchanged to SAM and COMT can catalyze another O-methylation. In the central nervous system (CNS), LD is broken down to homovanillic acid (HVA) by COMT and monoamine oxidase (MAO). When LD is administered alone, approximately only 1% of the oral dose reaches the CNS.12–14 When LD is administered with an AADC inhibitor (benserazide [BZ] or carbidopa [CD]), ~90% of LD is converted to 3-O-methyldopa (3-OMD) by COMT, which could compete with LD for transport through the BBB.15–17 The clinical significance of 3-OMD as a competitive antagonist to LD is questionable.16,17 3-OMD is considered as a biomarker of peripheral COMT inhibition.18 Currently, there are three COMT inhibitors that can be used in the treatment of PD, entacapone (ENT), tolcapone (TCP), and since its approval for medical use by the European Medicines Agency in June 2016, opicapone (OPC; Ongentys®, developed by Bial-Portela & Ca. S.A.).19,20 OPC is indicated as an adjunctive therapy to combinations of LD/DOPA decarboxylase inhibitors (DDCIs) in adult PD patients with end-of-dose motor fluctuations whose symptoms cannot be stabilized with these preparations. Peripheral COMT inhibitors, like OPC, increase the half-life of LD, whereas central inhibitors slow its metabolism within the CNS.9 Our review highlights the biochemical properties of OPC and summarizes preclinical investigations and clinical trials with this novel COMT inhibitor.
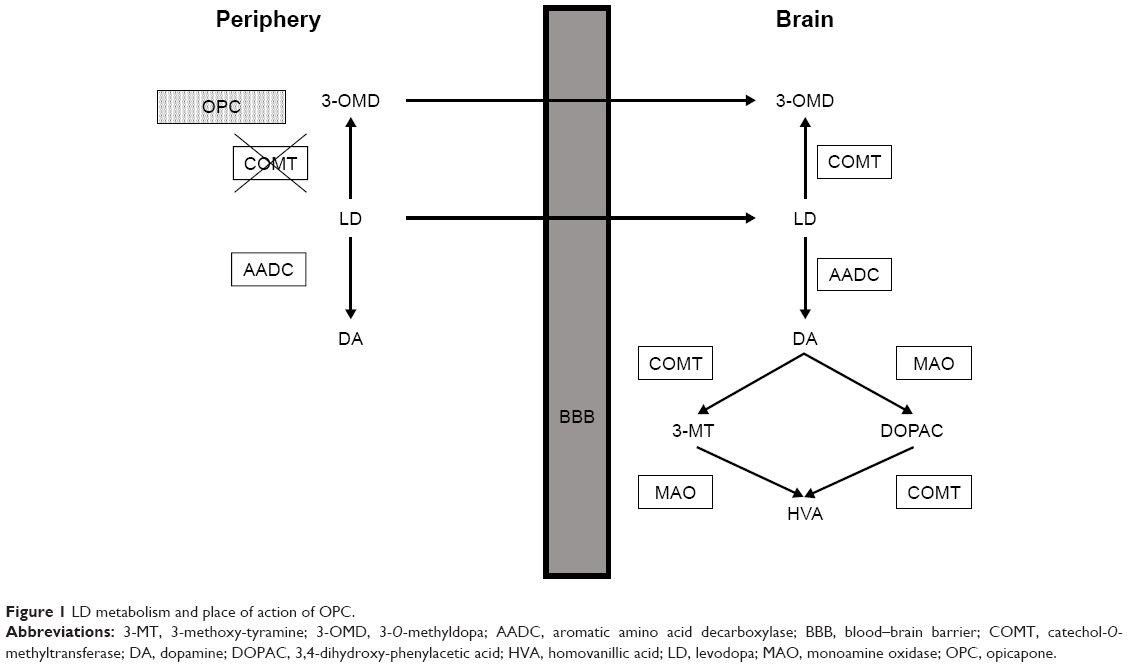 | Figure 1 LD metabolism and place of action of OPC. |
Pharmacokinetic and pharmacodynamic properties of OPC
OPC (2,5-dichloro-3-[5-(3,4-dihydroxy-5-nitrophenyl]-1,2-4-oxadiazol-3-yl)-4,6-dimethylpyridine 1-oxide, previously known as BIA 9-1067) is a novel third-generation nitrocatechol COMT inhibitor that has been approved for medical use by the European Medicines Agency in June 2016.20,21 Figure 2 shows the chemical structure of OPC. It is indicated as an adjunctive therapy to combinations of LD/DDCI in adult PD patients with end-of-dose motor fluctuations whose symptoms cannot be stabilized with these combinations. OPC has a pyridine N-oxide residue at position 3, providing high-affinity COMT inhibition and avoidance of cell toxicity.22 OPC has a very high protein-binding affinity in the plasma (>99%).23 Kiss et al22 found that OPC does not cross the BBB in rats and monkeys. An in vitro study using parallel artificial membrane permeability assay (PAMPA) also confirmed that OPC does not penetrate into the brain.24 Consequently, it was concluded that OPC inhibits only peripheral COMT enzymes. COMT is present in all mammalian tissues and has been shown to have the highest activity in the liver, kidney and cells of the gastrointestinal tract.25 Human erythrocytes also contain COMT and are generally used in clinical trials for the assessment of COMT activity. OPC has a remarkably high-affinity binding to COMT (dissociation constant is in the subpicomolar range).26 In comparison, the same constant is two magnitudes higher for TCP.
