")
Back to Journals » Journal of Inflammation Research » Volume 14
Spotlight on NLRP3 Inflammasome: Role in Pathogenesis and Therapies of Atherosclerosis
Authors Jiang C , Xie S, Yang G, Wang N
Received 21 October 2021
Accepted for publication 3 December 2021
Published 21 December 2021 Volume 2021:14 Pages 7143—7172
DOI https://doi.org/10.2147/JIR.S344730
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Chunteng Jiang,1,2 Santuan Xie,1 Guang Yang,3 Ningning Wang3
1Department of Internal Medicine, The Affiliated Zhongshan Hospital of Dalian University, Dalian, Liaoning, People’s Republic of China; 2Department of Cardiology and Pneumology, University Medical Center of Göttingen, Georg-August-University of Göttingen, Göttingen, Lower Saxony, Germany; 3Department of Food Nutrition and Safety, School of Public Health, Dalian Medical University, Dalian, Liaoning, People’s Republic of China
Correspondence: Guang Yang; Ningning Wang Email [email protected]; [email protected]
Abstract: Inflammation is an intricate biological response of body tissues to detrimental stimuli. Cardiovascular disease (CVD) is the leading cause of death worldwide, and inflammation is well documented to play a role in the development of CVD, especially atherosclerosis (AS). Emerging evidence suggests that activation of the NOD-like receptor (NLR) family and the pyridine-containing domain 3 (NLRP3) inflammasome is instrumental in inflammation and may result in AS. The NLRP3 inflammasome acts as a molecular platform that triggers the activation of caspase-1 and the cleavage of pro-interleukin (IL)-1β, pro-IL-18, and gasdermin D (GSDMD). The cleaved GSDMD forms pores in the cell membrane and initiates pyroptosis, inducing cell death and the discharge of intracellular pro-inflammatory factors. Hence, the NLRP3 inflammasome is a promising target for anti-inflammatory therapy against AS. In this review, we systematically summarized the current understanding of the activation mechanism of NLRP3 inflammasome, and the pathological changes in AS involving NLRP3. We also discussed potential therapeutic strategies targeting NLRP3 inflammasome to combat AS.
Keywords: cardiovascular disease, atherosclerosis, NLRP3 inflammasome, mechanisms, therapeutic strategies
Introduction
Cardiovascular disease (CVD) remains the dominant causal factor of mortality globally in spite of tremendous improvements in therapy and efficacy.1 Atherosclerosis (AS) is a leading cause of CVD, resulting in approximately 50% of deaths throughout the world. Four pathologic changes are considered to be associated with atherogenesis: (1) endothelial injury; (2) deposition of lipoproteins in the vessel wall; (3) adhesion, migration, and differentiation of monocytes into macrophages and formation of foam cells; (4) proliferation and migration of smooth muscle cells (SMCs).2 Notably, inflammatory response of these cells is the common basis for the physiopathologic changes throughout the initiation and progression of AS. It is now generally assumed that inflammatory processes and lipid metabolism work together in the formation of atherosclerotic plaques (Figure 1).3
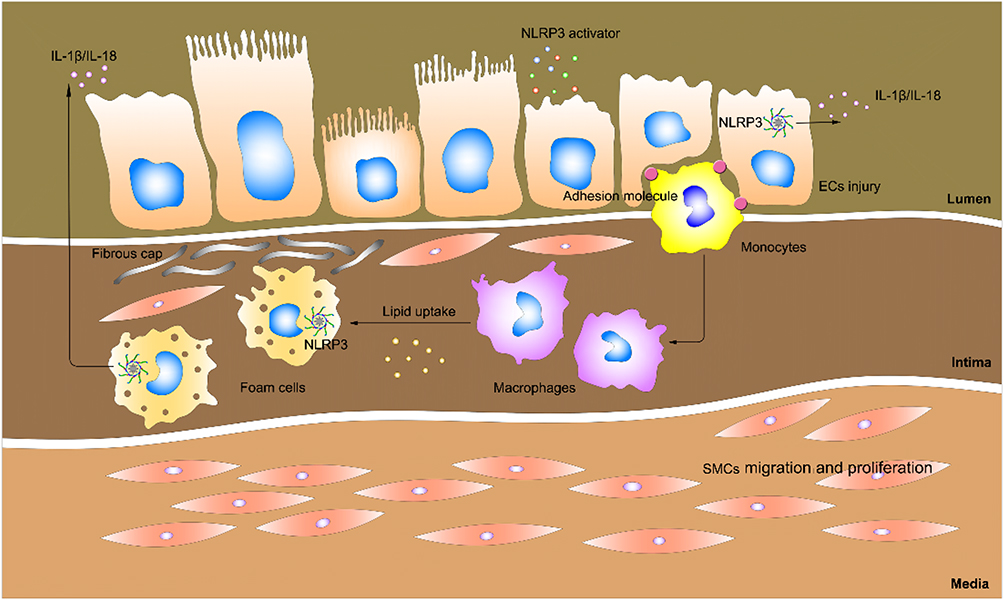 |
Figure 1 Schematic of atherogenesis. Risk factors such as high cholesterol, hyperglycemia, and smoking are contributors of atherogenesis. These activators gain access to the intima via damaged endothelial cells (ECs). Damaged ECs trigger NLRP3 inflammasome activation, IL-1β and IL-18 release, thus further leading to inflammation. On the other hand, damaged ECs express adhesion molecules that capture the monocytes, which get into the intima and differentiate into macrophages. Macrophages ingest lipid and turn into foams cells, which gradually die and release their contents (such as IL-1β and IL-18), giving rise to the inflammatory response. The inflammatory response stimulates the migration and proliferation of smooth muscles cells (SMCs), which aggregate in the plaques to form fibroproliferative lesions. |
Inflammation is a complex physiological and pathological process by which the body struggles to respond to injury and infection, as mediated by constituents of the innate immune system. The innate immune system acts as the first line of defense against non-self and danger signals from microbial invasion and tissue damage, and plays a central role in acute host defense. These danger signals can be recognized by pattern recognition receptors (PRRs), which are extensively expressed on the surface or in the cytosol in numerous types of cells, such as monocytes, macrophages, neutrophils, mast cells, dendritic cells, and natural killer cells. There are at least five groups of PRRs, including Toll-like receptors (TLRs), nucleotide-binding oligomerization domain-like receptors (NLRs), retinoic acid-inducible gene-I-like receptors (RLRs), C-type lectin receptors (CLRs), and absent-in-melanoma (AIM)-like receptors (ALRs).4 These activated PRRs subsequently oligomerize and assemble large multi-subunit complexes to activate signaling cascades that stimulate the recruitment of inflammatory mediators to eliminate pathogens, infected cells or tissues.5,6 Two types of molecular patterns can be identified by PRRs, including pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs).
To date, inflammasomes are considered to be activated by eight members of NLRs [including NLRP1, NLRP2, NLRP3, NLRP6, NLRP7, NLRP12, NLRC4, and NLR family apoptosis inhibitory protein (NAIP)] and AIM2, among which NLRP3 is most widely investigated. It recognizes PAMPs and DAMPs and forms the NLRP3 inflammasome molecular complex, which participates in innate immune response.7 In response to PAMPs or DAMPs, NLRP3 concatenates to caspase-1 or other caspase family members (such as caspase-4/5 and caspase-11) through apoptosis-associated speck-like protein which contains a caspase recruitment domain (ASC) and forms a macromolecular complex-NLRP3 inflammasome. The inflammasome then initiates the release of pro-inflammatory cytokines and launches pyroptotic cell death called pyroptosis. Indeed, hyper-regulated or abnormal NLRP3 inflammasome activity is implicated in the pathogenesis of AS.8,9
Up to now, a plenty of studies demonstrated that many risks and factors can lead to AS via activating NLRP3 inflammasome. However, there is no clear mechanism by which the NLRP3 inflammasome affects AS exactly. Therefore, understanding the mechanisms of NLRP3 inflammasome activation is vital in seeking reliable and effective therapies against AS. In this review, we summarize the current understanding of the molecular mechanisms of NLRP3 inflammasome activation, the role of NLRP3 inflammasome activation in the development of AS, as well as the potential strategies for inhibiting the activation of NLRP3 inflammasome in AS.
Overview of NLRP3 Inflammasome Activation
The NLRP3 inflammasome consists of a sensor (NLRP3), an adaptor (ASC) and an effector (caspase-1). NLRP3 is a macromolecular protein containing an amino-terminal pyrin domain (PYD) for recruitment of the adaptor protein ASC, a central nucleotide-binding domain (NBD) or oligomerization domain (NACHT) for enabling ATP-dependent oligomerization and activation, and a carboxy-terminal leucine-rich repeats (LRRs) motif that serves as the sensor domain.10 ASC is equipped with two protein interaction domains, an amino-terminal PYD and a carboxy-terminal caspase recruitment domain (CARD). The full-length caspase-1 possesses an amino-terminal CARD and two different sizes of catalytic structural domains (p20 and p10). Once stimulated, NLRP3 enables self-oligomerization via homotypic interactions between the NACHT domains. The self-oligomerized NLRP3 recruits ASCs via homotypic PYD-PYD interactions, which instigate the assembly of ASCs into large speckled structures.11 Thereafter, clustered ASCs recruit pro-caspase-1 via CARD-CARD interactions, promoting activation of caspase-1 autocatalysis which controls the maturation of pro-inflammatory cytokines, including pro-interleukin-1β and −18 (pro-IL-1β and −18), by direct cleavage.12 Besides, activated caspase-1 cleaves gasdermin D (GSDMD), which then leads to pore formation and induces cellular lysis and pyroptosis.13,14
Mechanisms of Canonical NLRP3 Inflammasome Activation
Canonical NLRP3 inflammasome activation necessitates two steps, priming (signal 1) and activation (signal 2) (Figure 2). With this dual imperative as a protective mechanism, the activation of inflammatory cells is strictly controlled. The priming process requires recognition of PAMPs and/or DAMPs by PRRs such as TLRs, TL-1R and cytokine receptors [eg, the tumor necrosis factor (TNF) receptor-TNFR1 and TNFR2], followed by activation of nuclear factor-κB (NF-κB), which then leads to transcriptional activation of NLRP3, pro-IL-1β and pro-IL-18.15,16 The activation process results in oligomerization of NLRP3 and assembly of NLRP3, ASC, and pro-caspase-1 to a complex, triggering caspase-1 activation, IL-1β, and IL-18 maturation and release.17 Despite being extensively studied, the mechanism of activation of NLRP3 inflammasome is still in debate so far. Several possible theories have been proposed, including ion fluxes, mitochondrial dysfunction, reactive oxygen species (ROS) overload, release of cathepsin from destabilized lysosomes, and trans-Golgi disintegration.
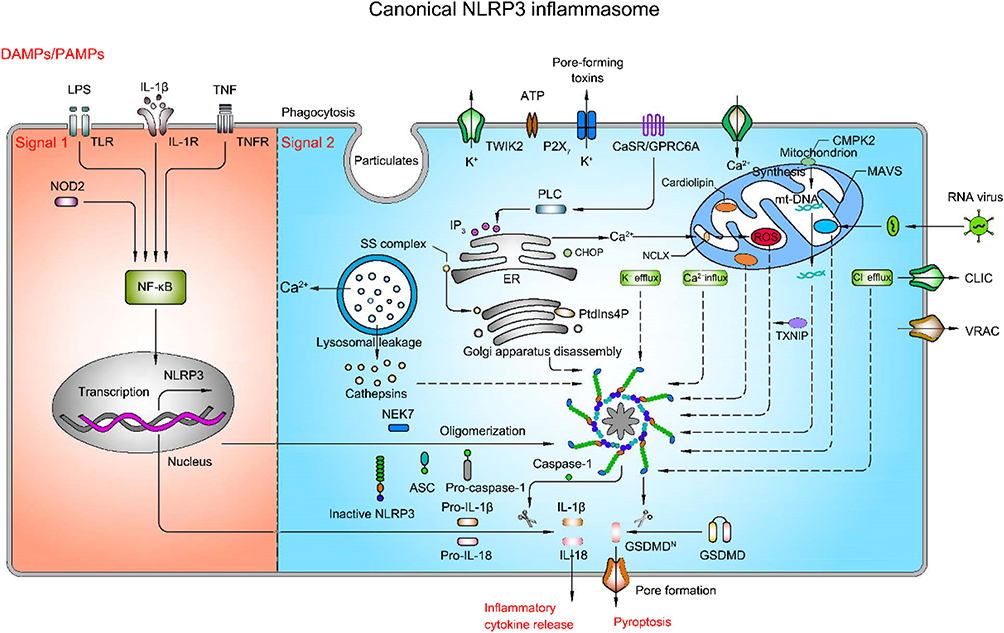 |
Figure 2 Mechanism of activation of canonical NLRP3 inflammasome pathway (priming-signal 1 and activation-signal 2). Priming process (saffron yellow background) is triggered by pattern recognition receptors (PRR), such as TLR4 and NOD2, or cytokine receptors, such as IL-1R and TNFR, and then activate NF-κB, in turn contributing to the transcription and translation of NLRP3, pro-IL-1β and pro-IL-18. Activation process (light blue background) is boosted by pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), such as ATP, particulates and pore-forming toxins, which activate a number of upstream signaling events. These signaling events include ion fluxes, lysosomal leakage, reactive oxygen species (ROS) production, the release of oxidized mitochondrial DNA (mtDNA), and trans-Golgi disintegration. In addition, other specific events activate the NLRP3 inflammasome as well. For instance, RNA virus triggers the NLRP3 inflammasome activation via mitochondrial antiviral signaling protein (MAVS). The translocation of sterol regulatory element-binding proteins (SREBPs)-SREBP cleavage-activating proteins (SCAP) complex (SS complex) from endoplasmic reticulum to Golgi also can initiate NLRP3 inflammasome assembly. Likewise, phosphatidylinositol-4-phosphate (PtdIns4P) on dispersed trans-Golgi network can trigger NLRP3 inflammasome activation. The NLRP3 inflammasome activation leads to the cleavage of caspase-1, which in turn cleaves pro-IL-1β and pro-IL-18 as well as gasdermin D (GSDMD). The N-terminal cleaved GSDMD embeds into the membrane, forming pores and triggering cell swelling and lysis called pyroptosis. |
Ion Fluxes
Ion fluxes [eg, potassium (K+) efflux, calcium (Ca2+) signaling, and chloride (Cl−) efflux] have been identified as vital events in NLRP3 activation.18–21
K+ Efflux
K+ efflux is a prevalent ionic event in NLRP3 inflammasome activation. The intracellular and extracellular K+ concentration gradients are controlled by Na+/K+ pump, a transmembrane ATPase that pumps Na+ out of the cells and simultaneously pumps K+ into the cells, resulting in a relative increase in K+ concentration in the cells. Exposure of Na+/K+ pump to NLRP3 agonists (eg, nigericin, K+/H+ ionophore, and extracellular ATP) breaks the balance and causes a reduction of K+ concentration in cells.18,22,23 It has been reported that ATP is a powerful activator that can reduce the intracellular K+ concentration by around 50%.24 Extracellular ATP activates ATP-gated cation channel P2X purinoceptor 7 (P2X7R) via regulation of the pannexin-1 hemichannel.25 Activated P2X7R triggers K+ efflux and results in NLRP3 inflammasome activation and IL-1β maturation in macrophages and monocytes.23,26
A recent study has shown that TWIK2, a two-pore domain potassium channel (K2P), co-regulates K+ efflux with P2X7R-mediated Ca2+ and Na+ influx, leading to the activation of NLRP3 inflammasome in ATP-induced macrophages.27 Of note, the NLRP3 inflammasome fails to be activated in TWIK2-deficient macrophages upon treatment with nigericin or imiquimod, suggesting that TWIK2 is an indispensable driver of NLRP3 inflammasome activation.27 NEK7, a member of the family of mammalian never-in-mitosis A (NIMA)-related kinase, acts as a critical mediator for NLRP3 inflammasome activation via inducing K+ efflux to facilitate NLRP3 oligomerization and activation.22 Mechanistically, NEK7 interplays with the leucine-rich repeat (LRR) and NACHT structural domains of NLRP3 through its catalytic structural domain to promote inflammasome activation in a manner independent of its kinase activity.22,28 DEAD-box helicase 3 X-linked (DDX3X), a newly identified gene that encodes the stress granule protein, interacts with NLRP3 and drives NLRP3 inflammasome activation by triggering K+ efflux under stress conditions.29 Therefore, a decrease of intracellular K+ concentration is an upstream signal for NLRP3 inflammasome activation. Interestingly, several chemical compounds, such as GB111-NH2, CL097, and imiquimod, are able to trigger NLRP3 inflammasome activation independent of K+ efflux.30,31 Activation of inflammasomes in mouse R258W macrophages carrying the mutant NLRP3 occurs only under LPS stimulation in the absence of K+ efflux.32 Taken together, these findings shed light on the importance of K+ efflux, but not a necessity in NLRP3 inflammasome activation.
