")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Spotlight on olaparib in the treatment of BRCA-mutated ovarian cancer: design, development and place in therapy
Authors Lorusso D, Tripodi E, Maltese G, Lepori S, Sabatucci I, Bogani G , Raspagliesi F
Received 2 August 2017
Accepted for publication 7 February 2018
Published 29 May 2018 Volume 2018:12 Pages 1501—1509
DOI https://doi.org/10.2147/DDDT.S124447
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sukesh Voruganti
Domenica Lorusso, Elisa Tripodi, Giuseppa Maltese, Stefano Lepori, Ilaria Sabatucci, Giorgio Bogani, Francesco Raspagliesi
Gynecologic Oncology Unit, Fondazione IRCCS, Istituto Nazionale dei Tumori, Milan, Italy
Abstract: Epithelial ovarian cancer is the sixth most common cancer among women worldwide and the first cause of death among gynecological malignancies. Most of the patients present recurrent disease and unfortunately cannot be cured. The unsatisfactory results obtained with salvage chemotherapy have elicited investigators to search for novel biological agents capable of achieving a better control of the disease. In the setting of homologous recombination deficiency, the DNA errors that occur cannot be accurately repaired, and the treatment with poly(ADP-ribose) polymerase (PARP) inhibition results in definitive cell death in a process called synthetic lethality. As a result of two positive clinical trials, Olaparib was approved in 2014 by U.S. Food and Drug Administration and European Medicines Agency as the first-in-class PARP inhibitor. Olaparib is effective and well tolerated in homologous recombination deficient patients. Several studies with Olaparib have been conducted in the recurrent setting either as maintenance in platinum-responsive patients or as a single agent. Ongoing trials are focused on the use of olaparib as maintenance in the first-line ovarian cancer setting alone or in combination with antiangiogenic agents. Future perspectives will probably investigate the association of olaparib with novel agents as check-point inhibitors and PI3K-AKT inhibitors. The PARP inhibitor era is just at the beginning.
Keywords: olaparib, ovarian cancer, PARP inhibitors, homologous recombination deficiency, BRCA mutation
Introduction
Epithelial ovarian cancer (EOC) is the sixth most common cancer among women worldwide and the first cause of death among gynecological malignancies.1
The standard management of early stage disease consists of comprehensive staging surgery, followed by adjuvant carboplatin (CBDCA) or CBDCA–paclitaxel (PTX) chemotherapy in high-risk cases.2,3 Cytoreductive surgery followed by PTX–CBDCA chemotherapy is the backbone of treatment for advanced EOC, whereas neoadjuvant chemotherapy followed by interval debulking surgery is indicated for women with poor clinical conditions or with a great amount of disease suggesting a low likelihood of obtaining an optimal cytoreduction (residual disease [RD] 0 or <1 cm).4
For advanced disease (FIGO stage IIIB–IV), bevacizumab, a humanized anti-VEGF monoclonal antibody, has been licensed by the European Medicines Agency (EMA) in combination with carboplatin and paclitaxel and in maintenance at the dose of 15 mg/kg for 15 months on the basis of two randomized clinical trials (GOG-218 and ICON-7) reporting that the combination of bevacizumab with chemotherapy translates into an increase in progression-free survival (PFS) without any differences in overall survival (OS).5,6
Approximately 13%–31% of patients with early EOC and 75%–80% of those with advanced disease relapse after a median interval of 11–29 months and 18–24 months, respectively.7
Patients with recurrent EOC receive second-line chemotherapy, mainly dependent on platinum-free interval, persistent toxicities, and the type of treatment previously received. Sequential single agents, such as weekly PTX, pegylated liposomal doxorubicin (PLD), and gemcitabine (GEM), are suggested for platinum-resistant patients;8 on the contrary, CBDCA doublets (ie, CBDCA in combination with PTX, GEM, or PLD), are used in patients with platinum-sensitive disease9–11 and the non-platinum combination of PLD+trabectedin is a therapeutic option for those who have partially platinum-sensitive disease as well as for those who do not fit for platinum rechallenge.12
Moreover, in this setting, two randomized trials reported that the combination of carboplatin–gemcitabine–bevacizumab or carboplatin–paclitaxel–bevacizumab administered until progression of disease, significantly increases PFS with an hazard ratio of 0.4813 with a nonsignificant trend in OS increase (hazard ratio 0.82).14
However, the unsatisfactory results obtained with salvage chemotherapy have elicited investigators to detect novel biological agents capable of achieving a better control of the disease.15 In the last two decades several changes in all fields of ovarian cancer management have occurred, from the diagnosis, to the treatment, to the translational research. Moreover, new drugs have been introduced in the treatment algorithm with the intent to increase the quantity and quality of life of ovarian cancer patients.