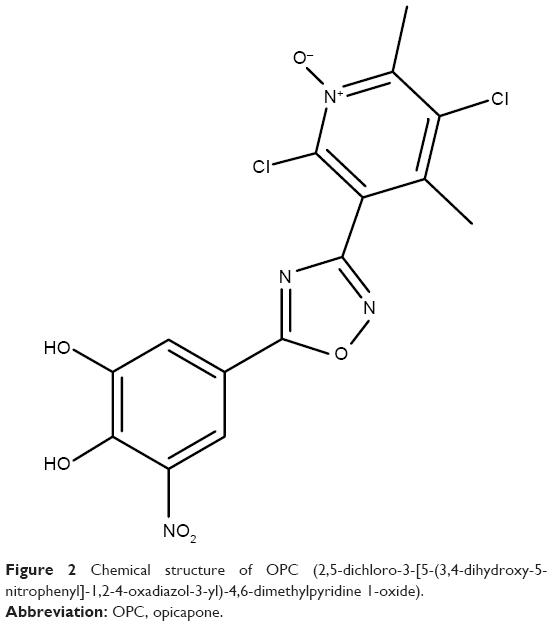 | Figure 2 Chemical structure of OPC (2,5-dichloro-3-[5-(3,4-dihydroxy-5-nitrophenyl]-1,2-4-oxadiazol-3-yl)-4,6-dimethylpyridine 1-oxide). |
Almeida et al27 found that after a single oral administration of various doses (10 mg, 25 mg, 50 mg, 100 mg, 200 mg, 400 mg, 800 mg and 1,200 mg) of OPC to healthy subjects, the terminal half-life ranged between 0.8 h (for 50 mg) and 3.2 h (for 1,200 mg). LD was given concomitantly and maximum LD concentration was detected between 1.5 h and 3.5 h. The maximum COMT inhibition was dose dependent and ranged between 36.1% (10 mg OPC) and 100% (200 mg–1,200 mg OPC). The inhibitory effect reached its peak between 0.7 h and 6.2 h post-dose.27 After 24 h of intake, OPC exerted COMT inhibitory effect ranging from 26.1% (10 mg) to 76.5% (800 mg). At 72 h post-dose, OPC still had COMT inhibitory effect: 5.9% (10 mg OPC) to 54.6% (800 mg).27 Systemic exposure to OPC was significantly lower after a high-fat, high-calorie meal compared to after fasting, implying that OPC should be taken before meals. The only metabolite detected in the urine of participants was BIA 9-1106.
Despite its relatively quick elimination from the circulation, OPC has a sustained COMT inhibitory effect. Rocha et al28 hypothesized that the long-lasting effect of OPC might be related to its binding to the complex of COMT–SAHcy, thus blocking the exchange of SAM to SAHcy and therefore hindering the O-methylation of another OPC molecule.
In nonclinical studies, the following five metabolites of OPC have been detected: BIA 9-1100 and BIA 9-1101 are inactive methylated metabolites, BIA 9-1103 is an inactive sulfated metabolite, BIA 9-1106 is an inactive glucuronide metabolite and BIA 9-1079 is an active COMT-inhibitor metabolite.27,28 OPC is mainly metabolized by sulfation to BIA 9-1103. Reduction to BIA 9-1079 and glucuronidation to BIA 9-1106 are alternate metabolic routes.27,28 A total of 70% of orally administered radiolabeled 14C-OPC was eliminated by feces in human beings,23 whereas 12% of the drug was excreted by urine. Therefore, hepatobiliary excretion seems to be the main elimination pathway for OPC. In patients with moderate liver impairment (Child-Pugh category B, scores 7–9), compared to healthy controls, increased bioavailability of OPC was detected, possibly due to decreased first-pass effect in the liver. No serious adverse events or treatment-related adverse events were found in either groups.23
Pinto et al29 have demonstrated that neither 50 mg nor 800 mg of OPC caused clinically significant changes in electrocardiogram (ECG) morphology and intervals. A study was conducted where healthy white and Japanese subjects were given 5 mg, 25 mg and 50 mg OPC or placebo (PLC).30 No statistically significant differences were found in the pharmacokinetics and pharmacodynamics of OPC between the two populations.