Ca2+ Signaling
Intracellular Ca2+ mobilization plays an important role in the assembly and activation of NLRP3 inflammasome evoked by numerous stimuli (such as ATP, nigericin, and particulate matters).33 Inhibition of Ca2+ signaling hampers NLRP3 inflammasome activation, but has no impact on the activation of AIM2 and NLRC4 inflammasomes.19,33 The endoplasmic reticulum (ER) is the main intracellular Ca2+ reservoir and plays an important role in Ca2+ signaling. Previous studies have revealed that inositol 1,4,5-trisphosphate receptor (IP3R), a Ca2+-release channel on the ER, induces the release of Ca2+ and activation of NLRP3 inflammasome in response to extracellular calcium and ATP stimulation.19 Conversely, pharmacological inhibition or knockdown of the IP3R prevents Ca2+ mobilization and NLRP3 inflammasome activation.19 Calcium-sensing receptor (CaSR) is a G-protein coupled receptor with the capacity to sense extracellular calcium concentration. CaSR activates the NLRP3 inflammasome and IL-1β secretion via phospholipase C (PLC), which catalyses IP3R production and leads to Ca2+ release from ER.19,34 Rossol et al have suggested that the release of Ca2+ by necrotic cells mobilizes ER Ca2+ storage and triggers the activation of NLRP3 inflammasome via CaSR and the G protein-coupled calcium-sensing receptor (GPRC6A).35
Another Ca2+ regulator, the C/EPB homologous protein (CHOP; also known as DDIT3), has been confirmed to be involved in the NLRP3 inflammasome activation.33 As revealed in the study of Lebeaupin et al, administration of LPS or tunicamycin results in ER stress and up-regulation of CHOP, while knockdown of Chop prevents the accumulation of active caspase-1 and IL-1β.36 However, the exact role of CHOP in NLRP3 inflammasome activation remains unknown. Additional studies have shown that the NLRP3 inflammasome activation induced by Ca2+ influx can be modulated by the transient receptor potential cation channels TRPM7 and TRPV2 during the process of regulatory volume reduction in macrophages.37
On the other hand, the opening of the cell membrane channel also initiates Ca2+ influx. In general, Ca2+ influx caused by the opening of the cell membrane channel is accompanied by K+ efflux, serving as a counterion to Ca2+ influx. Treatment with potassium on the extracellular side causes sustained calcium conduction from the extracellular side to the cytoplasmic side, rather than a transient calcium flux from intracellular stores, and thereby induces NLRP3 inflammasome activation in ATP-exposed J774A.1 macrophages,38 suggesting that K+ efflux and Ca2+ influx cooperate to activate the assembly of NLRP3 inflammasome. However, contradictory evidence has demonstrated that Ca2+ influx is not pivotal for NLRP3 inflammasome activation, as it occurs after NLRP3 inflammasome and caspase-1 activation.39 Hence, the role of Ca2+ influx in NLRP3 inflammasome activation remains controversial.
Cl− Efflux
Cl− is the second most abundant anion in the body and can be easily taken from salt (sodium chloride). Under physiological conditions, the Cl− concentration on the intracellular side (5–15 mM) is substantially lower than that on the extracellular side (110–130 mM).40 A study has shown that the reduction of extracellular Cl− concentration (from 130 mM to 9 mM) intensifies the maturation and secretion of IL-1β under ATP challenge via P2X7R-caspase-1-IL-1β signaling cascade in bone marrow-derived macrophages (BMDMs).41 However, Green et al have found that the level of extracellular IL-1β does not change under Cl− free conditions (with normal extracellular 5 mM K+), whereas IL-1β secretion is further enhanced in the combined absence of K+ and Cl− in BMDMs,20 indicating the synergistic role of K+ efflux and Cl− efflux in NLRP3 inflammasome activation. These contradictory results may be due to different treatment concentrations of Cl−.
Cl− channels [volume-regulated anion channel (VRAC) and chloride intracellular channel (CLIC)] have been verified to activate NLRP3 inflammasome.42,43 Pharmacological inhibition of VRAC and CLIC blocks Cl− efflux, which then suppresses NLRP3 inflammasome activation.42,43 Besides, silencing the expression of CLIC family members (including Clic1, Clic4, and Clic5) inhibits NLRP3 inflammasome activation and IL-1β secretion when exposed to NLRP3 agonist.43 Mechanistically, inhibition of CLICs prevents NEK7-NLRP3 interactions, suppresses NLRP3-ASC complex formation and ASC oligomerization,43 implying that Cl− efflux mediated by CLICs facilitates NEK-NLRP3 interactions to trigger NLRP3 inflammasome activation.
ROS, Mitochondrial Dysfunction, and mtDNA
The generation of ROS, mitochondrial dysfunction-derived ROS (mtROS) or DNA (mtDNA) by multiple NLRP3 activators is a core mechanism for the assembly and activation of NLRP3 inflammasome. Mitochondria and NADPH oxidases are two major cellular sources of ROS.
The hypothesis that ROS functions as a trigger of NLRP3 activation is proposed by Cruz et al. They have demonstrated that inhibition of NADPH oxidase-derived ROS decreases the expression of caspase-1 and IL-1β in ATP-treated alveolar macrophages.44 Knockdown of the p22phox (a subunit of NADPH oxidase) markedly diminishes IL-1β expression in asbestos or monosodium urate (MSU)-treated THP1 cells, suggesting that ROS over-generation is an upstream event of NLRP3 inflammasome activation. Of note, ROS habitually promotes NLRP3 inflammasome activation in conjunction with other upstream events. ROS has been reported to trigger Ca2+ influx via transient receptor potential melastatin 2 (TRPM2), and ultimately activate the NLRP3 inflammasome in liposomes-treated THP-1 cells.45 Tang et al have suggested that in the presence of NLRP3 agonists, ROS arouses the translocation of CLICs to the plasma membrane, which induces Cl− efflux and promotes NEK7-NLRP3 interaction, thereby activating NLRP3 inflammasome.43
Mitochondrial dysfunction triggers the activation of complex I and complex III of the electron transport chain, resulting in the production of mtROS. Both the mitochondrial complex I inhibitor rotenone and tebufenpyrad, and complex III inhibitor antimycin A can potentiate the NLRP3 induction, ASC speck formation and pro-IL-1β processing to IL-1β when exposed to NLRP3 agonists.46,47 Likewise, mtDNA has been regarded as a DAMP for NLRP3 inflammasome activation.48 In the event of oxidative stress, mtROS and Ca2+ cooperate to open mitochondrial permeability transition (MPT) pores,49 and the release of mtDNA into the cytoplasm is believed to be dependent on the MPT pores and mtROS.50 In particular, oxidized mtDNA can be released into the cytosol where it binds to and activates the NLRP3 inflammasome in a mtROS-dependent manner upon impairment of mitochondria.50,51 Cytidine/uridine monophosphate kinase 2 (CMPK2), a rate-limiting enzyme, provides deoxyribonucleotides for mtDNA synthesis. It has been shown that CMPK2-dependent mtDNA synthesis is required for the production of oxidized mtDNA fragments after exposure to NLRP3 activators, highlighting the importance of CMPK2 in NLRP3 inflammasome activation through mtDNA synthesis. Conversely, scavenging ROS/mtROS [eg, N-acetyl-L-cysteine (NAC), mito-apocynin and MitoQ] or inhibiting mtDNA release into cytoplasm can restrain NLRP3 inflammasome activation.46,52,53 These studies imply that mtROS and mtDNA produced by mitochondrial impairment is potential inducer of the NLRP3 inflammasome-mediated inflammatory response.
NCLX, the mitochondrial Na+/Ca2+ exchanger, undertakes an important role in the balance between Ca2+ influx and efflux across the mitochondrial inner membrane. In high glucose-induced ECs, the expression of NCLX increases, and silencing of NCLX enhances mitochondrial Ca2+ influx and increases ROS generation, thus leading to NLRP3 inflammasome activation,54 which sheds light on the self-protective role of NCLX in mitochondrial maintenance and stability during NLRP3 inflammasome activation. Moreover, as shown by Zhou et al, treatment with uric acid crystals contributes to the dissociation of thioredoxin-interacting protein (TXNIP) from thioredoxin in a ROS-sensitive manner, which subsequently binds to NLRP3 and results in activation of the NLRP3 inflammasome.55 These observations suggest the strong correlation between TXNIP and ROS in NLRP3 inflammasome activation.
A study discovered a novel mechanism for NLRP3 inflammasome activation; the mitochondria-specific phospholipid cardiolipin is proposed to be a necessary mediator for oxazolidinone antibiotic-induced NLRP3 inflammasome activation via directly binding to NLRP3 in a ROS-independent manner, and functions as a mitochondrial butting site for NLRP3 inflammasome assembly.56 A very fresh study suggested that the absence of IL-1α resulted in a decrease in caspase-1 activity and a consequent decrease in IL-1β and IL-18 secretion, concurrent with reduced mitochondrial damage; moreover, LPS priming of macrophages led to translocation of pro-IL-1α to mitochondria, where it directly interacted with mitochondrial cardiolipin.57 In addition, computational modeling revealed a likely cardiolipin binding motif in pro-IL-1α, similar to that found in LC3b. Further experiments demonstrated that binding of pro-IL-1α to cardiolipin in activated macrophages may interrupt cardiolipin-LC3b-dependent mitophagy, causing enhanced NLRP3 inflammasome activation and IL-1β production.57 Mitochondrial antiviral signaling protein (MAVS), an adaptor protein in the RNA sensing pathway, has been proven to play a critical role in the activation of the NLRP3 inflammasome during RNA virus infection and after stimulation with the synthetic RNA polyinosinic–polycytidylic acid.7,58 MAVS urges the recruitment of NLRP3 to mitochondria and enhances its oligomerization and activation,58 suggesting that NLRP3 inflammasome activation through the mitochondrial pathway is not entirely dependent on mtROS or mtDNA.
Lysosomal Leakage
Phagocytosis of particulate matters activates NLRP3 inflammasome by destroying lysosomes and releasing cathepsin, a lysosomal cysteine protease of the papain family, into the cytoplasm.59 The event of lysosomal acidification is prior to lysosomal swelling and impairment, and is vital for NLRP3 activation.60 For example, bafilomycin A1, a potent vacuolar H+-ATPase inhibitor, impedes lysosomal acidification and blocks NLRP3 inflammasome activation stimulated by particulate matters, such as silica and alum.60 To date, the cathepsin B, C, S, and Z have been identified to be involved in NLRP3 inflammasome activation.59,61–63 Utilization of cathepsin inhibitors (eg, cathepsin B inhibitor CA-074-Me and a broad cathepsin inhibitor K777) or knockdown of cathepsin blocks the NLRP3 inflammasome activation.62,64 Interestingly, impairment of lysosome triggers cathepsin D release and induces IL-1β secretion, but has no effect on NLRP3 and caspase-1 expression, suggesting that cathepsin D may be implicated in other inflammatory signaling pathways.65 However, the specific mechanism underlying cathepsin D and associated inflammatory signaling pathways remains unclear.
Of note, a generally accepted point is that only lysosomal damage and rupture may demand additional signals to fully activate the NLRP3 inflammasome. For instance, Leu-Leu-O-methyl ester is a soluble lysosomotropic agent that induces activation of the NLRP3 inflammasome via the synergistic effects of lysosome membrane permeabilization and increased K+ efflux.66 A recent study showed a transfer of Ca2+ from lysosomes to cytosol after exposure of U937 and THP-1 cells to high glucose for 48 h, which leads to reduction of lysosomal Ca2+ concentration, the release of cathepsin B, and subsequent NLRP3 inflammasome activation,67 suggesting that Ca2+ mobilization is an important upstream event for lysosomal damage-mediated NLRP3 inflammasome activation.
Trans-Golgi Disintegration
The crucial role of the Golgi apparatus in NLRP3 inflammasome activation has been increasingly identified. Zhang and associates firstly reported the role of Golgi in NLRP3 inflammasome activation. In response to NLRP3 activators stimulation, Golgi membranes are surrounded by mitochondria-associated endoplasmic reticulum membranes (MAMs), and diacyl glycerol (DAG) in Golgi increases, resulting in recruitment of protein kinase D (PKD).68 PKD further leads to phosphorylation of NLRP3, which is subsequently released from MAMs and ultimately participates in the assembly of NLRP3 inflammasome,68 suggesting that the relationship between Golgi and MAMs during NLRP3 inflammasome activation is associated with the accumulation of DAG.
The interaction of ER and Golgi is implicated in NLRP3 inflammasome activation. It has been proposed that perturbation of ER-Golgi trafficking restrains NLRP3 inflammasome-dependent caspase-1 activation, suggestive of the engagement of ER-Golgi trafficking in NLRP3 activation.69 ER-Golgi transport of the sterol regulatory element-binding proteins (SREBPs)-SREBP cleavage-activating proteins (SCAPs) complex may be a plausible explanation for the activation of NLRP3 inflammasome. Blockade of SCAP-SREBP2’s ER-to-Golgi translocation by interacting with SCAP significantly suppresses caspase-1 maturation and IL-1β or IL-18 secretion when treatment with NLRP3 agonists in LPS-primed BMDMs.70 Chen et al uncovered the role of phosphatidylinositol-4-phosphate (PtdIns4P) on dispersed trans-Golgi network (dTGN) in NLRP3 inflammasome activation.71 In case of the TGN disintegration under NLRP3 activators, dTGN recruits NLRP3 by ionic bonding between its conserved polybasic region and PtdIns4P, thereby activating the downstream signaling cascade.71 Conversely, disruption of the interaction between NLRP3 and PtdIns4P on the dTGN blocks NLRP3 aggregation, caspase-1 activation, and IL-1β secretion. Hence, disintegration of trans-Golgi undoubtedly induces NLRP3 inflammasome activation.
Other Mechanisms Regulate NLRP3 Inflammasome Activation
Autophagy
Autophagy, an intracellular homeostatic mechanism, is responsible for delivering cytosolic constituents, including cytosolic macromolecules and organelles, to lysosomes for degradation and amino acid recycling. In yeasts and mammals, there are two conjugation systems involving ubiquitin-like proteins that contribute to the expansion of the phagophore: Atg12-Atg5-Atg16L1 complex and Atg8/LC3 system. The mission of these two systems is primarily to mentor the elongation of double-membrane phagosomes and the completion of mature autophagosome formation.72,73 Nowadays, numerous studies have confirmed that autophagy is related to the activation of NLRP3 inflammasome, but the specific role of autophagy in NLRP3 inflammasome activation is still debateable (Figure 3).
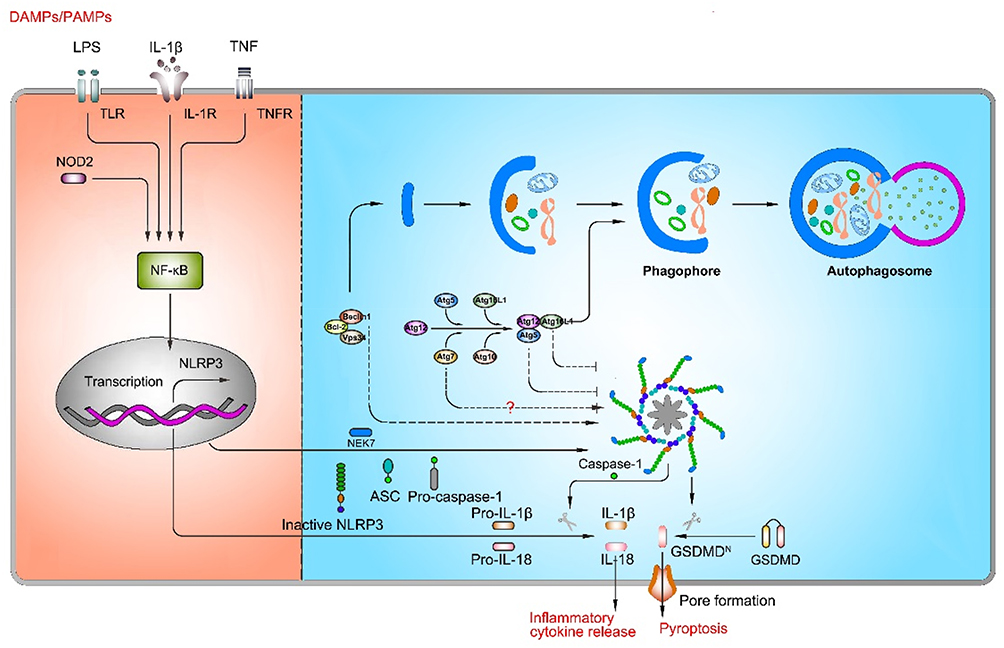 |
Figure 3 The role of autophagy in NLRP3 inflammasome activation. Autophagy plays a key role in NLRP3 inflammasome activation, but the role of autophagy remains controversial. On one hand, inhibition of autophagy-related genes (Atg5, Atg7, and Atg16L1) suppresses NLRP3 inflammasome activation; on the other hand, inhibition of autophagy-related genes (Atg7 and Beclin1) promotes NLRP3 inflammasome activation. |
The first study to explicitly highlight the relationship between autophagy and IL-1 family cytokines revealed that deficiency of the autophagy-related gene 16 like-1 (Atg16L1) in mouse fetal liver macrophages led to excessive secretion of IL-1 upon TLR3/TLR4 stimulation,74 suggesting that autophagy prevents NLRP3 inflammasome activation. Consistent with this result, Liu et al disclosed that retardation of autophagy by Sirtuin 3 (SIRT3) knockdown induced Atg5 acetylation, autophagosome immaturity and NLRP3 inflammasome activation.75 Similarly, Atg7 knockout triggers NLRP3 inflammasome activation and up-regulates caspase-1 expression upon stimulation by β-amyloid.76 These findings support the protective role of autophagy in the activation of NLRP3 inflammasome.