Homologous recombination repair defects: role of BRCA genes and PARPs
DNA is continuously subjected to injuries by environmental and endogenous exposures that cause a variety of DNA lesions, including double-strand breaks (DSBs) and single-strand breaks (SSBs).16 DNA repair systems are critical to maintain genomic integrity by allowing cells to replicate and survive.17 Homologous recombination repair (HRR) is the most important instrument of reparation of DSBs. The BRCA1/2 genes, together with several other genes, code proteins that are necessary for this process. When either BRCA1 or BRCA2 is defective, homologous recombination is dysfunctional and the reparation of DSBs is performed through alternative repair mechanisms such as nonhomologous end-joining (NHEJ) and single-strand repair.18,19 SSBs repair involves a variety of mechanisms such as base excision repair (BER) and nucleotide excision repair, all of which are supported by poly(ADP-ribose) polymerases (PARPs).20
PARPs constitute a family of 18 proteins.21 PARP1 and PARP2 are enzymes involved in SSBs and BER, which are activated by DNA damage and facilitate DNA repair.22 PARP1 becomes activated when an SSB occurs and, after binding to the damaged area, increases its catalytic activity and recruits various other proteins to the site of the DNA damage, initiating a repair complex. If a cell is not able to repair SSB before initiating replication, a single break is transformed into a double-strand during replication process.23
Several studies proposed the model of synthetic lethality, a process by which cancer cells are contemporarily targeted by the inactivation of two genes when the deficiency of either gene alone is nonlethal.23,24 This model can be applied to homologous recombination deficient (HRD) cells; in this case, in fact, PARP inhibitors inhibit the repair of DNA SSBs, thus transforming them into DNA DSBs. When homologous recombination is not functional (HRD), as it is in patients with BRCA mutations, the DNA DSBs cannot be repaired and the PARP inhibition ultimately results in cell death, as shown in Figure 1.25
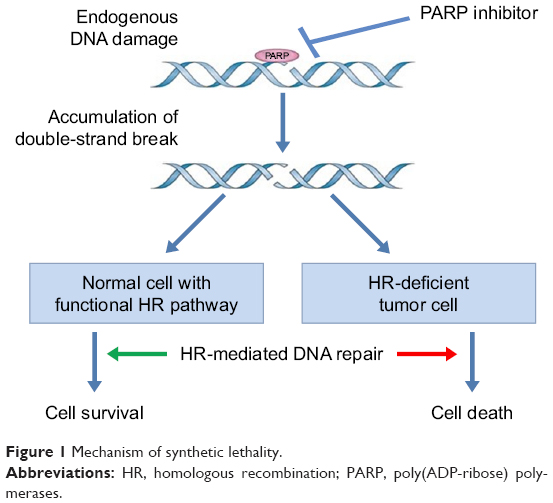 | Figure 1 Mechanism of synthetic lethality. |
This mechanism is an important therapeutic target, not just for PARP inhibitors, but for many chemotherapeutic agents and radiotherapy acting by inducing DNA damages. Platinum analogs, in fact, induce intrastrand and interstrand cross-links the reparation of which depends on nucleotide excision repair and by DSB formation.16 The reported elevated platinum sensitivity of BRCA-mutated EOC to platinum is believed to be related to the HRR defects. BRCA1 and BRCA2 are not the only genes involved in the HRD repair mechanism: other members of the Fanconi anemia family, such as RAD51C, RAD51D, and BRIP126–28 as well as ATM, CHEK1, CHEK2, and CDK12 also confer sensitivity to DNA damage and DNA repair inhibition.24,29–31 These mutations are responsible for what we actually call the BRCAness phenotype,32 a clinical situation in which, even in the absence of identified BRCA mutations, the disease present repeated platinum sensitivity, long natural history, and the potentiality to respond to PARP inhibitors treatment.
Ovarian cancer patients who present a germline or somatic BRCA1 and BRCA2 mutations have a better prognosis compared with BRCA wild-type patients, possibly because of the elevated response rate (RR) to platinum agents, but also to pegylated liposomal doxorubicin and trabectedin. The mutations appear particularly frequent (about 22%–26%) among high-grade serous ovarian cancer (HGSOC),33–37 which are also impaired by the presence of mutations of the tumor suppressor gene p53.38,39 Moreover, molecular analysis of HGSOC by The Cancer Genome Atlas revealed that around 50% of them present HRD.37 After sequencing 316 HGSOCs germline, BRCA1 and BRCA2 mutations were reported in 9% and 8% of patients, respectively, somatic additional mutations in BRCA1/2 in 3%, EMSY in 8%, PTEN in 7%, RAD51C in 3%, ATM/ATR in 2%, and Fanconi anemia genes in 5% of patients.37 The rates of somatic mutations reported in the literature are variable, and currently, the true prevalence of somatic mutations remains unknown; however it has been estimated between 5% and 8% of cases. This implies that for every five ovarian cancer patients with a germline BRCA mutation, there will be one patient with a somatic mutation.40
Sporadic EOCs with HR deficiency not linked to BRCA1-2 mutations have the same biological characteristics and clinical behavior as EOC with either germline or somatic BRCAm (“BRCAness” phenotype),41,42 and, as such, the potentiality to respond to the same drugs.