Animal studies with OPC
In one study, OPC was given orally to rats and resulted in significant reduction in COMT activity.31 The maximum plasma concentration was detected at 4 h following administration. OPC could not be detected after 8 h post-admission. When LD/BZ was given to rats 2 h and 24 h after administration of OPC, the maximum plasma concentration of LD increased by 1.6- and 1.4-fold, bioavailability of LD increased by 1.9- and 1.3-fold and 3-OMD exposure decreased by 6.3- and 1.6-fold, respectively.31 In the same study, adenosine triphosphate (ATP) concentrations and mitochondrial membrane potentials were measured after 24 h of incubation of human primary hepatocytes to investigate cytotoxic properties. Compared to TCP and ENT, OPC was the least potent compound to decrease mitochondrial membrane potential and ATP content of hepatocytes.31
In brain and liver homogenates of rats, OPC caused a more sustained COMT inhibitory effect compared to ENT and TCP.32 The medications were administered through a gastric tube 1 h after intake, and COMT inhibition reached 99%, 82% and 68%, respectively, with OPC, TCP and ENT. After 9 h of administration, OPC still showed a 91% reduction in COMT activity. No inhibitory effects of ENT could be detected and TCP produced only a 16% decrease in COMT activity.32
Bonifácio et al33 examined systemic and central bioavailability of LD and its metabolites in cynomolgus monkeys. Microdialysis probes were inserted into the substantia nigra, dorsal striatum and prefrontal cortex of monkeys. The animals received either vehicle or 100 mg/kg OPC for 14 days, and then they were challenged with a combination of LD and BZ (12/3 mg/kg). Blood samples and extracellular dialyzate from the brain were collected for measurements. Compared to PLC, a significant increase in LD levels was observed in the plasma and also in the dialyzate samples obtained from all three brain regions (1.7-, 1.4- and 2.3-fold increase in dorsal striatum, substantia nigra and prefrontal cortex, respectively).33 In addition, reduced 3-OMD exposure was observed (fivefold decrease in plasma and fivefold, sevenfold and 2.4-fold decrease in the dorsal striatum, substantia nigra and prefrontal cortex, respectively). Furthermore, erythrocyte COMT activity in the plasma decreased by ~76%–84%.33
Clinical trials in healthy subjects
Almeida et al27 investigated the pharmacokinetic and pharmacodynamic properties and tolerability of OPC after a single oral administration of various doses (10 mg, 25 mg, 50 mg, 100 mg, 200 mg, 400 mg, 800 mg and 1,200 mg). Systemic exposure to OPC and its metabolites showed an approximately dose-dependent pattern. Human erythrocyte COMT inhibition was also dose dependent. The half-life of COMT inhibition was 61.6 h. Detailed results of the trial are discussed in the second paragraph of the “Pharmacokinetic and pharmacodynamic properties of OPC” section of this article.
In a dose-escalation, double-blind, PLC-controlled study, healthy male subjects were given 5 mg, 10 mg, 20 mg or 30 mg OPC for 8 days.28 The pharmacokinetic and pharmacodynamic properties of OPC and its metabolites were investigated. The terminal half-life of OPC ranged from 1 h to 1.4 h. BIA 9-1103 was identified as the main metabolite. Its terminal half-life was between 25.1 h and 26.9 h. Although BIA 9-1079 is an active metabolite, it only represented <20% of systemic exposure to OPC. Therefore, it only has minor relevance in the therapeutic effect of OPC.28 COMT inhibition was found to be dose dependent. The maximum inhibition was detected between 3.8 h and 7.7 h after the first administration and 3.7 h and 5.3 h after the last intake of OPC. Long-lasting COMT inhibition was found. At 144 h after the last dose 16.3%, 21.3%, 31.4% and 20.3% decrease in COMT activity still remained with 5 mg, 10 mg, 20 mg and 30 mg OPC, respectively.28 Previously, the maximum COMT inhibition for 100 mg and 200 mg TCP and 200 mg ENT was found to be 72%, 80% and 65% respectively.34,35 COMT activity returned to baseline within ~18 h and 8 h after TCP and ENT, respectively.
In another study, healthy individuals received 25 mg, 50 mg, and 75 mg OPC or PLC for 11 days.36 On the 12th day, subjects in the PLC group received 200 mg ENT and LD/CD three times a day. Individuals in the OPC group were also administered LD and CD three times daily to assess the effects of OPC on LD pharmacokinetics. The minimum LD plasma concentration increased in all active treatment groups compared to the PLC arm (1.7- and 3.3-fold with 200 mg ENT and 75 mg OPC, respectively). All active treatments resulted in increased LD bioavailability and significant decrease in 3-OMD exposure in relation to the PLC group.36 Compared to ENT, OPC showed a more sustained effect on erythrocyte COMT inhibition. Adverse events were mild, the most common ones being nausea, vomiting, headache and dizziness. No serious adverse events were reported. It was concluded that OPC was more efficacious compared to ENT in increasing LD bioavailability.36
Table 1 summarizes the main findings of clinical trials in healthy subjects.
 | Table 1 Summary of clinical trials in healthy subjects |
Clinical trials in PD patients