Potential mechanisms by which autophagy inhibits NLRP3 inflammasomes may be relevant to the reduction of ASC, phosphorylation of NLRP3, and clearance of mtROS or damaged mitochondria. A study has demonstrated that hemin and its derivative, cobalt protoporphyrin (CoPP), significantly reduce IL-1β secretion in both human primary macrophages and BMDMs.77 Mechanistically, hemin and CoPP are regarded as autophagy activators to inhibit NLRP3 inflammasome assembly by reducing the amount of ASC. On the contrary, inhibition of autophagy by 3-methyladenine (3-MA) prevents the CoPP-induced depletion of ASC, implying that the ASC depletion is a result of increased autophagy.77 Spalinger et al have discovered that loss of protein tyrosine phosphatase non-receptor type 22 (PTPN22) results in an increase of phosphorylation of NLRP3, which is a signal for NLRP3 inflammasome inactivation. Moreover, NLRP3 inflammasome inactivation by the deletion of Ptpn22 is abrogated by inhibition of autophagy with 3-MA, wortmannin or bafilomycin A1, indicating that confinement of NLRP3 in the phagophores is necessary in NLRP3 phosphorylation-mediated NLRP3 inflammasome inactivation.78 Besides, sestrin 2 (SESN2), a stress-inducible protein, suppresses NLRP3 inflammasome activation by clearance of damaged mitochondria through inducing mitophagy in LPS and ATP-treated BMDMs.79
However, evidence referring to the detrimental role of autophagy in NLRP3 inflammasome activation has emerged. Houtman et al have discovered that Beclin1, a regulatory protein of autophagy, affects cytokine production of microglia. NLRP3, caspase-1, IL-1β and IL-18 expression are increased in microglia isolated from Beclin1+/− mice compared to wild type microglia exposed to LPS and ATP.80 Moreover, it has shown that mycotoxin zearalenone can induce autophagy and NLRP3 inflammasome activation, and inhibition of autophagy by 3-MA exacerbates the expression of NLRP3 and downstream caspase-1 and IL-1β in zearalenone-exposed rat insulinoma cells via the activation of NF-κB signaling pathway.81 Besides, our group have also proved that in ECs treated with acrolein, the activation of autophagy occurs prior to the activation of the NLRP3 inflammasome, and blockade of autophagy via pre-treatment with 3-MA or Atg7 knockdown aggravates NLRP3 inflammasome activation.82,83 These studies indicate that autophagy promotes NLRP3 inflammasome activation, and the possible explanations for the two contrary effects of autophagy on the NLRP3 inflammasome may be that the models triggering NLRP3 inflammasome activation or the degree of autophagy activation by various stimuli differ, thus further work is needed to address this discrepancy.
Post-Translational Modifications
Post-translation modifications (PTMs) are defined as any change of a protein or an amino acid sequence after translation and synthesis from ribosome, including phosphorylation, ubiquitination, sumoylation, glycosylation and acetylation.84,85 Emerging studies have identified that both the priming process and subsequent activation phase of NLRP3 can be regulated by PTMs.86 However, the regulatory mechanisms of these PTMs are complicated, and some PTMs have dual effects on NLRP3. Of these, phosphorylation is the most widely recognized PTM and involves over hundreds of kinases and phosphatases that dynamically modulate the proteome. The effects of phosphorylation on the NLRP3 inflammasome are discussed next (Figure 4).
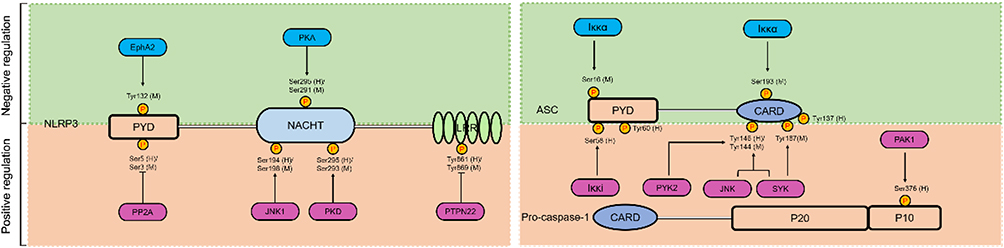 |
Figure 4 The role of phosphorylation in NLRP3 inflammasome activation. Phosphorylation plays a dual role in the activation of NLRP3 inflammasome. The pale green background represents phosphorylation of NLRP3 or ASC inhibits NLRP3 inflammasome activation, and the saffron yellow background represents phosphorylation of NLRP3 or pro-caspase-1 promote NLRP3 inflammasome activation. Activators and inhibitors of the NLRP3 inflammasome are represented by red and blue rounded rectangle, and phosphorylation sites on different domains of NLRP3 (LRR, NACHT, and PYD), ASC (PYD and CARD), and pro-caspase-1 (CARD, p10, and p20) from human and mouse species are indicated with (h) and (m), respectively. |
Phosphorylation of NLRP3 is first reported by Spalinger et al,78,87 (see “Autophagy”) who proposed that PTPN22 is a prerequisite for NLRP3 inflammasome activation, as Ptpn22 knockdown results in diminished IL-1β secretion.78 Dephosphorylation of Tyr861 (human) or Tyr869 (mouse) residue in the LRR domain of NLRP3 positively regulates NLRP3 inflammasome activation.87 In addition to these sites, dephosphorylation of Ser5 (human) or Ser3 (mouse) residue by protein phosphatase 2A (PP2A) in the PYD of NLRP3 also induces NLRP3 inflammasome activation.88 PP2A inhibition by okadaic acid and knockdown of Pp2a prevents ASC dissociation from IKKα and substantially suppresses NLRP3 inflammasome activation when exposure of BMDMs to nigericin.88,89 These studies reveal that dephosphorylation in the PYD structural domain may disrupt the interaction between NLRP3 and ASC, whereas maintaining a phosphorylated state induces the activation of NLRP3 inflammasome. Additionally, C-Jun N-terminal kinase 1 (JNK1)-mediated phosphorylation of NLRP3 at Ser198 (mouse) or Ser194 (human) in the NACHT domain is critical for the NLRP3 oligomerization, which promotes NLRP3 inflammasome assembly.90 Besides, phosphorylation of NLRP3 mediated by PKD is essential for the complete activation of the NLRP3 inflammasome. Activated PKD interacts with NACHT of NLRP3 and phosphorylates Ser295 (human) or Ser293 (mouse), resulting in NLRP3 discharge from MAMs to cytoplasm and eventually NLRP3 inflammasome maturation and activation.68
From above mentioned, phosphorylation of NLRP3 can trigger NLRP3 inflammasome assembly, and phosphorylation of ASC or caspase-1 can also induce NLRP3 inflammasome activation. Spleen associated tyrosine kinase (SYK) and JNK are able to active caspase-1 via the ASC-dependent NLRP3 inflammasomes. Phosphorylation of Tyr144 in mouse ASC is critical for speck formation and caspase-1 activation, which suggests that phosphorylation of ASC controls inflammasome activity through the formation of ASC specks.91 Lin et al also have reported that the phosphorylation of ASC both at Tyr144 (mouse) and Tyr187 (mouse) residues by SYK is critical to enhance for ASC oligomerization, pro-caspase-1 recruitment, and NLRP3 inflammasome formation.92 Intriguingly, SYK plays a cell type-specific role in NLRP3 inflammasome activation, given that this process is not indispensable in dendritic cells.91 Similarly, proline-rich tyrosine kinase 2 (PYK2) directly phosphorylates ASC at Tyr146 (human) to activate the NLRP3 inflammasome.93 Mambwe et al have uncovered that the phosphorylation of Tyr60 (human) and Tyr137 (human) of ASC is critical for NLRP3 inflammasome assembly by site-directed mutagenesis of ASC tyrosine residues, but exactly which upstream molecules contribute to the phosphorylation has not been determined.94 Furthermore, p21-activated kinase 1 (PAK1) immunoprecipitated from lysates of Helicobacter pylori LPS-challenged THP-1 cells induces phosphorylation of caspase-1 at Ser376 (human), resulting to caspase-1 activation.95 To conclude, these phosphorylation events function as inflammasome activators and are crucial for NLRP3 inflammasome or downstream activation.
Otherwise, phosphorylation events can also inhibit NLRP3 inflammasome activation. It has been reported that ephrin type-A receptor 2 (EphA2), a transmembrane tyrosine kinase, interacts with NLRP3 and induces its phosphorylation of Tyr132 (mouse) in the PYD domain, thereby inhibiting NLRP3 inflammasome assembly in murine airway epithelial cells during reovirus infection.96 Protein kinase A (PKA), a serine/threonine kinase, prevents NLRP3 inflammasome activation through the induction of Ser295 (human) or Ser291 (mouse) residue phosphorylation of NLRP3 in the NACHT.97,98 Studies have put forward that prostaglandin E2 (PGE2) and bile acids, as two protective factors can activate PKA signaling by up-regulation of cytoplasmic cyclic adenosine monophosphate (cAMP) levels, subsequent contribute to phosphorylation of the Ser295 residue of NLRP3, and inhibition of NLRP3 inflammasome activation.97,98 Pharmacological activation of PKA by forskolin causes an increase of cytoplasmic cAMP levels and inhibits NLRP3 inflammasome activation through phosphorylation of Ser295 residues in NLRP3.97 Iκκα dual phosphorylates Ser16 (mouse) and Ser193 (mouse) of ASC, leading to the confinement of ASC in the nucleus and inactivation of downstream NLRP3 inflammasome assembly.89 In contrast, Iκκi enables ASC binding and phosphorylation of the Ser58 (human) site during LPS-induced priming, which promotes ASC translocation from the nucleus to the perinuclear region.89 These studies demonstrate that phosphorylation events block NLRP3 inflammasome activation.
MicroRNAs
MircroRNAs (miRNAs), endogenous noncoding RNA, are 20–23 nucleotides in length and perform regulatory roles through base complementation with the 3′ untranslated region (3′ UTR) of protein-coding mRNAs.99 MiRNAs originate from long RNA hairpins precursor transcripts through the activity of the nucleases Drosha and Dicer, and dysregulation of miRNAs is correlated with the pathogenesis of many chronic inflammatory diseases. For instance, miR-7 targets NLRP3 inflammasome and modulates neuroinflammation in the pathogenesis of Parkinson’s disease.100 MiR-20a inhibits NLRP3 expression via targeting TXNIP in adjuvant-induced arthritis fibroblast-like synoviocytes.101 Several miRNAs, such as miR-9, miR-155, miR-30c-5p, miR-181a, miR-181b-5p, and miR-20a, regulate NLRP3 inflammasome activation in AS (see “NLRP3 inflammasome in AS” section).102–107 These findings strongly support the importance of miRNAs in regulating NLRP3 inflammasome.
Mechanisms of Non-Canonical NLRP3 Inflammasome Activation
The non-canonical NLRP3 inflammasome pathway is characterized by a requirement of caspase-11 (in mouse) or caspase-4/caspase-5 (in human) in the activation of the NLRP3 inflammasome complex and secretion of IL-1β and IL-18 (Figure 5) in response to specific infection caused by intracellular LPS of Gram-negative bacteria (eg, Escherichia coli, Citrobacter rodentium, and Vibrio cholerae).108 It is proposed that caspase-4/caspase-5 and caspase-11 have certain specificity and affinity for direct binding of their CARD domains with the lipid A moiety of LPS to induce NLRP3 inflammasome activation.108,109 Activated caspase-4/5 and caspase-11 cleave GSDMD, leading to pyroptosis in the absence of caspase-1.13 However, neither activated caspase-4/5 nor caspase-11 has the proteolytic activity processing pro-IL-1β and pro-IL-18. In general, caspase-4/5 or caspase-11-mediated pyroptosis works in collaboration with the NLPR3 inflammasome to promote inflammatory response. It has been revealed that caspase-11 instigates the extracellular release of ATP, followed by activation of the P2X7R and K+ efflux, which then results in the activation of the NLRP3 inflammasome and pyroptosis independent of GSDMD.110 Akin to the mechanisms of the canonical pathway, K+ efflux through the pannexin-1 channel is an upstream event of caspase-11-mediated NLRP3 inflammasome activation.110 Interestingly, stimulation of LPS directly induces the release of endogenous ATP from human monocytes and activates the P2X7R, thereby triggering the activation of the NLRP3 inflammasome and the maturation of IL-1β.111 Nevertheless, in mouse bone marrow-derived dendritic cells, exposure to LPS alone induces IL-1β secretion independent of the ATP-P2X7 pathway.111 Of note, deficiency of pannexin-1 in murine BMDMs deprives caspase-11-pannexin-1 axis, but still leads to K+ efflux and NLRP3 inflammasome activation independent of P2X7,110 indicating that ATP-P2X7-mediated K+ efflux is dispensable for the activation of the non-canonical NLRP3 inflammasome, and distinct cell types or germ lines may have different mechanisms for the activation of NLRP3 inflammasome.
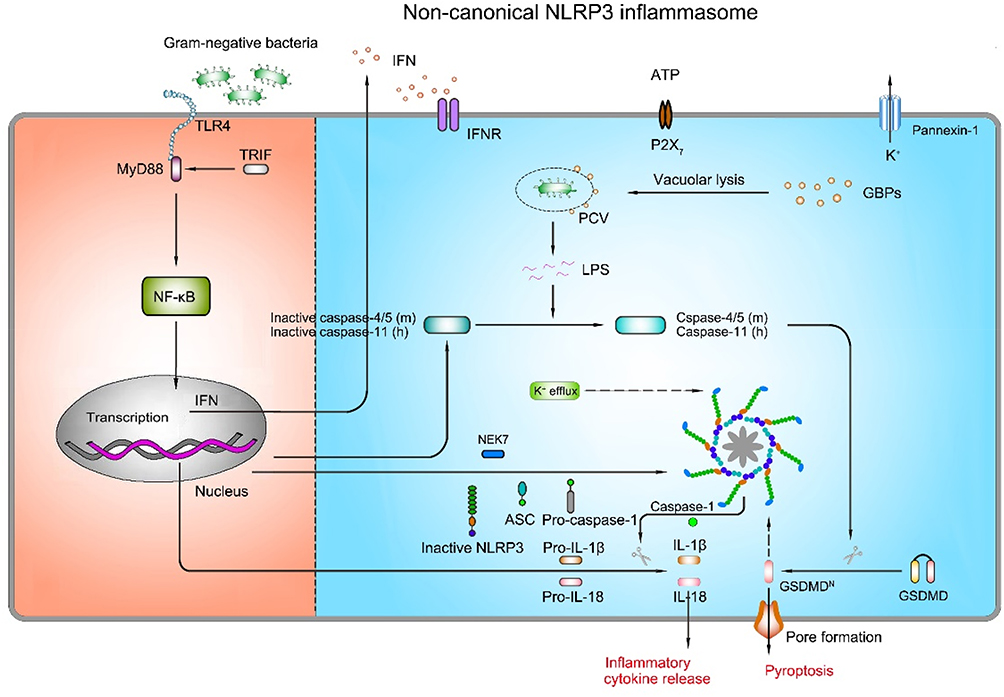 |
Figure 5 Mechanism of activation for non-canonical NLRP3 inflammasome pathway. The non-canonical NLRP3 inflammasome activation is induced by LPS of Gram-negative bacteria. Extracellular LPS induces the expression of pro-IL-1β, pro-IL-18, NLRP3, and type I interferon (IFN) via the TLR4/TRIF/MyD88-dependent pathway. IFN provides a feedback loop and activates type I interferon receptor (IFNR) to induce caspase-4/5 (mouse) or caspase-11 (human) expression. Guanylate-binding proteins (GBPs) are recruited to the pathogen-containing vacuole (PCV), where they mediate rupture of the PCV to permit the LPS release into the cytoplasm. Gram-negative bacteria deliver LPS into the cytosol. Cytosolic LPS binds to caspase-11 leading to caspase-11 activation. Activated caspase-11 then drives pyroptosis and activation of the non-canonical NLRP3 inflammasome. |
Studies have unveiled the role of type I interferon (IFN) signaling in the activation of non-canonical NLRP3 inflammasome.112,113 Recognition of Gram-negative bacterial LPS by TLR4 triggers TRIP-mediated IFN signaling, which then increases caspase-11 expression.113 Under the circumstance of Gram-negative bacterial infection, IFN signaling is activated and the expression of guanylate-binding proteins (GBPs) is increased. GBPs are recruited to the pathogen-containing vacuole (PCV), where they mediate rupture of the PCV and induce the LPS release into the cytoplasm to interact with caspase-11. After that, the combination of LPS with caspase-11 activates the proteolytic function of caspase-11, resulting in caspase 1-dependent IL-1β/18 processing. Besides, activated caspase-11 induces the cleavage of GSDMD, which triggers pyroptotic cell death.114 However, IFN inhibits NLRP3 inflammasome-dependent IL-1β secretion in response to urate crystals and ATP (in canonical NLRP3 inflammasome signaling),115 suggesting that IFN has dual effects in NLRP3 inflammasome activation. In conclusion, these data underline the importance of IFN signaling in the NLRP3 inflammasome surveillance of bacterial infections.
In addition to the LPS of Gram-negative bacteria, there is some evidence supporting the role of certain lipids as caspase-11 activators. Oxidized-1-palmitoyl-2-arachidonoyl-sn-glycerol-3-phosphorylcholine (Ox-PAPC, 100 µg/mL), an endogenous oxidized phospholipid, binds to caspase-11 and induces caspase-11-dependent IL-1β release in murine bone marrow-derived dendritic cells.116 However, it is interesting to note that although LPS triggers caspase-11-mediated IL-1β release as well as pyroptosis, Ox-PAPC only triggers IL-1β release, resulting in a hyper-activated status without cell death.116 These differences probably are attributed to the different binding sites of caspase-11 targeted by LPS and Ox-PAPC, and their discrepancy in terms of promoting caspase-11 enzyme activity. Intriguingly, exposure to different concentrations of Ox-PAPC (10 ng/well-200 ng/well) inhibits IL-1β release and pyroptosis in LPS-incubated BMDMs.117 Ox-PAPC competes with LPS to bind the CARD and catalytic domain of caspase-11 by employing caspase-11 mutants (caspase-11ΔCARD and caspase-11C254A) and pull-down assay,117 unraveling a virtually identical binding modality of caspase-11 by LPS and Ox-PAPC. Consequently, the role of Ox-PAPC is still unknown. Probably, low concentrations of Ox-PAPC can inhibit the activation of the NLRP3 inflammasome, and a toxic response arises with increasing concentrations, which in turn leads to the activation of the NLRP3 inflammasome. Moreover, stearoyl lysophosphatidylcholine (SLPC), another oxidized phospholipid, blocks LPS binding to caspase-11 and shields mice from lethal endotoxemia.118 PGE2 inhibits caspase-11-driven pyroptosis in BMDMs via interacting with caspase-11.119 In consequence, the exact role of endogenous lipid-based inflammatory mediators in the non-canonical signaling pathway is unclear.