Olaparib – the first PARP inhibitor approved
The first PARP enzyme was discovered over 50 years ago and the first drug capable of inhibiting PARP, 20 years later.43,44
In 2005, two outstanding studies, performed by two independent research groups, reported that BRCA1/2-deficient cells were 100–1,000-fold more sensitive to PARP inhibitors than wild-type cell,45,46 thus suggesting a particular sensitivity of BRCA-deficient cell lines to PARP inhibitors through a mechanism of “synthetic lethality”. Clinical trials were initiated to explore the clinical activity of PARP inhibitors in HRD-defective tumors.
Olaparib reported 47% RR in a Phase I trial in patients with breast, prostate, and ovarian cancers, harboring BRCA1 or BRCA2 mutations;47 moreover, a correlation between RR and platinum sensitivity was reported (RR 69% vs 45% vs 23% in platinum-sensitive, platinum-resistant, and platinum-refractory patients, respectively).48
Olaparib was approved in 2014 by the U.S. Food and Drug Administration (FDA) and EMA as the first-in-class PARP inhibitor on the basis of two prospective clinical trials (Study 19 and Study 42).49,50
On December 2014, the FDA approved olaparib capsules (Lynparza; AstraZeneca, Wilmington, DE, USA) for the treatment of patients with germline BRCA-mutated (gBRCAm) ovarian cancer who had received at least three previous chemotherapy lines.51
On October 2014, the EMA approved Lynparza as maintenance treatment in platinum-sensitive, BRCA-mutated (germline and/or somatic), high-grade serous EOC who were responding to the last platinum-based chemotherapy.52
Study 19 was an international, randomized, Phase II, double-blind, placebo-controlled trial in patients with relapsed platinum-sensitive, high-grade serous, ovarian, fallopian tube, or primary peritoneal cancers.49 In the study, 265 patients in complete or partial response to the last platinum-based chemotherapy were randomized to receive maintenance with olaparib (n=136) or placebo (n=129) after completion of at least four cycles of chemotherapy. The primary endpoint was PFS assessed by the investigators per Response Evaluation Criteria in Solid Tumors version 1.0 criteria. The trial reported a significant 3.6 months increase in median PFS in patients treated with olaparib with respect to placebo (median PFS 8.4 vs 4.8 months for patients treated with olaparib and placebo, respectively; HR 0.35; p<0.001).49 In the preplanned subgroup analysis, the 136 BRCA-mutated patients derived the greatest clinical benefit from olaparib with a significant improvement in PFS of 6.9 months (median PFS 11.2 vs 4.3 months for olaparib and placebo, respectively; HR 0.18; p<0.0001). The PFS improvement was confirmed by an independent radiological review.53 Study 19 pressed the fast-track EMA approval of olaparib as maintenance in Europe, but the approval was conditioned by the result of the ongoing randomized Phase III study, which were presented at International SGO Congress in March 2017. The confirmatory randomized Phase III SOLO-2 trial investigated olaparib tablets as maintenance in platinum-sensitive, germline BRCA-mutated ovarian cancer patients, who responded to the last platinum-based treatment. Two hundred ninety five eligible patients were randomly assigned to receive olaparib (n=196) or placebo (n=99) at the completion of at least four cycles of platinum-based chemotherapy. PFS was significantly longer in the olaparib arm than in the placebo arm (19.1 vs 5.5 months, respectively; HR 0.30; p<0.0001) with an acceptable toxicity profile and without any detrimental effect on patients’ quality of life.54
Study 42 was a single-arm, Phase II, prospective study on olaparib 400 mg BID in the treatment of patients with gBRCA1/2-mutated cancers; 193 out of 298 patients were heavily pretreated with mean number of 4.3 prior chemotherapy regimens.50 The overall RR was 26.2% in the general population and 31.1% in ovarian cancer setting. Prolonged (≥8 weeks) stable disease was observed in 42% of patients overall and in 40% of ovarian cancer patients.50
Recently, a 34% RR with a median response duration of 7.9 months, in a subgroup analysis of EOC patients who had received three or more previous chemotherapy lines, was reported.49 Study 42 sustained FDA approval of olaparib as a single agent in BRCA-mutated ovarian cancer patients who had received at least three previous chemotherapy lines.