In a phase II study, patients diagnosed with idiopathic PD, who had at least 1.5 h long OFF state in the waking day and a modified Hoehn–Yahr stage of <5 in the OFF state, received 25 mg, 50 mg, and 100 mg OPC or PLC once daily in addition to their previous 100/25 mg LD and CD or BZ therapy.37 The maximum plasma concentration of LD dose dependently increased after OPC intake compared to PLC. A marked decrease was observed in peak 3-OMD levels and also in the extent of exposure with 100 mg OPC. All active treatments inhibited the extent and rate of COMT activity in a dose-dependent, albeit disproportional, manner.37 Motor responses were also measured. A marked decrease in time to ON and time to best ON was detected. ON duration increased by 18% and 25% with 25 mg and 50 mg OPC, respectively. Adverse events were mostly mild to moderate in intensity.37
Ferreira et al38 investigated the effects of 28-day long administration of 5 mg, 15 mg and 30 mg OPC on LD pharmacokinetics, COMT inhibition and motor fluctuations in PD patients. The population of the study was similar to that described in the previous trial. All active treatments increased LD bioavailability in a dose-dependent manner, caused sustained dose-dependent but not proportional COMT inhibition and consequently a significantly lower exposure to 3-OMD. Based on the participants’ diaries, all active treatments caused a dose-dependent decrease in the absolute OFF time (15.6 min and 145 min with 5 mg and 30 mg OPC, respectively).38 Overall, 15 mg and 30 mg OPC reached statistical significance compared to PLC in improving ON time without dyskinesia. The investigators’ and patients’ global assessment of change was also favorable. No serious adverse events occurred during the study.38
BIPARK I was a phase III, randomized, double-blind, multicenter, PLC- and active-controlled trial that assessed the efficacy and safety profile of OPC.21 OPC was administered as an adjunct therapy to LD in PD patients with motor fluctuations. Participants were aged between 30 years and 83 years, had a clinical diagnosis of PD for at least 3 years, a Hoehn–Yahr stage of 1–3 during the ON state and had to have end-of-dose motor fluctuations for at least 4 weeks before screening. Initially, 600 patients met the inclusion criteria and were randomly assigned to five groups: patients either received PLC, 200 mg ENT or 5 mg, 25 mg, or 50 mg OPC for 14–15 weeks in addition to LD (administered three to eight times a day). OPC was given once daily and 1 h after the last LD dose. The primary end point was the change in the absolute time in the OFF state assessed by daily patient diaries.21 Key secondary end points included the change in the proportion of patients showing at least 1 h of reduction in the absolute time in the OFF state. Moreover, the proportion of patients achieving at least 1 h increase in the absolute time spent in the ON state was assessed. A significantly different reduction in the OFF time was reported for 50 mg OPC compared to PLC (−116.8 min and −56 min, respectively). ENT also showed a statistically different reduction in the absolute time spent in the OFF state (−96.3 min).21 These findings were in accordance with previous studies.39 Based on these results, the authors concluded that 50 mg OPC was superior to PLC and non-inferior to ENT. The 5 mg and 25 mg doses of OPC did not show significant reduction in the time spent in the OFF state compared to PLC. Regarding the secondary end points, a significantly higher proportion of patients showed at least 1 h reduction in the OFF state in the 25 mg and 50 mg OPC groups compared to PLC. In addition, the proportion of patients who had at least 1 h increase in the ON state was significantly higher in the 50 mg OPC arm. In all, 190 patients had treatment-related adverse events in the OPC groups.21 Most common adverse events were dyskinesia (44 patients among those receiving OPC), insomnia (16 patients) and constipation (11 patients taking OPC). Altogether, nine patients had serious adverse event in the OPC groups, including one who had a marked increase in the level of liver enzymes but no hyperbilirubinemia. No fatal events were reported. Therefore, OPC was found to have a favorable safety profile.21
In the BIPARK II study (as of yet, data are available only as abstract), which was a multinational, PLC-controlled, double-blind, parallel-group study, 25 mg and 50 mg OPC were administered once daily for 407 patients with PD showing end-of-dose motor fluctuations.40 The inclusion and exclusion criteria were similar to that of the BIPARK I study. Based on patients’ diaries, the reduction in the OFF time was significantly greater in both 25 mg and 50 mg OPC groups compared to PLC (1.7 h, 2.0 h and 1.1 h, respectively).The increase in the ON time with or without non-troublesome dyskinesia in both OPC groups was also greater than that in the PLC group (1.4 h, 1.43 h and 0.8 h in 25 mg and 50 mg OPC and PLC groups, respectively). OPC was found to be safe and well tolerated.40
Table 2 summarizes the main findings of clinical trials in PD patients.
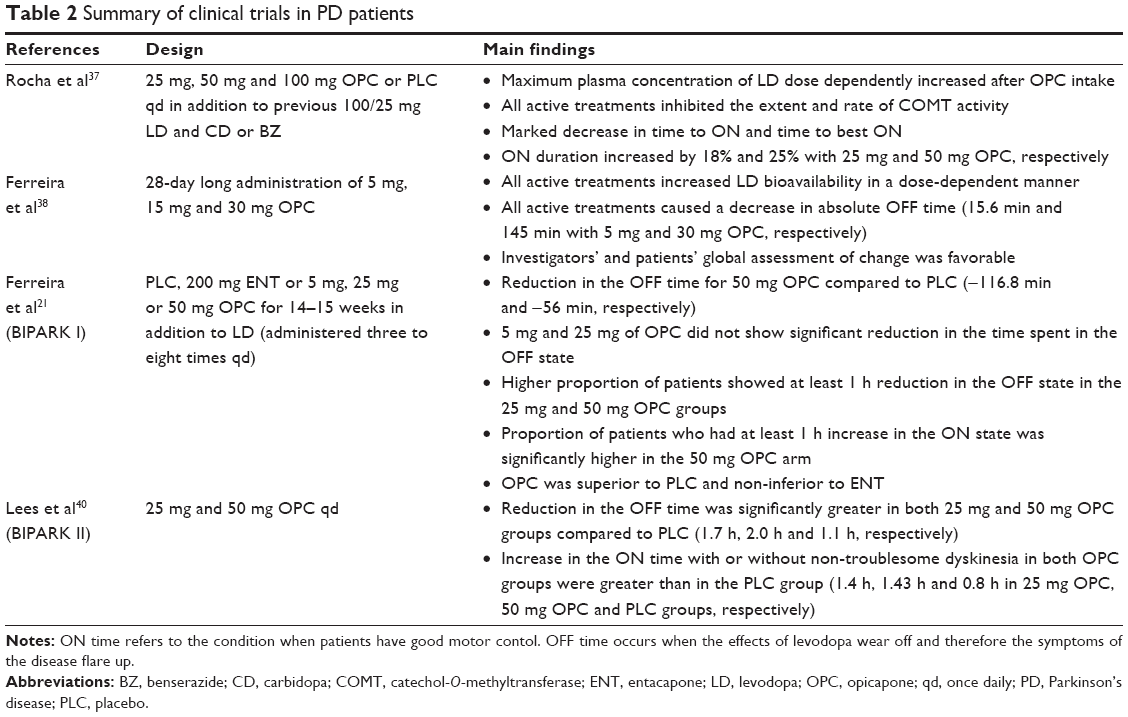 | Table 2 Summary of clinical trials in PD patients |
Pooled analysis and extension studies
Pooled analyses of the BIPARK I and BIPARK II studies have been carried out. Currently, only abstracts are available. The double-blind phases of both studies were pooled and assessed for hepatobiliary adverse events.41 No clinically relevant changes were observed in hepatic enzyme levels. No cases of severe hepatotoxic events were reported.