Mechanisms of Alternative NLRP3 Inflammasome Activation
As mentioned above, NLRP3 inflammasome activation triggers caspase-1 activation and IL-1β maturation through priming and activation signaling pathways. Nevertheless, NLRP3 inflammasome activation does not invariably comply with the two-step activation model. Stimulation with LPS alone induces caspase-1-dependent IL-1β maturation and secretion in human monocytes, but not in murine monocytes.120,121 This type of NLRP3 inflammasome activation is found to be mediated by the TRIF-RIPK1-FADD-caspase-8 axis in human and porcine peripheral blood mononuclear cells, but not murine cells.122 K+ efflux, ASC speck formation and pyroptosis are not required in this pathway.122 A recent study has suggested that apolipoprotein C3 (ApoC3) induces NLRP3 inflammasome activation in human monocytes through this signaling pathway via caspase-8 and dimerization of TLR-2 and TLR-4.123 Furthermore, they have unveiled that alternative inflammasome activation in human monocytes is mediated by the TLR adapter protein-SCIMP, which induces the activation of caspase-8 by triggering tyrosine kinase LYN/SYK-dependent calcium influx and the production of ROS.123 This specific activation pathway induced by TLR2 or TLR4 signaling pathway without involving other secondary activators, is referred to as alternative NLRP3 inflammasome activation (Figure 6).
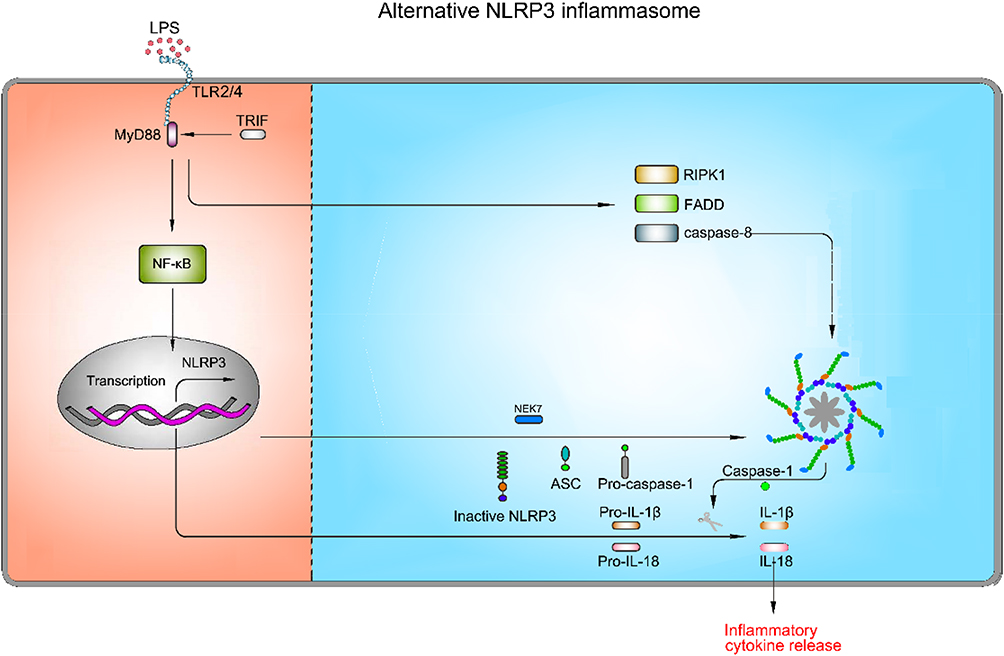 |
Figure 6 Mechanism of activation for alternative NLRP3 inflammasome pathway. The alternative NLRP3 inflammasome pathway is triggered by TLR2/TLR4. LPS binds to TLR2/TLR4, which results in up-regulation of pro-1β, pro-18, and NLRP3 via the NF-κB signaling pathway. However, this type of NLRP3 inflammasome activation is mediated by the TRIF-RIPK1-FADD-caspase-8 axis, whereas K+ efflux, ASC speck formation and pyroptosis are not required in this pathway. |
NLRP3 Inflammasome in AS
Endothelial cells (ECs), immune cells (such as monocytes, macrophages and dendritic cells), and SMCs are three types of cells involved in AS pathogenesis and are able to express components of NLRP3 inflammasome,124–126 which is emphasized by a large number of studies to play an important role in the pathogenesis of AS. Therein, IL-1β and IL-18 are NLRP3 inflammasome activation products that play a key role in atherogenesis. In murine as well as human atherosclerotic coronary arteries, IL-1β is produced by ECs, macrophages and SMCs.127,128 Compared to the ApoE−/− (knockout of apolipoprotein E) mice, ApoE−/− and IL-1β−/− (double knockout of apolipoprotein E and IL-1β) mice show an approximately 30% reduction in the size of atherosclerotic plaques, demonstrating that IL-1β is a deleterious regulator in atherogenesis. Surprisingly, one recent study showed the atheroprotective effect of IL-1β in mice with advanced atherosclerotic lesions, including promotion of cardiac outward remodelling and formation and maintenance of fibrous cap.129 Evidently, the role of IL-1β in AS depends on the stage of AS.
The role of NLRP3 inflammasome in AS is firstly reported by Duewell et al in 2010.124 They discovered that absence of Nlrp3, Asc or IL-1α/β in the bone marrow cells in atheroprone low-density lipoprotein receptor (Ldlr)−/− mice results in smaller size of atherosclerotic lesions. Consistent with this notion, in 2015, transplantation of caspase-1−/− or caspase-11−/− bone marrow into Ldlr−/− mice induced a significant decrease in the size of atherosclerotic plaques, after 12 weeks feeding of high-fat diets (HFD) and high-cholesterol diets (HCD).130 In recent years, several epidemiological studies have revealed the relationship between the activation of NLRP3 inflammasome and development of AS in human beings. High expression of NLRP3 in the aorta is observed in patients undergoing coronary artery bypass graft, and the expression of NLRP3 in ascending aortic tissues is dramatically higher than those without AS and is positively correlated with the severity of AS lesions and risk factors of AS.131 Moreover, compared to healthy arteries in patients, the transcription and translation levels of NLRP3, ASC and caspase-1, as well as IL-1β and IL-18 were significantly increased in carotid plaques, which was proposed in 2015.132 Besides, a study in 2016 has shown that in human atherosclerotic plaque samples, NLRP3 and ACS co-exist in CD68-positive macrophages, as well as in SMCs.133 Recently, evaluation of functional dependency of single-nucleotide polymorphisms in NLRP3 inflammasome activation in monocyte-enriched peripheral blood mononuclear cells reveals a significantly higher incidence of coronary heart disease in individuals carrying NLRP3 intronic variant rs10754555 than in non-carriers.134 However, a study published in 2016 found no significant association between NLRP3 polymorphisms and susceptibility to CVD in patients with pre-hypertension,135 possibly due to the small size of the cohort and the sample size needs to be increased to improve the authenticity of the results.
Numerous NLRP3 agonists drive NLRP3 inflammasome activation in various way. Herein, the role of NLRP3 inflammasome activation in ECs and monocytes/macrophages in AS is discussed as follows.
Vascular Endothelial Cells Injury
Covering the inner surface of the blood vessels as a single layer, ECs are responsible for transporting nutrients from the blood to surrounding tissues, providing them with essential nutrients for growth and metabolism.136 Lesions of the ECs is an inaugural step in AS, and both mechanical injury (eg, hemodynamic force) and chemical injury (eg, tobacco smoking, heavy metal exposure, hyperlipemia, and hyperglycemia) can cause EC damage.137–139 In ECs, these stimuli trigger NLRP3 inflammasome activation, which secretes IL-1β and IL-18 to promote further inflammatory processes.8 Activated caspase-1 has been reported to promote the expression of C-X-C motif chemokine ligand 16 (CXCL16) and its receptor CXCR6, resulting in the recruitment of T lymphocytes into endothelium, consequently stimulating and propelling the inflammatory response of ECs.140 Secreted IL-1β and IL-18 also foster the secretion of adhesion molecules [eg, E-selectin, intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1)] and secondary inflammatory factors [eg, IL-6 and C-reactive protein (CRP)], inducing a potent pro-inflammatory response.8 With this positive feedback regulation, the injury of ECs is further exacerbated. Besides, NLRP3 inflammasome-mediated pyroptosis has been identified as a potential cause of ECs death.
An increasing number of stimuli have been reported to activate NLRP3 inflammasome through different mechanisms (Table 1). The disturbed blood flow and associated oscillatory stress (OS) at the point of arterial curvature and branch points are physical risk factors for AS that can trigger inflammatory response in ECs. Xiao et al found that the expression of caspase-1 and IL-1β is significantly increased in both ECs and the lesser curvature of the aortic arch in C57BL/6 mice under OS stress.141 More recently, a similar study has proposed that m6A methyltransferase-METTL3 expression and m6A hypermethylation are increased as a response to disturbed flow and OS, while knockdown of Mettl3 prevents the atherogenic process and NLRP3 up-regulation in AS model induced by partial ligation of the carotid artery.142 Recent studies identified low shear stress as another physical risk factor for the onset and development of AS, which is involved in ECs injury. As shown by Xu et al, low shear stress suppresses miR-181b-5p expression, accompanied by the elevated expression of signal transducer and activator of transcription 3 (STAT3) and NLRP3-dependent pyroptosis in ECs.106 STAT3 works as a direct target of miR-181b-5p to regulate NLRP3 inflammasome activation. Introduction of miR-181b-5p or silencing of Stat3 can alleviate NLRP3 inflammasome-dependent pyroptosis.106
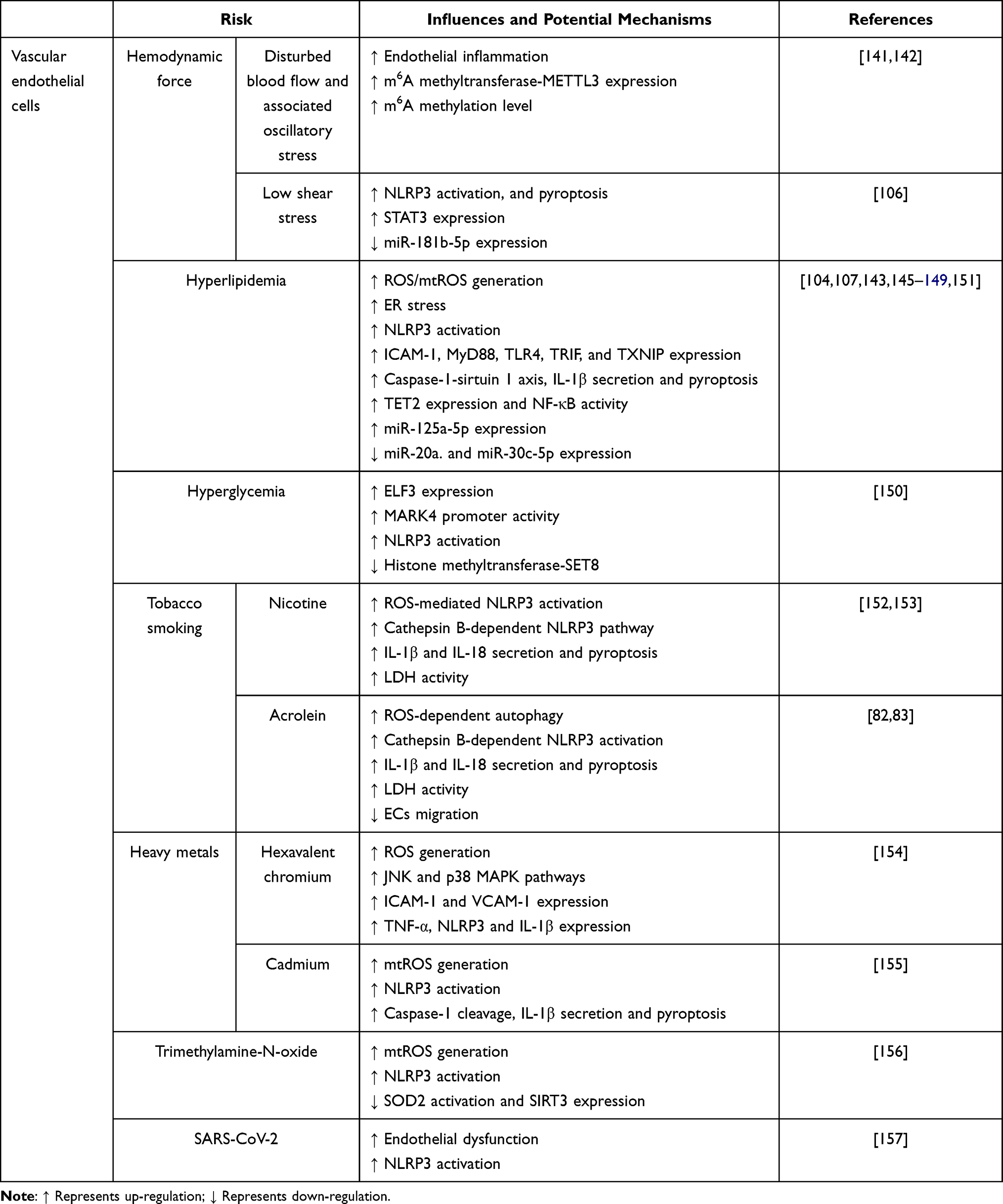 |
Table 1 The Role of Vascular Endothelial Cells (ECs) Injury in Atherogenesis |
Hyperlipidemia [eg, increase of total serum cholesterol, ApoC3, low-density lipoprotein cholesterol (LDL-C), and especially oxidized LDL (ox-LDL)] and hyperglycemia serve as hazard factors of AS and lead to NLRP3 inflammasome in ECs.143,144 Early hyperlipidemia facilitates ECs activation prior to monocyte recruitment via caspase-1-sirtuin 1 axis and ROS generation, followed by pyroptosis of ECs.143,145 Early AS can be attenuated in ApoE(-/-)/caspase-1(-/-) mice, indicative of a detrimental role of ApoE and caspase-1 in AS development. Another study has suggested that TNF-α receptor-associated factor-interacting protein with forkhead-associated domain (TIFA) regulates the priming and activation of NLRP3 inflammasome in ECs by generating ROS under hyperglycemia conditions.146 Moreover, LDL triggers ER stress and ROS generation, which then activates the NLRP3 inflammasome and ultimately leads to ECs injury.147 Similarly, Huang and co-workers found that exposure to ox-LDL in ECs or feeding mice with HFD triggers ROS generation, contributing to NLRP3 inflammasome activation and IL-1β secretion; whereas, endothelia-specific Nlrp3 knockout attenuates the severity of AS in HFD-fed mice.148 In addition, intraperitoneal administration of poloxamer 407-induced acute hypercholesterolemia in mice aggravates endothelial dysfunction in normal Nlrp3+/+ coronary arteries, which is markedly ameliorated in Nlrp3−/− mice, and exposure of ECs to cholesterol crystals induces NLRP3 inflammasome activation and pyroptosis.149 Besides, hyperglycemia is supposed to induce endothelial NLRP3 inflammasome activation via up-regulation of E74-like ETS transcription factor 3 (ELF3).150 Mechanistically, high glucose increases ELF3 expression, reduces histone methyltransferase-SET8 expression, and improves microtubule affinity-regulating kinase 4 (MARK4) promoter activity, thus leading to NLRP3 inflammasome activation.150 These findings provide new insights into the development of therapeutic strategies for AS.
An increasing number of miRNAs have been proved to be involved in AS development via ECs injury in the presence of ox-LDL. For instance, miR-125a-5p can mediate ox-LDL-induced pyroptosis in ECs via abnormal DNA methylation and mitochondrial dysfunction/ROS pathway. MiR-125a-5p down-regulates ten eleven translocation methylcytosine dioxygenase 2 (TET2) and promotes the activation of NF-κB, NLRP3 and caspase-1, which eventually results in pyroptosis.151 Moreover, miR-20a expression is decreased in ox-LDL-irritative ECs, specifically, it negatively regulates MyD88/NF-κB-mediated NLRP3 pathway by interacting with TLR4 and TXNIP,107 indicating a protective role of miR-20a in the activation of NLRP3 inflammasome. Likewise, miR-30c-5p plays a protective role in NLRP3 inflammasome activation as well. The expression of miR-30c-5p is decreased upon treatment of ox-LDL in ECs, while overexpression of miR-30c-5p protects ECs from ox-LDL-induced NLRP3 inflammasome activation and pyroptosis.104 These AS-associated miRNAs provide new vision for inhibition of AS development under ox-LDL stress.
Exogenous stimuli can also induce NLRP3 inflammasome activation and cause AS. Tobacco smoking is a major contributor of AS, in which nicotine, a chemical constituent of tobacco, results in larger atherosclerotic plaques and secretion of inflammatory cytokines in ApoE−/− mice fed with HFD.152 Exposure of ECs to nicotine activates the NLRP3 inflammasome and induces cell pyroptosis through the ROS signaling pathway. Nicotine induces cleavage of caspase-1, secretion of IL-1β and IL-18, and elevation of LDH activity, while silencing of Nlrp3, scavenging of ROS by NAC, or inhibition of caspase-1 by VX-765 suppresses these pathways.152 Furthermore, nicotine-induced ECs dysfunction can be triggered by cathepsin B-dependent NLRP3 inflammasome activation.153 We previously have found another chemical constituent of tobacco, acrolein, induces NLRP3 inflammasome-mediated cell pyroptosis via ROS-dependent autophagy and cathepsin B-dependent signaling pathway.82,83 Besides, exposure to heavy metals, such as hexavalent chromium and cadmium, are also believed to result in NLRP3 inflammasome activation and pyroptosis in ECs via ROS/mtROS signaling pathway.154,155 Trimethylamine-N-oxide (TMAO) has recently been recognized as a novel and independent risk factor that promoting AS by inducing vascular inflammation. It has been showed that TMAO activates NLRP3 inflammasome via engagement of mtROS signaling pathway.156 A very fresh study has suggested that SARS-CoV-2 attacks the immune system, which results in NLRP3 inflammasome activation and perpetuates a vicious cycle of deteriorated endothelial dysfunction that further promotes AS.157 Taken together, endothelial NLRP3 inflammasome activation plays an important role in AS development.