Phase II/III studies of olaparib in ovarian cancer are summarized in Table 1.50,53,55–58
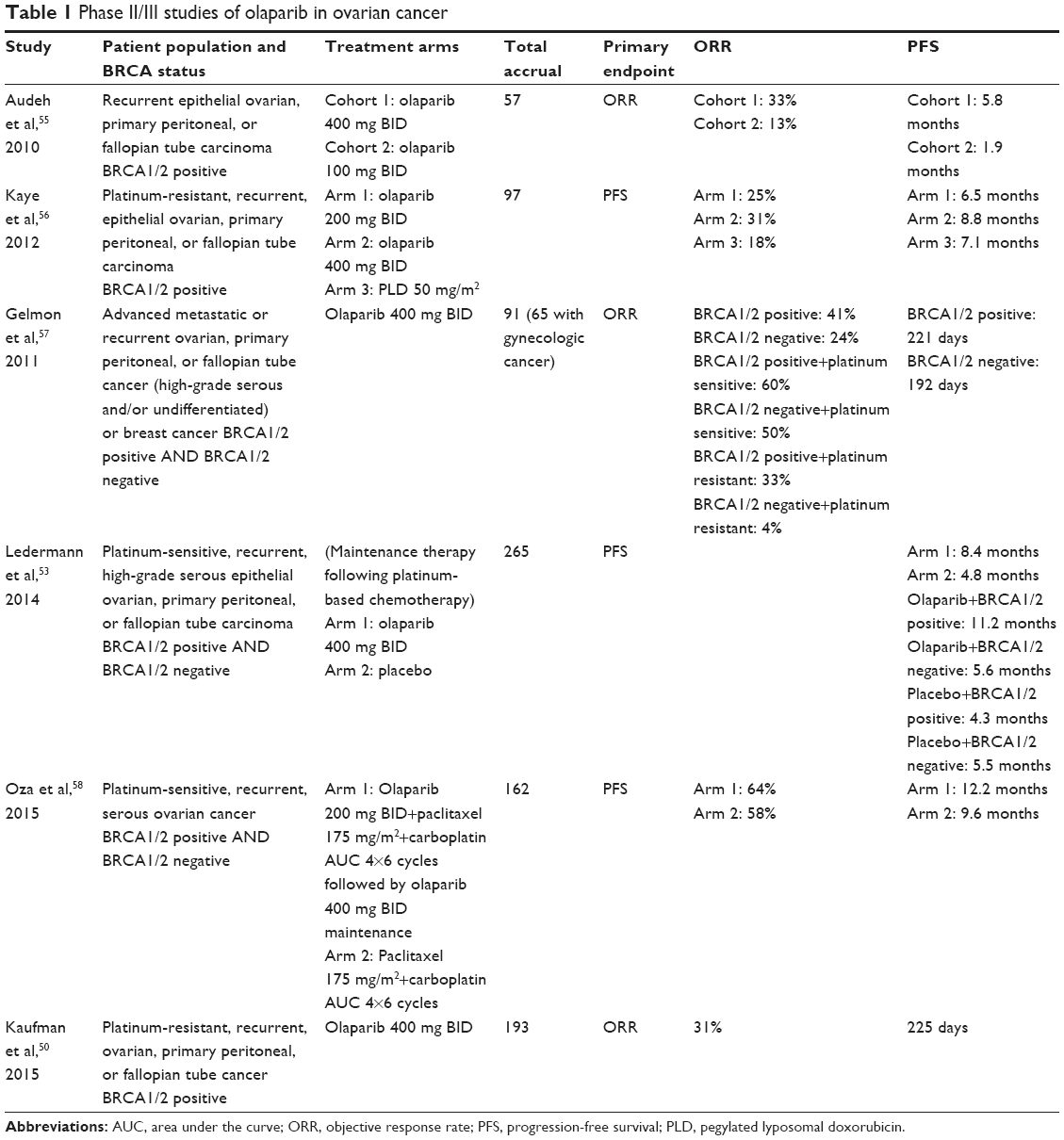 | Table 1 Phase II/III studies of olaparib in ovarian cancer |
Safety profile
Olaparib is generally well tolerated; adverse reactions are typically of mild or moderate severity (Common Terminology Criteria for Adverse Events grade 1 or 2) and, in most cases, are short term in nature, self-limiting, and do not require treatment discontinuation or dose reductions.
In the pivotal Phase II trial (Study 19), the most commonly reported adverse events (AEs) were nausea, fatigue, vomiting, and anemia.49,53 The tolerability profile of olaparib in patients with BRCAm cancer did not differ from that of the overall population.49 Subsequent analyses reported that typically AEs occurred within the first 4–8 weeks of treatment and were mainly grade 1 or 2, generally transient, and managed with supportive care without dose reductions.58,59 In the overall population, serious AEs (SAEs) were reported in 18% of patients receiving olaparib vs 9% of those who received placebo while SAEs causally related to olaparib and placebo in the investigator’s judgment were 5.9% and 0.8%, respectively.53
Treatment interruptions and dose reductions in the olaparib arm were reported in 28% and 23% of patients, respectively.49 After a median follow-up of 5.9 years, no new safety findings appeared.59 The SAEs leading to permanent discontinuation of treatment occurred in 6% and 2% of patients receiving olaparib and placebo, respectively.49,60,61
In the SOLO-2 study, the most commonly reported grade ≥3 AEs were anemia (19% in the olaparib group vs 2% in the placebo group), fatigue (4% vs 2%), and neutropenia (5% vs 4%). SAEs were experienced by 18% of patients in the olaparib group and 8% of patients in the placebo group, respectively. One patient in the olaparib group had an acute myeloid leukemia with death as outcome.54
In Study 42, grade 3 AEs were reported in 54.4% of patients (anemia and fatigue were the most common); in 30.9% of cases, they were considered drug related. SAEs were seen in 30.1% of patients; in about 10.0% they were considered causally related to olaparib. Nine patients died as a result of AEs; in 3.7% of patients the AEs led to treatment discontinuation and in 40.3% caused drug dose modification.50 The most frequently reported AEs in the trials involving olaparib are reported in Table 2.