It was found that OPC improved motor fluctuations regardless of concomitant DA agonist or MAO-B inhibitor therapy at baseline.42 Exploratory post hoc analysis showed that OPC improved motor fluctuations as early as in the first week of treatment (decreased the OFF time by 61 min and 75 min with 25 mg and 50 mg OPC, respectively) and had maximum and sustained beneficial effects after 2–3 weeks.43 It was also demonstrated that OPC treatment resulted in a significant improvement in motor fluctuations regardless of the baseline Hoehn–Yahr stage or disease duration.44 Patients older than 70 years also responded well to the OPC therapy.45 The mean OFF time was reduced by 1.40 h, 1.77 h and 2.26 h with PLC, 25 mg and 50 mg OPC treatment, respectively. Hallucinations (4.6%) and weight loss (4.6%) were more common in this subpopulation of patients, but still, OPC was found to be safe and well tolerated.
One-year open-label extension studies for both BIPARK I and BIPARK II were performed. To date, data are available only as abstracts. All participants received 25 mg OPC for 1 week, and then the investigators could freely adjust doses of both OPC (5 mg, 25 mg and 50 mg) and LD according to clinical response and safety concerns. After the double-blind phase, 495 patients continued the BIPARK I study for 1 year.46,47 The primary end point was the change in the OFF time based on patients’ diaries. In relation to the open-label extension baseline, the mean OFF time decreased by 34 min.46 Participants who previously received PLC and ENT showed a statistically significant reduction in the OFF time (65 min and 39 min, respectively) and increase in the ON time without troublesome dyskinesia (43 min and 46 min, respectively) compared to the open-label baseline. Those who were initially receiving OPC had a sustained beneficial effect regarding both ON and OFF times. Unified Parkinson’s Disease Rating Scale (UPDRS) II scores in ON and OFF states and UPDRS III score at the end point of the open-label phase (−2.2, −4.4 and −7.4 points, respectively) showed improvement compared to previous PLC treatment.47 The most common adverse events in the extension study were related to dopaminergic effects: dyskinesia (14.5%), decrease in the effect of the medication (12.1%), and worsening of parkinsonism (6.7%).45 A total of 11 deaths occurred during the trial; however, none was found to be related to OPC treatment.
A total of 367 patients participated in the 1-year open-label extension of the BIPARK II trial.48 The primary end point was the change in the OFF time compared to baseline based on patients’ diaries. A marked reduction in the OFF time (~1.5 h) was observed compared to the open-label baseline for patients previously taking PLC. The OFF time reductions (12.5%), percentage of the OFF time responders (67.5%) and increase in the ON time with or without non-troublesome dyskinesia (~1.7 h) were consistent with the results of the double-blind phase of the trial.48 The most common treatment-related adverse events were dyskinesia (21.5%), worsening symptoms of PD (17%), falls (9.1%), increased level of blood creatine phosphokinase (7.4%), insomnia (5.7%) and orthostatic hypotension (5.4%).49 These adverse events were mild to moderate. A fatal intracerebral hemorrhage occurred after traumatic brain injury in one of the participants. The other four deaths in the trial were unrelated to OPC treatment. One confirmed case of melanoma was reported, and 4% of subjects had impulse control disorders (ICDs). No effects of OPC on suicidality have been shown.49
Pooled analyses of the double-blind and open-label stages of BIPARK I and BIPARK II have been carried out. Currently, only abstracts are available. ICDs were detected in 1.5% of patients, the most common being pathological gambling. ICDs were mild to moderate in intensity.50 No increased risk for suicidality was found.
The results of both BIPARK I and II were pooled and analyzed for ECG abnormalities.51 A total of 3,060 ECGs were examined. Slight, albeit clinically irrelevant, increase in the QRS interval (1.6 ms and 0.9 ms with 25 mg and 50 mg OPC treatment, respectively) was observed.
Table 3 summarizes the results of extension studies and pooled analysis.