Notably, it has been proved that NLRP3 inflammasome activation up-regulates the expression of ICAM-1 and E-selectin in ECs after treatment with macrophage-derived microparticles (MPs).158 Knockdown of Nlrp3 in THP-1 cells reduces the activity of the MPs, and blockade of the IL-1 receptor (IL-1R) in MPs-treated ECs suppresses the expression of cell adhesion molecules.158 This study implies that NLRP3 inflammasome activation in ECs probably may be an intricate progress involving the engagement and regulation of multiple types of cells. The cross-talk between ECs and macrophages needs further investigation.
The Infiltration of Monocytes/Macrophages and the Formation of Foam Cells
Recruitment of monocytes/macrophages into the intima and subintima and formation of foam cells are pathological hallmarks of AS.159 The importance of innate and adaptive immune factors in AS is increasingly highlighted. Monocytes, as a type of immune cells, play an important role in the development and exacerbation of AS. Mechanistically, uptake of excessive oxidized lipoproteins followed by monocyte infiltration and lipid-rich foam cell formation results in the formation of early plaques called fatty streaks.160 Lipid-laden macrophages generate extracellular matrix (eg, collagen), and subsequently form a fibrous plaque called atheroma. Inflammatory macrophages in the atheroma enlarge the lesion area by secreting growth factors, cytokine, and proteases, which gradually destabilize the plaque and eventually lead to plaque disruption and thrombosis.161 In this process, monocytic NLRP3 inflammasome activation is a key event driving AS (Table 2).
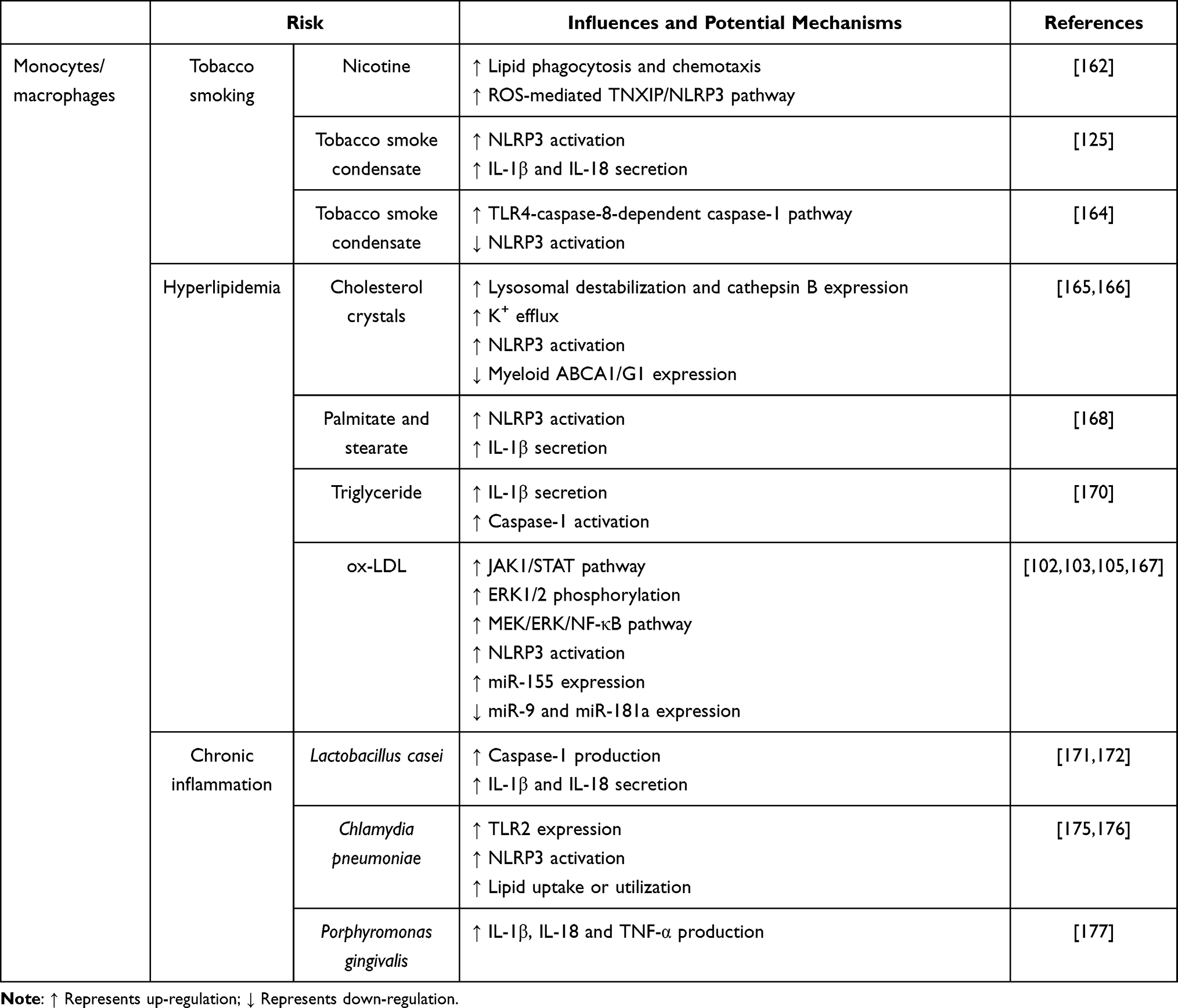 |
Table 2 The Role of Monocytes/Macrophages Infiltration in Atherogenesis |
Tobacco smoking not only induces NLRP3 inflammasome activation in ECs, but also in monocytes. A study recruiting 100 patients with coronary heart disease found that smokers have higher burden of plaque, and expression of TXNIP, IL-1β and IL-18 than non-smokers.162 Treatment of BMDMs with nicotine enhances lipid phagocytosis, chemotaxis, and ROS generation, which induces TXNIP/NLRP3 axis activation and pyroptosis. Nicotine intake by ApoE-/- mice fed HFD shows a similar result.162 Moreover, exposure to tobacco smoke condensate activates NLRP3 inflammasome in THP-cells, which is indicated by up-regulation of NLRP3 at both transcriptional and translational levels, and increase of IL-1β and IL-18 expression.125 After treatment with MyD88 inhibitor (NBP2-29328) in tobacco smoke condensate-exposed THP-1 cells, NLRP3, caspase-1, IL-1β and IL-18 expression are significantly attenuated via suppression of NF-κB, suggesting that MyD88/NF-κB pathway is an upstream regulator of NLRP3 inflammasome in smoking-induced AS.163 Intriguingly, NLRP3 inflammasome activation is inhibited by tobacco smoke condensate in a study, while caspase-1 is activated via TLR4-caspase-8-dependent pathway,164 demonstrating that tobacco smoking is a stimulator of caspase-1 activation, IL-1β and IL-18 secretion, and contributes to the development of AS via different inflammatory signal pathways.
Total serum cholesterol, LDL-C and ox-LDL can also induce macrophages to secrete inflammatory factors that aggravate AS. Cholesterol crystals are phagocytosed by macrophages, causing lysosomal destabilization and leakage of cathepsin B to cytoplasm, which then activates NLRP3 inflammasome.165 Besides, macrophages phagocytosis also causes K+ efflux that further activates NLRP3 inflammasome.165 Consistently, cholesterol is identified to induce NLRP3 inflammasome activation and atherogenesis both in monocytes and mice model.166 Importantly, cholesterol transporters ATP binding cassette A1 and G1 (ABCA1/G1) mediate cholesterol efflux to high-density lipoprotein (HDL), which then suppresses NLRP3 inflammasome activation. Therefore, ABCA1/G1 play a protective role against NLRP3 inflammasome activation,166 As such, stimulation with ox-LDL in THP-1 cells up-regulates P2X7R, NLRP3 and IL-1β expression.167 Moreover, the expression of P2X7R in the atherosclerotic lesions of the aortic sinuses is higher in ApoE−/− mice than in the wild type.167 Knockdown of P2x7r attenuates the development of AS in ApoE−/− mice,167 indicating that P2X7R serves as an NLRP3 driver and plays a key role in the development of AS. Other lipids [eg, the saturated fatty acid (palmitate and stearate) and its metabolic product ceramide and triglyceride], which are danger factors of AS, can also trigger macrophage NLRP3 inflammasome-dependent pyroptosis and accentuate AS development.168–170
Chronic inflammation induced by some components of bacteria is an additional risk factor that can lead to AS. As reported by Chen and co-workers, injection of Lactobacillus casei cell wall extract (LCWE) and feeding with HFD for 8 weeks in ApoE or Ldlr knockout mice shows a dramatic acceleration of AS.171 Besides, LCWE induces atherogenesis through activation of caspase-1 and secretion of IL-1β and IL-18 by CD11c+ macrophages, whereas deletion of caspase-1 or Nlrp3 inhibits atherogenesis.172 Chronic Chlamydia pneumoniae infection has been associated with AS development.173 It has been established that the atherogenic effects of Chlamydia rely on serum cholesterol and are specific for pneumoniae.174 Chlamydia pneumoniae induces macrophage foam cell formation predominantly via modulation of TLR2.175 Moreover, it has been shown that in addition to eliciting NLRP3 inflammasome activation, Chlamydia pneumoniae can also harness NLRP3 inflammasome activation for intracellular replication via increased lipid uptake or utilization of chlamydial inclusions.176 Porphyromonas gingivalis has been shown to accelerate development of atherosclerotic lesion in hyperlipidemic animals. Oral infection with Porphyromonas gingivalis significantly increases the area of atherosclerotic plaque covering the aortic sinus and alveolar bone loss in ApoE−/− mice.177 Stimulation with Porphyromonas gingivalis enhances IL-1β, IL-18 and TNF-α production in peritoneal macrophages, and gene expression of Nlrp3, pro-IL-1β, pro-IL-18 and pro-caspase-1 in gingiva and aorta.177 It can be seen from the above, chronic inflammation caused by some special bacteria is a potential risk of NLRP3 inflammasome-driven AS.
In addition to these regulators and risk factors, some miRNAs are deemed to affect monocyte/macrophage NLRP3 inflammasome activation in AS. MiR-9, a known target of inflammatory pathway, recently has been confirmed to be a negative regulator of NLRP3 inflammasome, thereby attenuating AS.102,178 It has been shown that miR-9 is down-regulated in peripheral blood mononuclear cells isolated from patients with acute coronary syndrome and ApoE−/− mice fed with HFD.178 In ox-LDL-stimulated human THP-1 derived macrophages, inhibition of miR-9 aggravates NLRP3 inflammasome assembly via activation of Janus kinase 1 (JAK1)-STAT.102 Moreover, as reported by Yin et al, miR-155 is remarkably up-regulated in intravascular plaques of ApoE−/− mice fed with HFD.103 Transfection of miR-155 promotes the ox-LDL-induced NLRP3 inflammasome activation in THP-1 macrophages and aggravates atherosclerosis in ApoE−/− mice through phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2).103 More recently, miR-181a has been suggested to be involved in atherogenesis. In the carotid of AS mice and ox-LDL-stimulated THP-1 macrophages, the expression of miR-181a is down-regulated, while the expression of meiosis-specific serine/threonine-protein kinase 1 (MEK1) and NLRP3 is up-regulated. Exogenous overexpression of miR-181a inhibits the activation of the MEK/ERK/NF-κB pathway and decreases the expression of NLRP3 inflammasome-related proteins, including NLRP3, caspase-1, IL-18 and IL-1β.105
Targeting the NLRP3 Inflammasome for the Treatment of AS
Owing to its crucial role in the pathogenesis of inflammatory response, NLRP3 is a promising therapeutic target for AS. To date, a number of advances have been achieved in identifying exogenous compounds that suppress NLRP3 inflammasome activation or IL-1 activity. Whilst most of them are still under pre-clinical studies, the canakinumab anti-inflammatory thrombosis outcome study (CANTOS) trial provides compelling evidence for their anti-inflammatory effect in AS.179 The mechanisms of NLRP3 inflammasome activation are quite complicated, and thus a diverse range of targets are used to develop therapeutic strategies, such as suppression of upstream signaling pathways, inhibition of NLRP3 inflammasome activation, blockade of NLRP3 inflammasome assembly, suppression of caspase-1 activation and IL-1 secretion. Herein, several typical pharmacological inhibitors against AS development will be discussed below (Table 3).
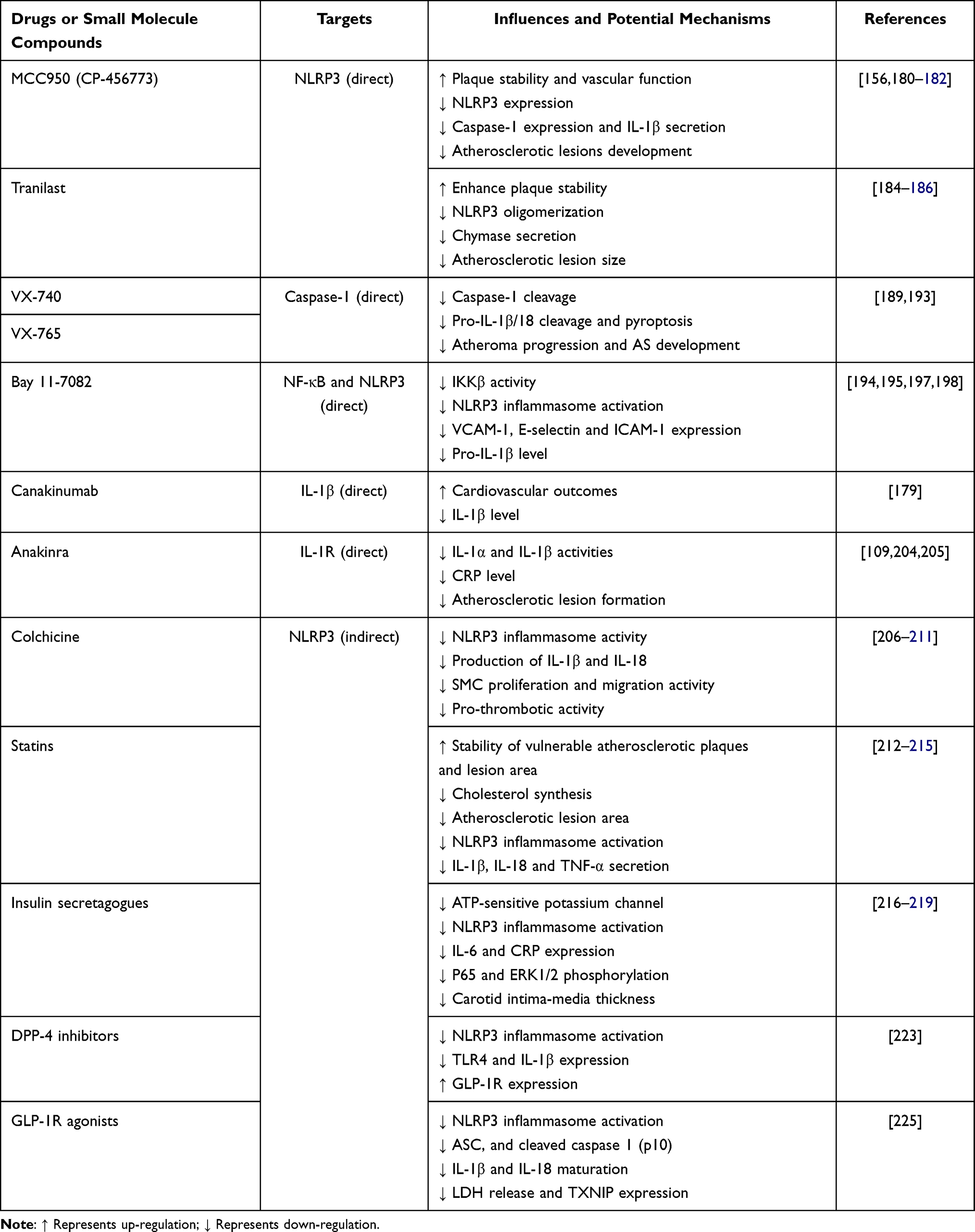 |
Table 3 Potential Drugs or Small Molecule Compounds for the Treatment of AS |
MCC950
MCC950 (also known as CP-456773), a diarylsulfonylurea-containing compound, inhibits ASC oligomerization as well as inflammasome assembly and activation, leading to a reduction in IL-1β production.180 MCC950 specifically inhibits the NLRP3 inflammasome activation at nanomolar concentrations to reduce the secretion of IL-1β, but not the NLRP1, NLRC4 or AIM2 inflammasomes,180 implying that MCC950 partially blocks IL-1β secretion and maintains immune response to certain extent. The potential therapeutic effect of MCC950 in AS has been demonstrated in several studies. MCC950 ameliorates IL-1β release in BMDMs and dendritic cells in response to LPS and cholesterol crystals stimulation, and reduces the development of atherosclerotic lesions in ApoE−/− mice.181 Sharma et al found remarkable down-regulation of the caspase-1 and IL-1β expression in vitro under high glucose or LPS challenge, a reduction in plaque development, an increase in plaque stability, and improvement in vascular function in vivo.182 Similarly, MCC950 alleviates AS development in TMAO-treated ECs and in aortas from ApoE−/− mice, as demonstrated by reduced expression of NLRP3 and IL-1β.156 These results suggest that MCC950 is a potential small compound that improves the development of AS by targeting the NLRP3 inflammasome.