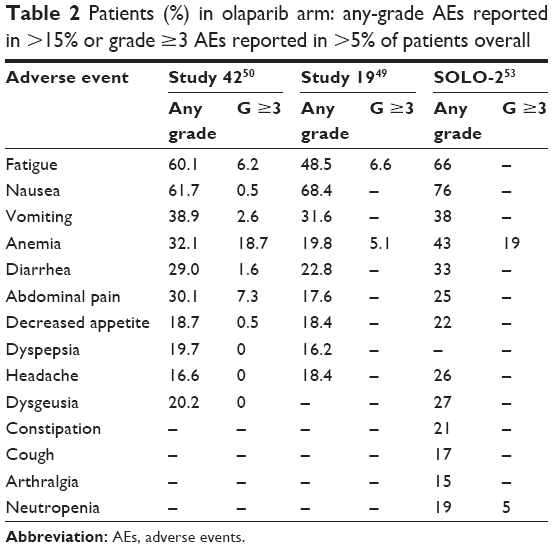 | Table 2 Patients (%) in olaparib arm: any-grade AEs reported in >15% or grade ≥3 AEs reported in >5% of patients overall |
Olaparib as maintenance or in combination with chemotherapy?
A synergism between PARP inhibitors and DNA-damaging agents such as cisplatin, carboplatin, or cyclophosphamide has been described.62,63 Thus, the combination of olaparib with other chemotherapy agents appears promising. In a randomized, open-label, Phase II study, patients with platinum-sensitive recurrent HGSOC were treated with carboplatin and paclitaxel in combination and maintenance with olaparib or chemotherapy alone. PFS was significantly increased in the experimental arm (12.2 months vs 9.6 months; p=0.0012), and the improvement was larger in patients with BRCA mutations. The increased toxicity reported in the combination arm and the shape of the curves which diverged only in the maintenance phase seem to suggest no additional benefit of the combination vs the maintenance only.58
Olaparib: future perspectives
In an attempt to move toward the first-line treatment in ovarian cancer patients, the recently concluded SOLO-1 trial was developed. SOLO-1 trial is a randomized Phase III study with olaparib as maintenance at the completion of first-line platinum–paclitaxel chemotherapy in FIGO stage III–IV, BRCA-mutated ovarian cancer patients. The results are awaited for Q2 2018.
The combination of PARP inhibitors with other molecular-targeted agents has been explored in clinical trials.
A synergism between PARP inhibitors and antiangiogenic agents has been reported,64 possibly due to the downregulation of DNA repair mechanism by antiangiogenic agents. A significant increase in PFS (17.7 vs 9.0 months; p=0.005) and RR (79.6% vs 47.8%; p=0.002) has been reported with the combination of olaparib plus cediranib vs olaparib alone in recurrent, platinum-sensitive, high-grade ovarian tumors.65
A Phase I study was performed combining olaparib and bevacizumab; patients with advanced cancers received increasing doses of olaparib (100, 200, and 400 mg BID, capsule formulation) in combination with bevacizumab 10 mg/kg IV every 2 weeks.66 A total of 12 patients were enrolled and the authors concluded that the combination of olaparib 400 mg BID and bevacizumab 10 mg/kg was well tolerated and it represented the maximum tolerated dose for future trials.66
PAOLA-1 is an ongoing ENGOT/GCIG Phase III trial evaluating olaparib (tablet formulation) vs placebo in combination with bevacizumab as maintenance treatment in patients with stage IIIB–IV high-grade serous or endometrioid ovarian cancers treated with standard first-line platinum-based chemotherapy plus bevacizumab.
Another interesting combination with a strong preclinical rationale is the association between PARP inhibitors and PI3K inhibitors. Juvekar et al reported an in vivo synergism between PI3K inhibitor BKM120 and olaparib in BRCA1-mutated breast tumors, thus suggesting an important role of PI3Kα in the DNA damage response.67
In the Phase I study of olaparib and BKM120, patients with either breast or ovarian cancer were enrolled; clinical benefit was observed in both gBRCAm and gBRCAwt patients but the combination required attenuation of the BKM120 dose. Randomized Phase II studies are needed to further define the efficacy of PI3K/PARP inhibitor combinations as compared with a PARP inhibitor alone.68
Michalarea et al recently presented data on the combination of olaparib with AZ5363, an AKT inhibitor.69 Common (>15%) G1-2 toxicities were nausea, vomiting, fatigue, diarrhea, and anemia. Based on tolerability, recommended Phase II dose for the combination was established at 640 mg BID 2/7 AZD+300 mg BID olaparib.69
Recently, the causes of resistance to PARP inhibitors have been investigated: unfortunately, in most of the cases, the resistance is due to unknown mechanisms, in 15% to a BRCA mutation reversion, and in 10% to TP53BP1 gene mutations, which have opposing activity to BRCA1 in preventing DNA resection and promoting NHEJ.70
The future goal of the research will be the identification of biomarkers that can predict the response to PARP inhibitors.