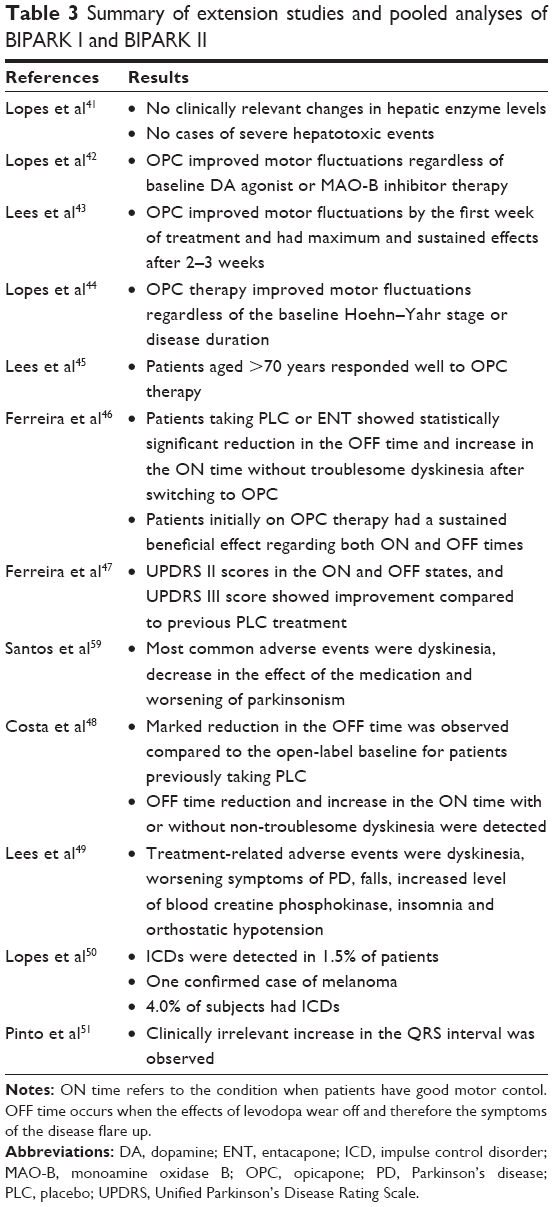 | Table 3 Summary of extension studies and pooled analyses of BIPARK I and BIPARK II |
Discussion
OPC is a promising new peripheral COMT inhibitor that has been approved for adjunctive therapy to LD/DDCI in PD patients showing motor fluctuations. Preclinical and clinical studies have shown that OPC, compared to the previous two COMT inhibitors, is more efficacious, has a significantly longer effect and is less toxic. Therefore, if long-term results will show similar beneficial effects, it should become the first-choice COMT inhibitor. Although ENT is a safe medication, its efficacy is limited and requires frequent administration.52 TCP compared to ENT is more efficacious and has a more sustained effect on COMT activity. However, its safety profile is unfavorable. Three cases of fatal hepatotoxicity were previously reported.53–55 Owing to its hepatotoxic effect, the administration of TCP requires frequent liver function monitoring and therefore it is only recommended to a few selected patients among those showing motor fluctuations and poor motor control.56
The recommended dose for OPC is 50 mg once daily taken at bedtime at least 1 h before or after intake of LD therapy.57 Since it has >24 h long COMT inhibitory effect, it can be expected that coadministration of this novel drug to LD/DDCI medications will result in better control of motor and maybe even nonmotor symptoms of PD patients. It is also possible that the required doses for LD/DDCI medications can be reduced due to the sustained effects of OPC and should allow for better compliance from the patients.
OPC therapy is thus far only recommended for patients who have been taking LD therapy for years and are inevitably showing signs of motor fluctuations. In the future, it would be interesting to see the effects of OPC treatment before the occurrence of motor fluctuations. In the STRIDE-PD study, early initiation of ENT treatment in addition to LD/CD did not delay the occurrence of dyskinesia compared to LD/CD alone.58 In fact, the time of onset of dyskinesia was shorter and the frequency of dyskinesia was increased in the LD/CD/ENT group compared to the LD/CD arm. The authors hypothesized that these findings were probably due to higher LD dose equivalents in the first group and that continuous dopaminergic stimulation was not achieved. Still, we believe that there is a possibility that OPC can delay the onset of the adverse effects of LD therapy by providing steadier and continuous plasma LD concentrations.
Acknowledgment
This work was supported by the project GINOP-2.3.2-15-2016-00034, the Hungarian Brain Research Program NAP (Grant Nos KTIA_13_NAP-A-III/9 and KTIA_13_NAP-A-II/17) and the MTA-SZTE Neuroscience Research Group of the Hungarian Academy of Sciences and the University of Szeged.
Disclosure
The authors report no conflicts of interest in this work.
References
Samii A, Nutt JG, Ransom BR. Parkinson’s disease. Lancet. 2004;363(9423):1783–1793. | ||
Lewitt PA. Levodopa for the treatment of Parkinson’s disease. N Engl J Med. 2008;359(23):2468–2476. | ||
Zádori D, Klivényi P, Toldi J, Fülöp F, Vécsei L. Kynurenines in Parkinson’s disease: therapeutic perspectives. J Neural Transm (Vienna). 2012;119(2):275–283. | ||
Klivényi P, Vécsei L. Novel therapeutic strategies in Parkinson’s disease. Eur J Clin Pharmacol. 2010;66(2):119–125. | ||
Zádori D, Szalárdy L, Toldi J, Fülöp F, Klivényi P, Vécsei L. Some molecular mechanisms of dopaminergic and glutamatergic dysfunctioning in Parkinson’s disease. J Neural Transm (Vienna). 2013;120(4):673–681. | ||
Dézsi L, Vécsei L. Safinamide for the treatment of Parkinson’s disease. Expert Opin Investig Drugs. 2014;23(5):729–742. | ||
Obál I, Majláth Z, Toldi J, Vécsei L. Mental disturbances in Parkinson’s disease and related disorders: the role of excitotoxins. J Parkinsons Dis. 2014;4(2):139–150. | ||
Rascol O, Perez-Lloret S, Ferreira JJ. New treatments for levodopa-induced motor complications. Mov Disord. 2015;30(11):1451–1460. | ||
Devos D, Moreau C. Opicapone for motor fluctuations in Parkinson’s disease. Lancet Neurol. 2016;15(2):127–128. | ||
Olanow CW, Schapira AH. Therapeutic prospects for Parkinson disease. Ann Neurol. 2013;74(3):337–347. | ||
Weinshilboum RM, Otterness DM, Szumlanski CL. Methylation pharmacogenetics: catechol O-methyltransferase, thiopurine methyltransferase, and histamine N-methyltransferase. Annu Rev Pharmacol Toxicol. 1999;39:19–52. | ||