Tranilast
Tranilast, an analogue of tryptophan metabolite, has been approved since 1982 in Korea and Japan for the treatment of allergies, asthma and hypertrophic scars.183 It has been reported that tranilast binds to the NACHT domain of NLRP3 and prevents the assembly of the NLRP3 inflammasome by blocking NLRP3 oligomerization.184 A study has suggested that mice fed with HDL diets are susceptible to plaque vulnerability by increasing the activity and secretion of chymase, while treatment with tranilast significantly inhibits the secretion of chymase, thereby enhancing plaque stability.185 A very recent study has elaborated the protective role of tranilast in AS by targeting NLRP3 inflammasome, in which tranilast markedly suppresses NLRP3 inflammasome activation in Ldlr−/− and ApoE−/− macrophages.186 In vivo, Ldlr−/− and ApoE−/− mice receiving western diets for 12 weeks show a significant reduction in atherosclerotic lesion size after treatment with tranilast, along with a significant decrease in macrophage content and expression of IL-1β in plaques.186 Therefore, tranilast is a promising therapeutic agent for the treatment of NLRP3-related inflammatory diseases such as AS.
VX-740 and VX-765
VX-740 (Pralnacasan) and its analogue VX-765 are oral active prodrugs hydrolyzed by plasma esterases to produce a potent and selective caspase-1 inhibitor.187,188 These two small compounds covalently modify the catalytic cysteine residues at the caspase-1 active sites, thus hindering the cleavage of caspase-1 and processing pro-IL-1β/18.189 VX-740 has been shown to be effective in treating rheumatoid arthritis (RA) and osteoarthritis (OA) both in murine models and phase IIa clinical trials. However, severe hepatotoxicity was observed after long-term application, and therefore further studies have been discontinued.190 Nowadays, VX-765 has become the most commonly exploited caspase-1 inhibitor and has been used for treating various diseases such as psoriasis, epilepsy, RA and AS.191–193 Simultaneously, in Phase II trials for patients with epilepsy and RA, VX-765 exhibits significant neuroprotective and anti-inflammatory effects with a good pharmacokinetic profile.191,192 In addition, VX-765 dramatically reduces pyroptosis and IL-1β expression in ox-LDL-incubated SMCs and inhibits the progression of atheroma and the development of AS in ApoE−/− mice fed with western diets as well.193 Therefore, VX-765 is a promising agent for NLRP3-related diseases.
Bay 11-7082
Bay 11-7082, a phenyl vinyl sulfone, not only blocks the NF-κB signaling pathway by inhibiting the kinase activity of IKKβ, but also selectively inhibits NLRP3 inflammasome, whereas it has no effects on NLRP1, NLRC4, and AIM2 inflammasomes.194–196 Bay 11-7082 suppresses NLRP3 inflammasome activation in the presence of ATP, nigericin, and MSU in macrophages due to the alkylation of cysteine residues of the NLRP3 ATPase region.196 In TNF-α-stimulated ECs, Bay 11-7082 reduces the mRNA levels of VCAM-1, E-selectin, and ICAM-1.197 In addition, pre-treatment with Bay 11-7082 suppresses the expression of NLRP3 and pro-IL-1β in CRP-treated ECs.198 Taken together, Bay 11-7082 shows a good inhibitory effect on NLRP3 inflammasome activation.
Canakinumab
The CANTOS trail has identified IL-1β as a therapeutic target for AS, as its neutralization with antibody-canakinumab (at a dose of 150 mg every 3 months) selectively reduces the probabilities of AS-related cardiovascular events significantly, independent of the reduction in lipid levels.179 However, The CANTOS trail does not show a decline in all-cause mortality, and presences side effects such as leucopenia and a higher rate of fatal infection.179 Although interference with IL-1β improves cardiovascular outcomes clinically, heralding a new era of anti-inflammatory treatment for AS, further studies should identify the subgroups of the patient with inflammation specifically driven AS, and who would benefit most from anti-inflammatory therapy. For instance, as mentioned above, IL-1β functions as a protective factor in advanced atherosclerotic lesions of mice. Treatment of AS with canakinumab at this stage is apparently inappropriate. Therefore, accurate staging of the AS provides important guidance for clinical intervention.
Anakinra
Anakinra, a human IL-1R antagonist, blocks the IL-1R to inhibit the activities of IL-1α and IL-1β. It has recently been approved for the treatment of adult-onset Still’s disease, deficiency of the IL-1R antagonist, rheumatoid arthritis, and cryopyrin-associated periodic syndromes.199–202 Due to the large numbers of patients having received daily treatment with anakinra for over 15 years, the safety of anakinra compared to other biologics has been well established.203 However, it has not been approved for the treatment of AS. In a phase II clinical trial, the patients with acute coronary syndromes received 14 days anakinra show a reduction of CRP expression levels but no improvement in clinical symptoms.204 Animal models of AS have shown a reduction of atherosclerotic lesion formation upon treatment with anakinra,172,205 suggesting that anakinra might be a potential drug against AS.
Colchicine
Colchicine is a broadly available, safe and affordable drug that is used primarily in gouty arthritis and familial Mediterranean fever. More recently, it has been illustrated that colchicine acts as an effective anti-inflammatory agent by blocking the activity of NLRP3 inflammasome and suppressing the secretion of IL-1β and IL-18 under NLRP3 agonists stress.206,207 Pre-clinical data shows a beneficial effect of colchicine on the development of atherogenesis. Colchicine contributes to the diminution of SMC proliferation and migration activity in atherosclerotic plaques and inhibits the pro-thrombotic activity of ox-LDL in ECs.208,209 In patients with the acute coronary syndrome who received short-term colchicine treatment, the expression of IL-1β and caspase-1 is significantly down-regulated compared with the untreated patients,210,211 elucidating that colchicine has a certain atheroprotective property.
Statins
Statins stand out for their ability of inhibiting cholesterol synthesis, which makes them effective agents for the treatment of AS. A 3-hydroxy-3-methylglutaryl coenzyme A inhibitor, known as atorvastatin, is shown to enhance the stability of vulnerable atherosclerotic plaques and reduce aortic lesion area in AS mice model.212 In addition, atorvastatin inhibits NLRP3 inflammasome activation, inflammatory cytokines (IL-1β, TNF-α, and IL-18) secretion and enhances autophagy in ox-LDL-treated RAW264.7 cells.212 Furthermore, atorvastatin exerts an anti-inflammatory effect by inhibiting NLRP3 inflammasome activation due to its suppression of TLR4/MyD88/NF-κB pathway in THP-1 cells stimulated by phorbol 12-myristate 13-acetate (PMA).213
In addition to atorvastatin, rosuvastatin and simvastatin also show potency for the treatment of AS. In acute coronary syndrome patients, administration of high dose rosuvastatin modulates the inflammatory process of AS by down-regulation of cathepsin-B, NLRP3, IL-1β and IL-18.214 Treatment with simvastatin suppresses NLRP3 inflammasome activation and IL-1β release triggered by cholesterol crystals in human peripheral blood mononuclear cells.215 These studies highlight the protective role of statins against AS.
Insulin Secretagogues
To date, studies have confirmed that some insulin secretagogues can prevent the progression of AS. Glyburide, a sulfonylurea drug widely used in the treatment of type 2 diabetes mellitus (T2DM), blocks the ATP-sensitive potassium channel (KATP), to release insulin.216 Glyburide has been reported to prevent crystal-induced NLRP3 inflammasome activation in BMDMs, and this inhibition effect seems to be specific to NLRP3 inflammasome as it cannot prevent the IL-1β secretion from other NLRs.217 It has been shown that administration of glyburide remarkedly ameliorates the progression of AS and reduces the production of inflammatory cytokines as well as phosphorylation of p65 and ERK1/2 in LPS-stimulated RAW264.7 cells.218 A clinical trial has also revealed that receiving repaglinide or glyburide for 12 months reduces postprandial glucose peak, serum IL-6 and CRP level, and carotid intima-media thickness,219 suggesting that these two hypoglycemic drugs have anti-AS effects to some extent.
Dipeptidyl Peptidase 4 (DPP-4) Inhibitors and Glucagon Like Peptide 1 Receptor (GLP-1R) Agonists
DPP-4 inhibitors and GLP-1R agonists are novel groups of medicine used in T2DM. DPP inhibitors protect the incretin hormone GLP-1 and glucose dependent insulinotropic polypeptide (GIP) from enzymatic degradation by DPP-4 to promote diet-stimulated insulin secretion from pancreatic β cells.220 Beyond the hypoglycemic effect, DPP-4 inhibitors have been shown the beneficial effects on body weight, blood pressure, postprandial lipemia, inflammatory markers, oxidative stress, and endothelial function in patients with T2DM.221,222 Besides, DPP-4 inhibitors have been supposed to exert anti-AS function to a certain extent. Sitagliptin and NVPDPP728, two kinds of DPP-4 inhibitors, can induce a significant reduction in NLRP3, TLR4 and IL-1β expression and increasement in GLP-1R expression in ox-LDL-induced THP-1 cells.223 GLP-1R agonists exhibit cardiovascular benefits as well.224 Dulaglutide, a newly developed GLP-1R agonist, possesses a protective effect against high glucose- induced activation of the NLRP3 inflammasome in endothelial cells.225 These findings reveal the potential anti-AS effects of DPP-4 inhibitors and GLP-1R agonists.
Conclusion and Perspective
Shedding light on the molecular mechanisms in charge of NLRP3 inflammasome activation is essential for improving our understanding of the pathogenesis of AS in association with the NLRP3 inflammasome. Numerous DAMPs and PAMPs trigger canonical, non-canonical and alternative NLRP3 inflammasome activation through a variety of signaling and cellular events (Figure 7). Upon activation, it enhances the cleavage property of pro-caspase-1, the release of the inflammatory cytokines (IL-1β and IL-18), and induces inflammatory cell death (pyroptosis). These discoveries provide new insights into the understanding of NLRP3 inflammasome activation.
 |
Figure 7 Possible factors that are involved in NLRP3 inflammasome activation. Many risks influence different cells so as to affect human bodies by activating NLRP3 inflammasome. Note: Created with BioRender.com. |
It is well accepted that the progression of AS is implicated in the NLRP3 inflammasome. ECs injury, the infiltration of monocytes/ macrophages, and the formation of foam cells are the main pathologic changes of AS, and the risk factors of AS (eg, hemodynamic force, hyperlipidemia, hyperglycemia, and tobacco smoking) are acting on these cells and leading to AS via activation of NLRP3 inflammasome. Hence, inhibition of NLRP3 inflammasome activation is a possible therapeutic strategy for the treatment of AS. The current treatment for AS in clinical is primarily lipid-lowering therapy. In recent, anti-IL-1β therapy has been confirmed to be extremely effective in the treatment of AS, indirectly implying that activation of the NLRP3 inflammasome is a major driver of AS. Besides, some small-molecule inhibitors that directly inhibit NLRP3 inflammasome and caspase-1 activation could be a promising therapeutic strategy to combat AS, but some have serious side effects and are still in clinical trials. Further studies should focus on the development of specific small-molecule inhibitors of NLRP3 inflammasome with improved pharmacokinetic properties to ameliorate the progression of AS.
Abbreviations
3-MA, 3-methyladenine; ABCA1/G1, ATP binding cassette A1 and G1; ALRs, absent-in-melanoma-like receptors; AIM, absent-in-melanoma; ApoC3, apolipoprotein C3; ApoE, apolipoprotein E; AS, atherosclerosis; BMDMs, bone marrow-derived macrophages; cAMP, cyclic adenosine monophosphate; CANTOS, canakinumab anti-inflammatory thrombosis outcome study; CARD, caspase recruitment domain; CaSR, calcium-sensing receptor; CHOP, C/EPB homologous protein; CLIC, chloride intracellular channel; CLRs, C-type lectin receptors; CMPK2, Cytidine/uridine monophosphate kinase 2; CoPP, cobalt protoporphyrin; CRP, C-reactive protein; CVD, cardiovascular disease; CXCL16, C-X-C motif chemokine ligand 16; DAG, diacyl glycerol; DAMPs, damage-associated molecular patterns; dTGN, dispersed trans-Golgi network; DDX3X: DEAD-box helicase 3 X-linked; ECs, endothelial cells; DPP-4, dipeptidyl peptidase 4; ELF3, E74-like ETS transcription factor 3; ER, endoplasmic reticulum; ERK1/2, extracellular signal-regulated kinase 1/2; GBPs, guanylate-binding proteins; GLP-1R, glucagon like peptide 1 receptor; GPRC6A, G protein-coupled calcium-sensing receptor; GSDMD, gasdermin D; HCD, high-cholesterol diets; HFD, high-fat diets; ICAM-1, intercellular adhesion molecule-1; IFN, type I interferon; IL-1β/18, interleukin-1β/18; IL-1R, IL-1 receptor; IP3R, inositol 1,4,5-trisphosphate receptor; JAK1, Janus kinase 1; JNK-1, C-Jun N-terminal kinase 1; LCWE, Lactobacillus casei cell wall extract; LDL-C, low-density lipoprotein cholesterol; LDLR, low-density lipoprotein receptor; LRR, leucine-rich repeat; MAMs, mitochondria-associated endoplasmic reticulum membranes; MARK4, microtubule affinity-regulating kinase 4; MAVS, mitochondrial antiviral signaling protein; MEK1, meiosis-specific serine/threonine-protein kinase 1; MPT, mitochondrial permeability transition; NAC, N-acetyl-L-cysteine; NACHT, oligomerization domain; NAIP, NLR family apoptosis inhibitory protein; NCLX, mitochondrial Na+/Ca2+ exchanger; NEK7, never-in-mitosis A-related kinase 7; NF-κB, nuclear factor-κB; NLRP3, NOD-like receptor family and the pyridine-containing domain 3; OA, osteoarthritis; OS, oscillatory stress; ox-LDL, oxidized LDL; Ox-PAPC, Oxidized-1-palmitoyl-2-arachidonoyl-sn-glycerol-3-phosphorylcholine; P2X7R, P2X purinoceptor 7; PAK1, p21-activated kinase 1; PAMPs, pathogen-associated molecular patterns; PCV, pathogen-containing vacuole; PGE2, prostaglandin E2; PtdIns4P, phosphatidylinositol-4-phosphate; PTPN22, protein tyrosine phosphatase non-receptor type 22; PKA, protein kinase A; PKD, protein kinase D; PLC, phospholipase C; PP2A, protein phosphatase 2A; PRRs, pattern recognition receptors; PTMs, post-translation modifications; PYD, pyrin domain; PYK2, proline-rich tyrosine kinase 2; RA, rheumatoid arthritis; RLRs, retinoic acid-inducible gene-I-like receptors; ROS, reactive oxygen species; SCAPs, SREBP cleavage-activating proteins; SESN2, sestrin; SIRT3, sirtuin 3; SLPC, stearoyl lysophosphatidylcholine; SMCs, smooth muscle cells; SOD2, superoxide dismutase 2; SREBPs, sterol regulatory element-binding proteins; STAT3, signal transducer and activator of transcription 3; SYK, spleen associated tyrosine kinase; TET2, ten eleven translocation methylcytosine dioxygenase 2; TIFA, TNF-α receptor-associated factor-interacting protein with forkhead-associated domain; TLRs, Toll-like receptors; TMAO, trimethylamine-N-oxide; TNF, tumor necrosis factor; TRPM2, transient receptor potential melastatin 2; TWIK2, two-pore domain potassium channel; TXNIP, thioredoxin-interacting protein; VCAM-1, vascular cell adhesion molecule-1; VRAC, volume-regulated anion channel.
Acknowledgments
This work is supported by National Natural Science Foundation of China (Grant No. 82003476 to Ningning Wang), (Grant No. 81872623 to Guang Yang) and China Scholarship Council (CSC, Grant/Award No. 202008080101 to Chunteng Jiang). The authors thank cartographers Zhongsai Ye and Xiaoye Zhuang for helping us to draw such fantastic mechanical illustrations, and thank associate professor Dr. Ruoxu Gu for helping us to polish the review.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. Guang Yang and Ningning Wang are co-correspondence authors.