In 2014, Lee et al proposed that tumor FOXO3a expression may represent a predictor of response to the combination therapy of carboplatin plus olaparib in mBRCA patients with ovarian or breast cancer.71
Other preclinical data report that the abundance of PARP may reflect cellular DNA repair deficiencies, thus constituting a universal predictive biomarker for the response to PARP inhibitors.72 PARP binding protein overexpression is reported in pancreatic cancers, where it induces genetic instability and PARP hyperactivation, thus suggesting that it might predict both HRD and sensitivity to PARP inhibition.73
The characterization and real application of such biomarkers in clinical practice need to be verified in prospective trials.
Other PARP inhibitors
Other PARP inhibitors have been developed. Veliparib, a PARP1 and PARP2 inhibitor, reported 20% and 35% RR in platinum-resistant and platinum-sensitive, BRCA-mutated ovarian cancer patients, respectively.62 A Phase III study on veliparib as maintenance therapy in first-line treatment of ovarian cancer is ongoing (NCT02470585).
The ARIEL2 Phase II study tested rucaparib as a single agent in the treatment of BRCA-mutated, BRCA-wild type, and BRCA-like patients selected according to the genome-wide loss of heterozygosity (LOH) next generation sequencing test. RR was 69% vs 39% vs 11% in BRCA-mutated, BRCA-like, and BRCA-wild type ovarian cancer patients.74,75
Based on this study, rucaparib received FDA approval for the treatment of BRCA-mutated ovarian cancer patients (either germline or somatic) who had previously received two or more chemotherapy lines.
ARIEL3 (NCT 01968213) is a Phase III trial designed to evaluate the efficacy of Rucaparib vs placebo as maintenance treatment after platinum-based chemotherapy in women with relapsed, platinum-sensitive, high-grade serous or endometrioid ovarian cancer. Responses to treatment will be analyzed based on homologous recombination (HR) status of tumor samples evaluated by LOH test. The study has been recently completed and the results were presented at ESMO 2017. The most robust clinical outcomes were observed among patients with a germline or somatic BRCA mutation. The median PFS for patients treated with rucaparib was 16.6 months (95% CI 13.4–22.9) vs 5.4 months (95% CI 3.4–6.7) in the placebo arm. The median PFS for the HRD patients treated with rucaparib was 13.6 months (95% CI 10.9–16.2) vs 5.4 months (95% CI 5.1–5.6) among those who received placebo. The median PFS of the intention to treat population was 10.8 months (95% CI 8.3–11.4) vs 5.4 months (95% CI 5.3–5.5) for rucaparib- and placebo-treated patients, respectively. The most common grade ≥3 AEs in the rucaparib group were anemia (18.8%) and liver enzyme increase (10.5%).76
Niraparib maintenance treatment showed a significant improvement in PFS with respect to placebo in recurrent, platinum-sensitive, BRCA-mutated, ovarian cancer patients responsive to the last platinum-based treatment (21.0 vs 5.5 months; p<0.001).73,74 Of note, PFS was significantly increased also in patients without germline BRCA mutation (9.3 vs 3.9 months; HR 0.45), thus suggesting that platinum sensitivity, and not BRCA mutation, represents the most performing predictive biomarker for HRD.67 Niraparib received FDA approval (March 2017) for maintenance treatment of platinum-sensitive, recurrent ovarian cancer regardless of BRCA mutations.77–79
Conclusion
Recurrent ovarian cancer represents a challenging clinical situation – cure is almost impossible and prolongation of survival at the prize of an acceptable toxicity and no detrimental effect on quality of life of the patients are the goals of treatments. In this scenario, the availability of new drugs will further contribute to the process of chronicization of the disease. PARP inhibitors represent very interesting drugs interfering with mechanism of DNA repair in patients harboring HRD deficiency (a process called synthetic lethality). The drugs reported outstanding clinical activity with manageable toxicity profile in all the setting of disease where they have been employed. Olaparib is the first-in-class PARP inhibitor approved in Europe as maintenance in BRCA-mutated, platinum-responsive patients and in the USA as a single agent in BRCA-mutated patients who have received at least three previous chemotherapy lines. The future of olaparib is to upgrade into the management of newly diagnosed disease either alone (SOLO-1 trial ongoing) or in combination with bevacizumab (PAOLA-1 trial ongoing) and in non-gBRCA patients in the light of the continuous discovery of several genes involved in BRCA-independent HRD impairment. Other interesting combinations are with immunotherapy and PI3K-AKT inhibitors. The exciting sensation is that these new compounds are here to stay and to change the natural history of disease at least in a subset of patients – actually 15% of patients initially enrolled in Study 19 are still receiving olaparib after >5 years. The PARP inhibitor era is just at the beginning.