Hauser RA. Levodopa: past, present, and future. Eur Neurol. 2009;62(1):1–8. | ||
Kaakkola S. Clinical pharmacology, therapeutic use and potential of COMT inhibitors in Parkinson’s disease. Drugs. 2000;59(6):1233–1250. | ||
Khor SP, Hsu A. The pharmacokinetics and pharmacodynamics of levodopa in the treatment of Parkinson’s disease. Curr Clin Pharmacol. 2007;2(3):234–243. | ||
Gomes P, Soares-da-Silva P. Interaction between L-DOPA and 3-O-methyl-L-DOPA for transport in immortalised rat capillary cerebral endothelial cells. Neuropharmacology. 1999;38(9):1371–1380. | ||
Nutt JG, Woodward WR, Gancher ST, Merrick D. 3-O-methyldopa and the response to levodopa in Parkinson’s disease. Ann Neurol. 1987;21(6):584–588. | ||
Gervas JJ, Muradás V, Bazán E, Aguado EG, de Yébenes JG. Effects of 3-OM-dopa on monoamine metabolism in rat brain. Neurology. 1983;33(3):278–282. | ||
Nissinen E, Mannisto PT. Biochemistry and pharmacology of catechol-O-methyltransferase inhibitors. Int Rev Neurobiol. 2010;95:73–118. | ||
Goncalves D, Alves G, Soares-da-Silva P, Falcão A. Bioanalytical chromatographic methods for the determination of catechol-O-methyltransferase inhibitors in rodents and human samples: a review. Anal Chim Acta. 2012;710:17–32. | ||
Scott LJ. Opicapone: a review in Parkinson’s disease. Drugs. 2016;76(13):1293–1300. | ||
Ferreira JJ, Lees A, Rocha JF, et al. Opicapone as an adjunct to levodopa in patients with Parkinson’s disease and end-of-dose motor fluctuations: a randomised, double-blind, controlled trial. Lancet Neurol. 2016;15(2):154–165. | ||
Kiss LE, Ferreira HS, Torrão L, et al. Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase. J Med Chem. 2010;53(8):3396–3411. | ||
Rocha JF, Santos A, Falcão A, et al. Effect of moderate liver impairment on the pharmacokinetics of opicapone. Eur J Clin Pharmacol. 2014;70(3):279–286. | ||
Bicker J, Alves G, Fortuna A, Soares-da-Silva P, Falcão A. A new PAMPA model using an in-house brain lipid extract for screening the blood-brain barrier permeability of drug candidates. Int J Pharm. 2016;501(1–2):102–111. | ||
Karhunen T, Tilgmann C, Ulmanen I, Julkunen I, Panula P. Distribution of catechol-O-methyltransferase enzyme in rat tissues. J Histochem Cytochem. 1994;42(8):1079–1090. | ||
Palma PN, Bonifácio MJ, Loureiro AI, Soares-da-Silva P. Computation of the binding affinities of catechol-O-methyltransferase inhibitors: multisubstrate relative free energy calculations. J Comput Chem. 2012;33(9):970–986. | ||
Almeida L, Rocha JF, Falcão A, et al. Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel catechol-O-methyltransferase inhibitor, in healthy subjects: prediction of slow enzyme-inhibitor complex dissociation of a short-living and very long-acting inhibitor. Clin Pharmacokinet. 2013;52(2):139–151. | ||
Rocha JF, Almeida L, Falcão A, et al. Opicapone: a short lived and very long acting novel catechol-O-methyltransferase inhibitor following multiple dose administration in healthy subjects. Br J Clin Pharmacol. 2013;76(5):763–775. | ||
Pinto R, l’Hostis P, Patat A, et al. Evaluation of opicapone on cardiac repolarization in a thorough QT/QTc study. Clin Pharmacol Drug Dev. 2015;4(6):454–462. | ||
Falcão A, Rocha JF, Santos A, et al. Opicapone pharmacokinetics and pharmacodynamics comparison between healthy Japanese and matched white subjects. Clin Pharmacol Drug Dev. 2016;5(2):150–161. | ||
Bonifácio MJ, Torrão L, Loureiro AI, Palma PN, Wright LC, Soares-da-Silva P. Pharmacological profile of opicapone, a third-generation nitrocatechol catechol-O-methyl transferase inhibitor, in the rat. Br J Pharmacol. 2015;172(7):1739–1752. | ||
Bonifácio MJ, Torrao L, Loureiro AI, Wright LC, Soares-da-Silva P. Opicapone: characterization of a novel peripheral long-acting catechol-O-methyltransferase inhibitor. Parkinsonism Relat Disord. 2012;18(suppl 2):S125. | ||
Bonifácio MJ, Sutcliffe JS, Torrão L, Wright LC, Soares-da-Silva P. Brain and peripheral pharmacokinetics of levodopa in the cynomolgus monkey following administration of opicapone, a third generation nitrocatechol COMT inhibitor. Neuropharmacology. 2014;77:334–341. | ||
Dingemanse J, Jorga KM, Schmitt M, et al. Integrated pharmacokinetics and pharmacodynamics of the novel catechol-O-methyltransferase inhibitor tolcapone during first administration to humans. Clin Pharmacol Ther. 1995;57(5):508–517. | ||
Keränen T, Gordin A, Karlsson M, et al. Inhibition of soluble catechol-O-methyltransferase and single-dose pharmacokinetics after oral and intravenous administration of entacapone. Eur J Clin Pharmacol. 1994;46(2):151–157. | ||