Disclosure
Francesco Raspagliesi was the principal investigator of the SOLO-1 trial. Domenica Lorusso was the principal investigator of NOVA and ARIEL3 trials. The other authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. | ||
ICON1 Collaborators. Optimal treatment of early-stage ovarian cancer. Ann Oncol. 2014;25:1165–1171. | ||
Trimbos JB. Surgical treatment of early-stage ovarian cancer. Best Pract Res Clin Obstet Gynaecol. 2017;41:60–70. | ||
Wright AA, Bohlke K, Armstrong DK, et al. Neoadjuvant chemotherapy for newly diagnosed, advanced ovarian cancer: society of Gynecologic Oncology and American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2016;34:3460–3473. | ||
Burger RA, Brady MF, Bookman MA, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Eng J Med. 2011;365:2473–2478. | ||
Perren TJ, Swart AM, Pfisterer J, et al. A Phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365:2484–2489. | ||
Gadducci A, Fuso L, Cosio S, et al. Are surveillance procedures of clinical benefit for patients treated for ovarian cancer? A retrospective Italian multicentric study. Int J Gynecol Cancer. 2009;19:367–374. | ||
Pujade-Lauraine E, Hilpert F, Weber B, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. J Clin Oncol. 2014;32:1302–1308. | ||
Parmar MK, Ledermann JA, Colombo N, et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet. 2003;361:2099–2106. | ||
Pfisterer J, Plante M, Vergote I, et al. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J Clin Oncol. 2006;24:4699–4707. | ||
Pujade-Lauraine E, Wagner U, Aavall-Lundqvist E, et al. Pegylated liposomal doxorubicin and carboplatin compared with paclitaxel and carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J Clin Oncol. 2010;28:3323–3329. | ||
Poveda A, Ray-Coquard I, Romero I, Lopez-Guerrero JA, Colombo N. Emerging treatment strategies in recurrent platinum-sensitive ovarian cancer: focus on trabectedin. Cancer Treat Rev. 2014;40:366–375. | ||
Aghajanian C, Blank SV, Goff BA, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30(17):2039–2045. | ||
Coleman RL, Brady MF, Herzog TJ, et al. Bevacizumab and paclitaxel–carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18(6):779–791. | ||
Rodriguez-Freixinos V, Mackay HJ, Karakasis K, Oza AM. Current and emerging treatment options in the management of advanced ovarian cancer. Expert Opin Pharmacother. 2016;17:1063–1076. | ||
Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. | ||
Bouwman P, Jonkers J. The effects of deregulated DNA damage signaling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12:587–598. | ||
Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7(4):a016600. | ||
Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A. 2011;108:3406–3411. | ||
Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. | ||
Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. | ||
Heale JT, Ball AR, Schmiesing JA, et al. Condensin I interacts with the PARP-1-XRCC1 complex and functions in DNA single-strand break repair. Mol Cell. 2006;21:837–848. | ||
Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5(4):387e93. | ||
Friend SH, Oliff A. Emerging uses for genomic information in drug discovery. N Engl J Med. 1998;338(2):125e6. | ||
Ledermann JA, Drew Y, Kristeleit RS. Homologous recombination deficiency and ovarian cancer. Eur J Cancer. 2016;60:49–58. | ||
Loveday C, Turnbull C, Ramsay E, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011;43:879–882. | ||
Meindl A, Hellebrand H, Wiek C, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42:410–414. | ||
Rafnar T, Gudbjartsson DF, Sulem P, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011;43:1104–1107. | ||
Bajrami I, Frankum JR, Konde A, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74:287–297. | ||
McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. | ||
Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20:764–775. | ||
Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110–120. | ||
Pharoah PD, Easton DF, Stockton DL, Gayther S, Ponder BA. Survival in familial, BRCA1-associated, and BRCA2-associated epithelial ovarian cancer. United Kingdom Coordinating Committee for Cancer Research (UKCCCR) Familial Ovarian Cancer Study Group. Cancer Res. 1999;59:868–871. | ||
Liu J, Cristea MC, Frankel P, et al. Clinical characteristics and outcomes of BRCA-associated ovarian cancer: genotype and survival. Cancer Genet. 2012;205:34–41. | ||
Bolton KL, Chenevix-Trench G, Goh C, et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA. 2012;307:382–390. | ||
Alsop K, Fereday S, Meldrum C, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012;30:2654–2663. | ||
Bell D, Berchuck A, Birrer M, et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. | ||
Vang R, Levine DA, Soslow RA, Zaloudek C, ShihIe M, Kurman RJ. Molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinoma: a re-review of cases lacking TP53 mutations in the cancer genome Atlas Ovarian Study. Int J Gynecol Pathol. 2016;35:48–55. | ||
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8(3):193e204. | ||
Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27:1449–1455. | ||
Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–819. | ||
Murata S, Zhang C, Finch N, Zhang K, Campo L, Breuer EK. Predictors and modulators of synthetic lethality: an update on PARP inhibitors and personalized medicine. Bio Med Res Int. 2016;2016:2346585. | ||
Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963;11:39–43. | ||
Purnell MR, Whish WJ. Novel inhibitors of poly(ADP-ribose) synthetase. Biochem J. 1980;185(3):775–777. | ||
Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. | ||
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. | ||
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. | ||
Fong PC, Yap TA, Boss DS, et al. Poly (ADP) ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–2519. | ||
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366:1382–1392. | ||
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250. | ||
Kim G, Ison G, McKee AE, et al. FDA Approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res. 2015;21(19):4257–4261. | ||
The European Medicines Agency; 2014. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003726/human_med_001831.jsp&mid=WC0b01ac058001d124. Accessed July 15, 2017. | ||
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15:852–861. | ||
Pujade-Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274–1284. | ||
Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–251. | ||
Kaye SB, Lubinski J, Matulonis U, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2mutations and recurrent ovarian cancer. J Clin Oncol. 2012;30:372–379. | ||
Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12:852–861. | ||
Oza AM, Cibula D, Benzaquen AO, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015;16:87–97. | ||
Banerjee S, Ledermann J, Matulonis U, et al. Management of nausea and vomiting during treatment with the capsule (CAP) and tablet (TAB) formulations of the PARP inhibitor Olaparib. Proceedings of European Cancer Congress; 25–29 September 2015, Vienna, Austria; abst 2759. | ||
Matulonis U, Friedlander M, Du Bois A, et al. Frequency, severity and timing of common adverse events (AEs) with maintenance Olaparib in patients (pts) with platinum sensitive relapsed serous ovarian cancer (PSR SOC). J Clin Oncol. 2015;33(15Suppl):5550–5550. | ||
Ledermann JA, Harter P, Gourley C, et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving Olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double- blind, phase 2 trial. Lancet Oncol. 2016;17:1579–1589. | ||
Donawho CK, Luo Y, Luo Y, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–2737. | ||
Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079–17084. | ||
Yadav A, Kumar B, Teknos TN, Kumar P. Sorafenib enhances the antitumor effects of chemoradiation treatment by downregulating ERCC-1 and XRCC-1 DNA repair proteins. Mol Cancer Ther. 2011;10:1241–1251. | ||
Liu JF, Barry WT, Birrer M, et al. Combination cediranib and Olaparib versus Olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014;15:1207–1214. | ||
Dean E, Middleton M, Pwint T, et al. Phase I study to assess the safety and tolerability of Olaparib in combination with bevacizumab in patients with advanced solid tumours. Br J Cancer. 2012;106:468–474. | ||
Juvekar A, Burga LN, Hu H, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2(11):1048–1063. | ||
Matulonis U, Wulf G, Barry W, et al. Phase 1 of oral BKM120 or BYL719 and Olaparib for high-grade serous ovarian cancer or triple-negative breast cancer: final results of the BKM120 plus Olaparib cohort. Cancer Res. 2015;75(15):CT324. | ||
Michalarea V, Lorente D, Lopez J, et al. Accelerated phase I trial of two schedules of the combination of the PARP inhibitor Olaparib and AKT inhibitor AZD5363 using a novel intrapatient dose escalation design in advanced cancer patients. Cancer Res. 2015;75(15):CT323. | ||
Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19(11):1381–1388. | ||
Lee JM, Hays JL, Annunziata CM, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J Natl Cancer Inst. 2014;106(6):dju089. | ||
Michels J, Vitale I, Saparbaev M, Castedo M, Kroemer G. Predictive biomarkers for cancer therapy with PARP inhibitors. Oncogene. 2014;33(30):3894–3907. | ||
Michels J, Vitale I, Galluzzi L, et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res. 2013;73:2271–2280. | ||
McNeish IA, Oza AM, Coleman RL, et al. Results of ARIEL2: a phase 2 trial to prospectively identify ovarian cancer patients likely to respond to rucaparib using tumor genetic analysis. J Clin Oncol. 2015;33(15):5508. | ||
Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75–87. | ||
Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–1961. | ||
Jones P, Wilcoxen K, Rowley M, et al. Niraparib: a poly(ADP-ribose) polymerase (PARP) inhibitor for the treatment of tumors with defective homologous recombination. J Med Chem. 2015;58:3302–3314. | ||
Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375:2154–2164. | ||
Oda K, Tanikawa M, Sone K, Mori-Uchino M, Osuga Y, Fujii T. Recent advances in targeting DNA repair pathways for the treatment of ovarian cancer and their clinical relevance. Int J Clin Oncol. 2017;22(4):611–618. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.