Rocha JF, Falcão A, Santos A, et al. Effect of opicapone and entacapone upon levodopa pharmacokinetics during three daily levodopa administrations. Eur J Clin Pharmacol. 2014;70(9):1059–1071. | ||
Rocha JF, Ferreira JJ, Falcão A, et al. Effect of 3 single-dose regimens of opicapone on levodopa pharmacokinetics, catechol-O-methyltransferase activity and motor response in patients with Parkinson disease. Clin Pharmacol Drug Dev. 2016;5(3):232–240. | ||
Ferreira JJ, Rocha JF, Falcão A, et al. Effect of opicapone on levodopa pharmacokinetics, catechol-O-methyltransferase activity and motor fluctuations in patients with Parkinson’s disease. Eur J Neurol. 2015;22(5):815–825. | ||
Deane KH, Spieker S, Clarke CE. Catechol-O-methyltransferase inhibitors for levodopa-induced complications in Parkinson’s disease. Cochrane Database Syst Rev. 2004;4:CD004554. | ||
Lees A, Ferreira JJ, Costa R, et al. Efficacy and safety of opicapone, a new COMT-inhibitor, for the treatment of motor fluctuations in Parkinson’s disease patients: BIPARK-II study [abstract no. 1038]. J Neurol Sci. 2013;333(suppl 1):e116. | ||
Lopes N, Ferreira J, Lees A, et al. Hepatic safety of opicapone in Parkinson’s disease patients. Mov Disord. 2015;30(suppl 1):S101. | ||
Lopes N, Ferreira J, Lees A, et al. Exploratory efficacy of opicapone in combination with dopamine agonists or MAO-B inhibitors on the treatment of motor fluctuations in Parkinson’s disease. Mov Disord. 2015;30(suppl 1):S101. | ||
Lees A, Ferreira J, Reichmann H, et al. Onset and stabilization of treatment effects in fluctuating Parkinson’s disease patients: exploratory by-week efficacy analysis of pooled phase III studies. Mov Disord. 2016;31(suppl 2):S642. | ||
Lopes N, Ferreira J, Lees A, et al. Exploratory efficacy of opicapone in fluctuating Parkinson’s disease patients at different stages of symptom progression. Mov Disord. 2016;31(suppl 2):S642. | ||
Lees A, Ferreira J, Lopes N, et al. Efficacy and safety of opicapone in patients over 70 years with Parkinson’s disease and motor fluctuations. Mov Disord. 2015;30(suppl 1):S99. | ||
Ferreira J, Lees A, Tolosa E, et al. Switching double-blind opicapone, entacapone or placebo to open-label opicapone: efficacy results of the 1-year extension of study BIPARK I. Mov Disord. 2016;31(suppl 2):S633. | ||
Ferreira J, Lees A, Rascol O, et al. Activities of daily living and motor scores of the UPDRS in fluctuating Parkinson’s disease treated with opicapone. Mov Disord. 2016;31(suppl 2):S643. | ||
Costa R, Oliveira C, Pinto R, et al. One-year open-label efficacy and safety of opicapone in Parkinson’s disease BIPARK-II study. Mov Disord. 2014;29(suppl 1):S233. | ||
Lees A, Ferreira J, Costa R, et al. 1-year safety of opicapone in patients with Parkinson’s disease and motor fluctuations. Mov Disord. 2015;30(suppl 1):S99. | ||
Lopes N, Ferreira J, Lees A, et al. Evaluation of impulse control disorders in fluctuating Parkinson’s disease patients under opicapone treatment. Mov Disord. 2016;31(suppl 2):S643. | ||
Pinto R, Vaz-da-Silva M, Lopes N, et al. Cardiac safety of opicapone in patients with Parkinson’s disease: analysis of the centralized phase III ECG dataset. Mov Disord. 2015;30(suppl 1):S112. | ||
Dingemanse J. Issues important for rational COMT inhibition. Neurology. 2000;55(11 suppl 4):S24–S27. Discussion S28–S32. | ||
Keating GM, Lyseng-Williamson KA. Tolcapone: a review of its use in the management of Parkinson’s disease. CNS Drugs. 2005;19(2):165–184. | ||
Assal F, Spahr L, Hadengue A, Rubbia-Brandt L, Burkhard PR. Tolcapone and fulminant hepatitis. Lancet. 1998;352(9132):958. | ||
Colosimo C. The rise and fall of tolcapone. J Neurol. 1999;246(10):880–882. | ||
Movement Disorder Society. Management of Parkinson’s disease: an evidence-based review. Mov Disord. 2002;17(suppl 4):S1–S166. | ||
European Medicines Agency [homepage on the Internet]. Ongentys 25 mg Hard Capsules: Summary of Product Characteristics. 2016. Available from: http://www.ema.europa.eu. Accessed October 12, 2016. | ||
Stocchi F, Rascol O, Kieburtz K, et al. Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: the STRIDE-PD study. Ann Neurol. 2010;68(1):18–27. | ||
Santos A, Ferreira J, Lees A, et al. Safety of opicapone in fluctuating Parkinson’s disease patients: results of the 1-year extension of study BIPARK I. Mov Disord. 2016;31(suppl 2):641. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.