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Ezzati M, Obermeyer Z, Tzoulaki I, Mayosi BM, Elliott P, Leon DA. Contributions of risk factors and medical care to cardiovascular mortality trends. Nat Rev Cardiol. 2015;12(9):508–530. doi:10.1038/nrcardio.2015.82
2. Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15(2):104–116. doi:10.1038/nri3793
3. Libby P, Hansson GK. Inflammation and immunity in diseases of the arterial tree: players and layers. Circ Res. 2015;116(2):307–311. doi:10.1161/CIRCRESAHA.116.301313
4. Kim YK, Shin JS, Nahm MH. NOD-like receptors in infection, immunity, and diseases. Yonsei Med J. 2016;57(1):5–14. doi:10.3349/ymj.2016.57.1.5
5. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4(3):a006049–a006049. doi:10.1101/cshperspect.a006049
6. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi:10.1016/j.cell.2010.01.022
7. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153(2):348–361. doi:10.1016/j.cell.2013.02.054
8. Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res. 2018;122(12):1722–1740. doi:10.1161/CIRCRESAHA.118.311362
9. Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: its regulation and involvement in atherosclerosis. J Cell Physiol. 2018;233(3):2116–2132. doi:10.1002/jcp.25930
10. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. doi:10.1016/j.cell.2010.01.040
11. Cai X, Chen J, Xu H, et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell. 2014;156(6):1207–1222. doi:10.1016/j.cell.2014.01.063
12. Li Y, Fu TM, Lu A, et al. Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc Natl Acad Sci U S A. 2018;115(43):10845–10852. doi:10.1073/pnas.1810524115
13. Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi:10.1038/nature15514
14. Kesavardhana S, Kanneganti TD. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol. 2017;29(5):201–210. doi:10.1093/intimm/dxx018
15. McKee CM, Coll RC. NLRP3 inflammasome priming: a riddle wrapped in a mystery inside an enigma. J Leukoc Biol. 2020;108(3):937–952. doi:10.1002/JLB.3MR0720-513R
16. Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–791. doi:10.4049/jimmunol.0901363
17. Lamkanfi M, Kanneganti TD. Nlrp3: an immune sensor of cellular stress and infection. Int J Biochem Cell Biol. 2010;42(6):792–795. doi:10.1016/j.biocel.2010.01.008
18. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–1153. doi:10.1016/j.immuni.2013.05.016
19. Lee GS, Subramanian N, Kim AI, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492(7427):123–127. doi:10.1038/nature11588
20. Green JP, Yu S, Martín-Sánchez F, et al. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc Natl Acad Sci U S A. 2018;115(40):E9371–e9380. doi:10.1073/pnas.1812744115
21. Scambler T, Jarosz-Griffiths HH, Lara-Reyna S, et al. ENaC-mediated sodium influx exacerbates NLRP3-dependent inflammation in cystic fibrosis. eLife. 2019;8. doi:10.7554/eLife.49248
22. He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530(7590):354–357. doi:10.1038/nature16959
23. Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–232. doi:10.1038/nature04515
24. Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem. 1994;269(21):15195–15203. doi:10.1016/S0021-9258(17)36591-2
25. Kanneganti TD, Lamkanfi M, Kim YG, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. 2007;26(4):433–443. doi:10.1016/j.immuni.2007.03.008
26. Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J. 1995;14(8):1607–1614. doi:10.1002/j.1460-2075.1995.tb07149.x
27. Di A, Xiong S, Ye Z, et al. The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity. 2018;49(1):56–65.e54. doi:10.1016/j.immuni.2018.04.032
28. Shi H, Wang Y, Li X, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2016;17(3):250–258. doi:10.1038/ni.3333
29. Samir P, Kesavardhana S, Patmore DM, et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature. 2019;573(7775):590–594. doi:10.1038/s41586-019-1551-2
30. Groß CJ, Mishra R, Schneider KS, et al. K(+) efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity. 2016;45(4):761–773. doi:10.1016/j.immuni.2016.08.010
31. Sanman LE, Qian Y, Eisele NA, et al. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. eLife. 2016;5:e13663. doi:10.7554/eLife.13663
32. Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. 2009;30(6):860–874. doi:10.1016/j.immuni.2009.04.012
33. Murakami T, Ockinger J, Yu J, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2012;109(28):11282–11287. doi:10.1073/pnas.1117765109
34. Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci. 2013;126(Pt 13):2903–2913. doi:10.1242/jcs.124388
35. Rossol M, Pierer M, Raulien N, et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun. 2012;3:1329. doi:10.1038/ncomms2339
36. Lebeaupin C, Proics E, de Bieville CH, et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015;6(9):e1879. doi:10.1038/cddis.2015.248
37. Compan V, Baroja-Mazo A, López-Castejón G, et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity. 2012;37(3):487–500. doi:10.1016/j.immuni.2012.06.013
38. Yaron JR, Gangaraju S, Rao MY, et al. K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis. 2015;6(10):e1954. doi:10.1038/cddis.2015.277
39. Katsnelson MA, Rucker LG, Russo HM, Dubyak GR. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol. 2015;194(8):3937–3952. doi:10.4049/jimmunol.1402658
40. Feraille E, Dizin E. Coordinated control of ENaC and Na+, K+-ATPase in renal collecting duct. J Am Soc Nephrol. 2016;27(9):2554–2563. doi:10.1681/ASN.2016020124
41. Verhoef PA, Kertesy SB, Lundberg K, Kahlenberg JM, Dubyak GR. Inhibitory effects of chloride on the activation of caspase-1, IL-1beta secretion, and cytolysis by the P2X7 receptor. J Immunol. 2005;175(11):7623–7634. doi:10.4049/jimmunol.175.11.7623
42. Daniels MJ, Rivers-Auty J, Schilling T, et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun. 2016;7:12504. doi:10.1038/ncomms12504
43. Tang T, Lang X, Xu C, et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun. 2017;8(1):202. doi:10.1038/s41467-017-00227-x
44. Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282(5):2871–2879. doi:10.1074/jbc.M608083200
45. Zhong Z, Zhai Y, Liang S, et al. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun. 2013;4:1611. doi:10.1038/ncomms2608
46. Sarkar S, Malovic E, Harishchandra DS, et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinson’s Dis. 2017;3:30. doi:10.1038/s41531-017-0032-2
47. Jabaut J, Ather JL, Taracanova A, Poynter ME, Ckless K. Mitochondria-targeted drugs enhance Nlrp3 inflammasome-dependent IL-1β secretion in association with alterations in cellular redox and energy status. Free Radic Biol Med. 2013;60:233–245. doi:10.1016/j.freeradbiomed.2013.01.025
48. Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi:10.1038/nature08780
49. Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787(11):1395–1401. doi:10.1016/j.bbabio.2009.06.009
50. Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. doi:10.1038/ni.1980
51. Shimada K, Crother TR, Karlin J, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. doi:10.1016/j.immuni.2012.01.009
52. Han Y, Xu X, Tang C, et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: the role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 2018;16:32–46. doi:10.1016/j.redox.2018.02.013
53. Dashdorj A, Jyothi KR, Lim S, et al. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013;11:178. doi:10.1186/1741-7015-11-178
54. Zu Y, Wan LJ, Cui SY, Gong YP, Li CL. The mitochondrial Na(+)/Ca(2+) exchanger may reduce high glucose-induced oxidative stress and nucleotide-binding oligomerization domain receptor 3 inflammasome activation in endothelial cells. J Geriatr Cardiol. 2015;12(3):270–278. doi:10.11909/j.issn.1671-5411.2015.03.003
55. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–140. doi:10.1038/ni.1831
56. Iyer SS, He Q, Janczy JR, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39(2):311–323. doi:10.1016/j.immuni.2013.08.001
57. Dagvadorj J, Mikulska-Ruminska K, Tumurkhuu G, et al. Recruitment of pro-IL-1α to mitochondrial cardiolipin, via shared LC3 binding domain, inhibits mitophagy and drives maximal NLRP3 activation. Proc Natl Acad Sci U S A. 2021;118(1). doi:10.1073/pnas.2015632118
58. Park S, Juliana C, Hong S, et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol. 2013;191(8):4358–4366. doi:10.4049/jimmunol.1301170
59. Campden RI, Zhang Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch Biochem Biophys. 2019;670:32–42. doi:10.1016/j.abb.2019.02.015
60. Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9(8):847–856. doi:10.1038/ni.1631
61. Bruchard M, Mignot G, Derangère V, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. 2013;19(1):57–64. doi:10.1038/nm.2999
62. Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple cathepsins promote Pro-IL-1β synthesis and NLRP3-mediated IL-1β activation. J Immunol. 2015;195(4):1685–1697. doi:10.4049/jimmunol.1500509
63. Oleszycka E, Moran HB, Tynan GA, et al. IL-1α and inflammasome-independent IL-1β promote neutrophil infiltration following alum vaccination. FEBS J. 2016;283(1):9–24. doi:10.1111/febs.13546
64. Bauer C, Duewell P, Mayer C, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 2010;59(9):1192–1199. doi:10.1136/gut.2009.197822
65. Edye ME, Lopez-Castejon G, Allan SM, Brough D. Acidosis drives damage-associated molecular pattern (DAMP)-induced interleukin-1 secretion via a caspase-1-independent pathway. J Biol Chem. 2013;288(42):30485–30494. doi:10.1074/jbc.M113.478941
66. Katsnelson MA, Lozada-Soto KM, Russo HM, Miller BA, Dubyak GR. NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: roles for K+ efflux and Ca2+ influx. Am J Physiol Cell Physiol. 2016;311(1):C83–c100. doi:10.1152/ajpcell.00298.2015
67. Tseng HHL, Vong CT, Kwan YW, Lee SM, Hoi MPM. Lysosomal Ca(2+) signaling regulates high glucose-mediated interleukin-1β secretion via transcription factor EB in human monocytic cells. Front Immunol. 2017;8:1161. doi:10.3389/fimmu.2017.01161
68. Zhang Z, Meszaros G, He WT, et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med. 2017;214(9):2671–2693. doi:10.1084/jem.20162040
69. Hong S, Hwang I, Gim E, et al. Brefeldin A-sensitive ER-Golgi vesicle trafficking contributes to NLRP3-dependent caspase-1 activation. FASEB J. 2019;33(3):4547–4558. doi:10.1096/fj.201801585R
70. Guo C, Chi Z, Jiang D, et al. Cholesterol homeostatic regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity. 2018;49(5):842–856.e847. doi:10.1016/j.immuni.2018.08.021
71. Chen J, Chen ZJ. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature. 2018;564(7734):71–76. doi:10.1038/s41586-018-0761-3
72. Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9(9):859–864. doi:10.1038/embor.2008.163
73. Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–156. doi:10.1146/annurev-biochem-052709-094552
74. Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–268. doi:10.1038/nature07383
75. Liu P, Huang G, Wei T, et al. Sirtuin 3-induced macrophage autophagy in regulating NLRP3 inflammasome activation. Biochim Biophys Acta Mol Basis Dis. 2018;1864(3):764–777. doi:10.1016/j.bbadis.2017.12.027
76. Cho MH, Cho K, Kang HJ, et al. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10(10):1761–1775. doi:10.4161/auto.29647
77. Nurmi K, Kareinen I, Virkanen J, et al. Hemin and cobalt protoporphyrin inhibit NLRP3 inflammasome activation by enhancing autophagy: a novel mechanism of inflammasome regulation. J Innate Immun. 2017;9(1):65–82. doi:10.1159/000448894
78. Spalinger MR, Lang S, Gottier C, et al. PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy. 2017;13(9):1590–1601. doi:10.1080/15548627.2017.1341453
79. Kim MJ, Bae SH, Ryu JC, et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy. 2016;12(8):1272–1291. doi:10.1080/15548627.2016.1183081
80. Houtman J, Freitag K, Gimber N, Schmoranzer J, Heppner FL, Jendrach M. Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3. EMBO J. 2019;38(4). doi:10.15252/embj.201899430
81. Wang X, Jiang L, Shi L, et al. Zearalenone induces NLRP3-dependent pyroptosis via activation of NF-κB modulated by autophagy in INS-1 cells. Toxicology. 2019;428:152304. doi:10.1016/j.tox.2019.152304
82. Jiang C, Jiang L, Li Q, et al. Acrolein induces NLRP3 inflammasome-mediated pyroptosis and suppresses migration via ROS-dependent autophagy in vascular endothelial cells. Toxicology. 2018;410:26–40. doi:10.1016/j.tox.2018.09.002
83. Jiang L, Luo S, Qiu T, et al. Bidirectional role of reactive oxygen species during inflammasome activation in acrolein-induced human EAhy926 cells pyroptosis. Toxicol Mech Methods. 2021;31:680–689.
84. Vu LD, Gevaert K, De Smet I. Protein language: post-translational modifications talking to each other. Trends Plant Sci. 2018;23(12):1068–1080. doi:10.1016/j.tplants.2018.09.004
85. Liu D, Zeng X, Li X, et al. Advances in the molecular mechanisms of NLRP3 inflammasome activators and inactivators. Biochem Pharmacol. 2020;175:113863. doi:10.1016/j.bcp.2020.113863
86. Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489. doi:10.1038/s41577-019-0165-0
87. Spalinger MR, Kasper S, Gottier C, et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest. 2016;126(5):1783–1800. doi:10.1172/JCI83669
88. Stutz A, Kolbe CC, Stahl R, et al. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J Exp Med. 2017;214(6):1725–1736. doi:10.1084/jem.20160933
89. Martin BN, Wang C, Willette-Brown J, et al. IKKα negatively regulates ASC-dependent inflammasome activation. Nat Commun. 2014;5:4977. doi:10.1038/ncomms5977
90. Song N, Liu ZS, Xue W, et al. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol Cell. 2017;68(1):185–197.e186. doi:10.1016/j.molcel.2017.08.017
91. Hara H, Tsuchiya K, Kawamura I, et al. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol. 2013;14(12):1247–1255. doi:10.1038/ni.2749
92. Lin YC, Huang DY, Wang JS, et al. Syk is involved in NLRP3 inflammasome-mediated caspase-1 activation through adaptor ASC phosphorylation and enhanced oligomerization. J Leukoc Biol. 2015;97(5):825–835. doi:10.1189/jlb.3HI0814-371RR
93. Chung IC, OuYang CN, Yuan SN, et al. Pyk2 activates the NLRP3 inflammasome by directly phosphorylating ASC and contributes to inflammasome-dependent peritonitis. Sci Rep. 2016;6:36214. doi:10.1038/srep36214
94. Mambwe B, Neo K, Javanmard Khameneh H, et al. Tyrosine dephosphorylation of ASC modulates the activation of the NLRP3 and AIM2 inflammasomes. Front Immunol. 2019;10:1556. doi:10.3389/fimmu.2019.01556
95. Basak C, Pathak SK, Bhattacharyya A, Mandal D, Pathak S, Kundu M. NF-kappaB- and C/EBPbeta-driven interleukin-1beta gene expression and PAK1-mediated caspase-1 activation play essential roles in interleukin-1beta release from Helicobacter pylori lipopolysaccharide-stimulated macrophages. J Biol Chem. 2005;280(6):4279–4288. doi:10.1074/jbc.M412820200
96. Zhang A, Xing J, Xia T, et al. EphA2 phosphorylates NLRP3 and inhibits inflammasomes in airway epithelial cells. EMBO Rep. 2020;21(7):e49666. doi:10.15252/embr.201949666
97. Mortimer L, Moreau F, MacDonald JA, Chadee K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat Immunol. 2016;17(10):1176–1186. doi:10.1038/ni.3538
98. Guo C, Xie S, Chi Z, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. 2016;45(4):802–816. doi:10.1016/j.immuni.2016.09.008
99. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi:10.1016/j.cell.2009.01.002
100. Zhou Y, Lu M, Du RH, et al. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol Neurodegener. 2016;11:28. doi:10.1186/s13024-016-0094-3
101. Li XF, Shen WW, Sun YY, et al. MicroRNA-20a negatively regulates expression of NLRP3-inflammasome by targeting TXNIP in adjuvant-induced arthritis fibroblast-like synoviocytes. Joint Bone Spine. 2016;83(6):695–700. doi:10.1016/j.jbspin.2015.10.007
102. Wang Y, Han Z, Fan Y, et al. MicroRNA-9 inhibits NLRP3 inflammasome activation in human atherosclerosis inflammation cell models through the JAK1/STAT signaling pathway. Cell Physiol Biochem. 2017;41(4):1555–1571. doi:10.1159/000470822
103. Yin R, Zhu X, Wang J, et al. MicroRNA-155 promotes the ox-LDL-induced activation of NLRP3 inflammasomes via the ERK1/2 pathway in THP-1 macrophages and aggravates atherosclerosis in ApoE-/- mice. Ann Palliat Med. 2019;8(5):676–689. doi:10.21037/apm.2019.10.11
104. Li P, Zhong X, Li J, et al. MicroRNA-30c-5p inhibits NLRP3 inflammasome-mediated endothelial cell pyroptosis through FOXO3 down-regulation in atherosclerosis. Biochem Biophys Res Commun. 2018;503(4):2833–2840. doi:10.1016/j.bbrc.2018.08.049
105. Song J, Yang S, Yin R, Xiao Q, Ma A, Pan X. MicroRNA-181a regulates the activation of the NLRP3 inflammatory pathway by targeting MEK1 in THP-1 macrophages stimulated by ox-LDL. J Cell Biochem. 2019;120(8):13640–13650. doi:10.1002/jcb.28637
106. Xu X, Yang Y, Wang G, et al. Low shear stress regulates vascular endothelial cell pyroptosis through miR-181b-5p/STAT-3 axis. J Cell Physiol. 2021;236(1):318–327. doi:10.1002/jcp.29844
107. Chen M, Li W, Zhang Y, Yang J. MicroRNA-20a protects human aortic endothelial cells from Ox-LDL-induced inflammation through targeting TLR4 and TXNIP signaling. Biomed Pharmacother. 2018;103:191–197. doi:10.1016/j.biopha.2018.03.129
108. Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. doi:10.1038/nature10558
109. Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–192. doi:10.1038/nature13683
110. Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 requires the Pannexin-1 channel and the Purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. 2015;43(5):923–932. doi:10.1016/j.immuni.2015.10.009
111. He Y, Franchi L, Núñez G. TLR agonists stimulate Nlrp3-dependent IL-1β production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J Immunol. 2013;190(1):334–339. doi:10.4049/jimmunol.1202737
112. Rathinam VA, Vanaja SK, Waggoner L, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150(3):606–619. doi:10.1016/j.cell.2012.07.007
113. Gurung P, Malireddi RK, Anand PK, et al. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-β (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem. 2012;287(41):34474–34483. doi:10.1074/jbc.M112.401406
114. Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. 2016;165(4):792–800. doi:10.1016/j.cell.2016.03.046
115. Guarda G, Braun M, Staehli F, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34(2):213–223. doi:10.1016/j.immuni.2011.02.006
116. Zanoni I, Tan Y, Di Gioia M, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science (New York, NY). 2016;352(6290):1232–1236. doi:10.1126/science.aaf3036
117. Chu LH, Indramohan M, Ratsimandresy RA, et al. The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat Commun. 2018;9(1):996. doi:10.1038/s41467-018-03409-3
118. Li W, Zhang W, Deng M, et al. Stearoyl lysophosphatidylcholine inhibits endotoxin-induced Caspase-11 activation. Shock (Augusta, Ga). 2018;50(3):339–345. doi:10.1097/SHK.0000000000001012
119. Zasłona Z, Flis E, Wilk MM, et al. Caspase-11 promotes allergic airway inflammation. Nat Commun. 2020;11(1):1055. doi:10.1038/s41467-020-14945-2
120. Piccini A, Carta S, Tassi S, Lasiglié D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A. 2008;105(23):8067–8072. doi:10.1073/pnas.0709684105
121. Netea MG, Nold-Petry CA, Nold MF, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009;113(10):2324–2335. doi:10.1182/blood-2008-03-146720
122. Gaidt MM, Ebert TS, Chauhan D, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. 2016;44(4):833–846. doi:10.1016/j.immuni.2016.01.012
123. Zewinger S, Reiser J, Jankowski V, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol. 2020;21(1):30–41. doi:10.1038/s41590-019-0548-1
124. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi:10.1038/nature08938
125. Mehta S, Dhawan V. Exposure of cigarette smoke condensate activates NLRP3 inflammasome in THP-1 cells in a stage-specific manner: an underlying role of innate immunity in atherosclerosis. Cell Signal. 2020;72:109645. doi:10.1016/j.cellsig.2020.109645
126. Wen C, Yang X, Yan Z, et al. Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int J Cardiol. 2013;168(3):2242–2247. doi:10.1016/j.ijcard.2013.01.211
127. Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol. 1996;16(8):1000–1006. doi:10.1161/01.ATV.16.8.1000
128. Bhaskar V, Yin J, Mirza AM, et al. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E-deficient mice. Atherosclerosis. 2011;216(2):313–320. doi:10.1016/j.atherosclerosis.2011.02.026
129. Gomez D, Baylis RA, Durgin BG, et al. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat Med. 2018;24(9):1418–1429. doi:10.1038/s41591-018-0124-5
130. Hendrikx T, Jeurissen ML, van Gorp PJ, et al. Bone marrow-specific caspase-1/11 deficiency inhibits atherosclerosis development in Ldlr(-/-) mice. FEBS J. 2015;282(12):2327–2338. doi:10.1111/febs.13279
131. Zheng F, Xing S, Gong Z, Xing Q. NLRP3 inflammasomes show high expression in aorta of patients with atherosclerosis. Heart Lung Circ. 2013;22(9):746–750. doi:10.1016/j.hlc.2013.01.012
132. Shi X, Xie WL, Kong WW, Chen D, Qu P. Expression of the NLRP3 inflammasome in carotid atherosclerosis. J Stroke Cerebrovasc Dis. 2015;24(11):2455–2466. doi:10.1016/j.jstrokecerebrovasdis.2015.03.024
133. Paramel Varghese G, Folkersen L, Strawbridge RJ, et al. NLRP3 inflammasome expression and activation in human atherosclerosis. J Am Heart Assoc. 2016;5(5). doi:10.1161/JAHA.115.003031
134. Schunk SJ, Kleber ME, März W, et al. Genetically determined NLRP3 inflammasome activation associates with systemic inflammation and cardiovascular mortality. Eur Heart J. 2021;42(18):1742–1756. doi:10.1093/eurheartj/ehab107
135. Zhao X, Gu C, Yan C, et al. NALP3-inflammasome-related gene polymorphisms in patients with prehypertension and coronary atherosclerosis. Biomed Res Int. 2016;2016:7395627. doi:10.1155/2016/7395627
136. Cersosimo E, DeFronzo RA. Insulin resistance and endothelial dysfunction: the road map to cardiovascular diseases. Diabetes Metab Res Rev. 2006;22(6):423–436. doi:10.1002/dmrr.634
137. Peng Z, Shu B, Zhang Y, Wang M. Endothelial response to pathophysiological stress. Arterioscler Thromb Vasc Biol. 2019;39(11):e233–e243. doi:10.1161/ATVBAHA.119.312580
138. Klein CL, Köhler H, Kirkpatrick CJ. Increased adhesion and activation of polymorphonuclear neutrophil granulocytes to endothelial cells under heavy metal exposure in vitro. Pathobiology. 1994;62(2):90–98. doi:10.1159/000163883
139. Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. 2017;120(4):713–735. doi:10.1161/CIRCRESAHA.116.309326
140. Sheikine Y, Sirsjö A. CXCL16/SR-PSOX–a friend or a foe in atherosclerosis? Atherosclerosis. 2008;197(2):487–495. doi:10.1016/j.atherosclerosis.2007.11.034
141. Xiao H, Lu M, Lin TY, et al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. 2013;128(6):632–642. doi:10.1161/CIRCULATIONAHA.113.002714
142. Chien CS, Li JY, Chien Y, et al. METTL3-dependent N(6)-methyladenosine RNA modification mediates the atherogenic inflammatory cascades in vascular endothelium. Proc Natl Acad Sci U S A. 2021;118(7). doi:10.1073/pnas.2025070118
143. Koka S, Xia M, Chen Y, et al. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. 2017;13:336–344. doi:10.1016/j.redox.2017.06.004
144. Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011;14(5):575–585. doi:10.1016/j.cmet.2011.07.015
145. Yin Y, Li X, Sha X, et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler Thromb Vasc Biol. 2015;35(4):804–816. doi:10.1161/ATVBAHA.115.305282
146. Lin TY, Wei TW, Li S, et al. TIFA as a crucial mediator for NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2016;113(52):15078–15083. doi:10.1073/pnas.1618773114
147. Hang L, Peng Y, Xiang R, Li X, Li Z. Ox-LDL causes endothelial cell injury through ASK1/NLRP3-mediated inflammasome activation via endoplasmic reticulum stress. Drug Des Devel Ther. 2020;14:731–744. doi:10.2147/DDDT.S231916
148. Huang D, Gao W, Zhong X, Ge J. NLRP3 activation in endothelia promotes development of diabetes-associated atherosclerosis. Aging. 2020;12(18):18181–18191. doi:10.18632/aging.103666
149. Zhang Y, Li X, Pitzer AL, Chen Y, Wang L, Li PL. Coronary endothelial dysfunction induced by nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3 inflammasome activation during hypercholesterolemia: beyond inflammation. Antioxid Redox Signal. 2015;22(13):1084–1096. doi:10.1089/ars.2014.5978
150. Wang J, Shen X, Liu J, et al. High glucose mediates NLRP3 inflammasome activation via upregulation of ELF3 expression. Cell Death Dis. 2020;11(5):383. doi:10.1038/s41419-020-2598-6
151. Zhaolin Z, Jiaojiao C, Peng W, et al. OxLDL induces vascular endothelial cell pyroptosis through miR-125a-5p/TET2 pathway. J Cell Physiol. 2019;234(5):7475–7491. doi:10.1002/jcp.27509
152. Wu X, Zhang H, Qi W, et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018;9(2):171. doi:10.1038/s41419-017-0257-3
153. Zhang Y, Chen Y, Zhang Y, Li PL, Li X. Contribution of cathepsin B-dependent Nlrp3 inflammasome activation to nicotine-induced endothelial barrier dysfunction. Eur J Pharmacol. 2019;865:172795. doi:10.1016/j.ejphar.2019.172795
154. Cao X, Wang S, Bi R, Tian S, Huo Y, Liu J. Toxic effects of Cr(VI) on the bovine hemoglobin and human vascular endothelial cells: molecular interaction and cell damage. Chemosphere. 2019;222:355–363. doi:10.1016/j.chemosphere.2019.01.137
155. Chen H, Lu Y, Cao Z, et al. Cadmium induces NLRP3 inflammasome-dependent pyroptosis in vascular endothelial cells. Toxicol Lett. 2016;246:7–16. doi:10.1016/j.toxlet.2016.01.014
156. Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc. 2017;6(9). doi:10.1161/JAHA.117.006347
157. Liu Y, Zhang HG. Vigilance on new-onset atherosclerosis following SARS-CoV-2 infection. Front Med. 2020;7:629413. doi:10.3389/fmed.2020.629413
158. Wang JG, Williams JC, Davis BK, et al. Monocytic microparticles activate endothelial cells in an IL-1β-dependent manner. Blood. 2011;118(8):2366–2374. doi:10.1182/blood-2011-01-330878
159. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340(2):115–126. doi:10.1056/NEJM199901143400207
160. Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol. 2009;27:165–197. doi:10.1146/annurev.immunol.021908.132620
161. Tabas I, Lichtman AH. Monocyte-macrophages and T cells in atherosclerosis. Immunity. 2017;47(4):621–634. doi:10.1016/j.immuni.2017.09.008
162. Mao C, Li D, Zhou E, Zhang J, Wang C, Xue C. Nicotine exacerbates atherosclerosis through a macrophage-mediated endothelial injury pathway. Aging. 2021;13(5):7627–7643. doi:10.18632/aging.202660
163. Mehta S, Dhawan V. Molecular insights of cigarette smoke condensate-activated NLRP3 inflammasome in THP-1 cells in a stage-specific atherogenesis. Int Immunopharmacol. 2020;88:107013. doi:10.1016/j.intimp.2020.107013
164. Buscetta M, Di Vincenzo S, Miele M, Badami E, Pace E, Cipollina C. Cigarette smoke inhibits the NLRP3 inflammasome and leads to caspase-1 activation via the TLR4-TRIF-caspase-8 axis in human macrophages. FASEB J. 2020;34(1):1819–1832. doi:10.1096/fj.201901239R
165. Rajamäki K, Lappalainen J, Oörni K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5(7):e11765. doi:10.1371/journal.pone.0011765
166. Westerterp M, Fotakis P, Ouimet M, et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation. 2018;138(9):898–912. doi:10.1161/CIRCULATIONAHA.117.032636
167. Peng K, Liu L, Wei D, et al. P2X7R is involved in the progression of atherosclerosis by promoting NLRP3 inflammasome activation. Int J Mol Med. 2015;35(5):1179–1188. doi:10.3892/ijmm.2015.2129
168. L’Homme L, Esser N, Riva L, et al. Unsaturated fatty acids prevent activation of NLRP3 inflammasome in human monocytes/macrophages. J Lipid Res. 2013;54(11):2998–3008. doi:10.1194/jlr.M037861
169. Salminen A, Ojala J, Kaarniranta K, Kauppinen A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: impact on the aging process and age-related diseases. Cell Mol Life Sci. 2012;69(18):2999–3013. doi:10.1007/s00018-012-0962-0
170. Son SJ, Rhee KJ, Lim J, Kim TU, Kim TJ, Kim YS. Triglyceride-induced macrophage cell death is triggered by caspase-1. Biol Pharm Bull. 2013;36(1):108–113. doi:10.1248/bpb.b12-00571
171. Chen S, Lee Y, Crother TR, et al. Marked acceleration of atherosclerosis after Lactobacillus casei-induced coronary arteritis in a mouse model of Kawasaki disease. Arterioscler Thromb Vasc Biol. 2012;32(8):e60–71. doi:10.1161/ATVBAHA.112.249417
172. Wakita D, Kurashima Y, Crother TR, et al. Role of interleukin-1 signaling in a mouse model of Kawasaki Disease-Associated Abdominal Aortic Aneurysm. Arterioscler Thromb Vasc Biol. 2016;36(5):886–897. doi:10.1161/ATVBAHA.115.307072
173. Campbell LA, Kuo CC. Chlamydia pneumoniae–an infectious risk factor for atherosclerosis? Nat Rev Microbiol. 2004;2(1):23–32. doi:10.1038/nrmicro796
174. Hu H, Pierce GN, Zhong G. The atherogenic effects of chlamydia are dependent on serum cholesterol and specific to Chlamydia pneumoniae. J Clin Invest. 1999;103(5):747–753. doi:10.1172/JCI4582
175. Cao F, Castrillo A, Tontonoz P, Re F, Byrne GI. Chlamydia pneumoniae–induced macrophage foam cell formation is mediated by Toll-like receptor 2. Infect Immun. 2007;75(2):753–759. doi:10.1128/IAI.01386-06
176. Itoh R, Murakami I, Chou B, et al. Chlamydia pneumoniae harness host NLRP3 inflammasome-mediated caspase-1 activation for optimal intracellular growth in murine macrophages. Biochem Biophys Res Commun. 2014;452(3):689–694. doi:10.1016/j.bbrc.2014.08.128
177. Yamaguchi Y, Kurita-Ochiai T, Kobayashi R, Suzuki T, Ando T. Activation of the NLRP3 inflammasome in Porphyromonas gingivalis-accelerated atherosclerosis. Pathog Dis. 2015;73(4). doi:10.1093/femspd/ftv011
178. Zhang R, Song B, Hong X, Shen Z, Sui L, Wang S. microRNA-9 inhibits vulnerable plaque formation and vascular remodeling via suppression of the SDC2-dependent FAK/ERK signaling pathway in mice with atherosclerosis. Front Physiol. 2020;11:804. doi:10.3389/fphys.2020.00804
179. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–1131. doi:10.1056/NEJMoa1707914
180. Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–255. doi:10.1038/nm.3806
181. van der Heijden T, Kritikou E, Venema W, et al. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler Thromb Vasc Biol. 2017;37(8):1457–1461. doi:10.1161/ATVBAHA.117.309575
182. Sharma A, Choi JSY, Stefanovic N, et al. Specific NLRP3 inhibition protects against diabetes-associated atherosclerosis. Diabetes. 2021;70(3):772–787.
183. Darakhshan S, Pour AB. Tranilast: a review of its therapeutic applications. Pharmacol Res. 2015;91:15–28. doi:10.1016/j.phrs.2014.10.009
184. Huang Y, Jiang H, Chen Y, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med. 2018;10(4). doi:10.15252/emmm.201708689
185. Guo T, Chen WQ, Zhang C, Zhao YX, Zhang Y. Chymase activity is closely related with plaque vulnerability in a hamster model of atherosclerosis. Atherosclerosis. 2009;207(1):59–67. doi:10.1016/j.atherosclerosis.2009.04.014
186. Chen S, Wang Y, Pan Y, et al. Novel role for tranilast in regulating NLRP3 ubiquitination, vascular inflammation, and atherosclerosis. J Am Heart Assoc. 2020;9(12):e015513. doi:10.1161/JAHA.119.015513
187. MacKenzie SH, Schipper JL, Clark AC. The potential for caspases in drug discovery. Curr Opin Drug Discov Devel. 2010;13(5):568–576.
188. Wannamaker W, Davies R, Namchuk M, et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J Pharmacol Exp Ther. 2007;321(2):509–516. doi:10.1124/jpet.106.111344
189. Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological inhibitors of the NLRP3 inflammasome. Front Immunol. 2019;10:2538. doi:10.3389/fimmu.2019.02538
190. Fischer U, Schulze-Osthoff K. Apoptosis-based therapies and drug targets. Cell Death Differ. 2005;12(Suppl 1):942–961. doi:10.1038/sj.cdd.4401556
191. Bassil F, Fernagut PO, Bezard E, et al. Reducing C-terminal truncation mitigates synucleinopathy and neurodegeneration in a transgenic model of multiple system atrophy. Proc Natl Acad Sci U S A. 2016;113(34):9593–9598. doi:10.1073/pnas.1609291113
192. Strand V, Sokolove J. Randomized controlled trial design in rheumatoid arthritis: the past decade. Arthritis Res Ther. 2009;11(1):205. doi:10.1186/ar2555
193. Li Y, Niu X, Xu H, et al. VX-765 attenuates atherosclerosis in ApoE deficient mice by modulating VSMCs pyroptosis. Exp Cell Res. 2020;389(1):111847. doi:10.1016/j.yexcr.2020.111847
194. Zhao J, Zhang H, Huang Y, et al. Bay11-7082 attenuates murine lupus nephritis via inhibiting NLRP3 inflammasome and NF-κB activation. Int Immunopharmacol. 2013;17(1):116–122. doi:10.1016/j.intimp.2013.05.027
195. Irrera N, Vaccaro M, Bitto A, et al. BAY 11-7082 inhibits the NF-κB and NLRP3 inflammasome pathways and protects against IMQ-induced psoriasis. Clin Sci. 2017;131(6):487–498. doi:10.1042/CS20160645
196. Juliana C, Fernandes-Alnemri T, Wu J, et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem. 2010;285(13):9792–9802. doi:10.1074/jbc.M109.082305
197. Catalán U, Fernández-Castillejo S, Pons L, et al. Alpha-tocopherol and BAY 11-7082 reduce vascular cell adhesion molecule in human aortic endothelial cells. J Vasc Res. 2012;49(4):319–328. doi:10.1159/000337466
198. Bian F, Yang XY, Xu G, Zheng T, Jin S. CRP-induced NLRP3 inflammasome activation increases LDL transcytosis across endothelial cells. Front Pharmacol. 2019;10:40. doi:10.3389/fphar.2019.00040
199. Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult-onset Still’s disease. Autoimmun Rev. 2014;13(7):708–722. doi:10.1016/j.autrev.2014.01.058
200. Kaufman MB. FDA approves new rituximab biosimilar & anakinra to treat a rare disease; 2021. Available from: https://www.the-rheumatologist.org/article/fda-approves-new-rituximab-biosimilar-anakinra-to-treat-a-rare-disease/.
201. Mertens M, Singh JA. Anakinra for rheumatoid arthritis. Cochrane Database Syst Rev. 2009;1:Cd005121. doi:10.1002/14651858.CD005121.pub3
202. Jesus AA, Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes. Annu Rev Med. 2014;65:223–244. doi:10.1146/annurev-med-061512-150641
203. Dinarello CA, Simon A, Van der meer JWM. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11(8):633–652. doi:10.1038/nrd3800
204. Morton AC, Rothman AM, Greenwood JP, et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA Heart Study. Eur Heart J. 2015;36(6):377–384. doi:10.1093/eurheartj/ehu272
205. Liu M, Yan M, Lv H, et al. Macrophage K63-linked ubiquitination of YAP promotes its nuclear localization and exacerbates atherosclerosis. Cell Rep. 2020;32(5):107990. doi:10.1016/j.celrep.2020.107990
206. Doss HM, Dey C, Sudandiradoss C, Rasool MK. Targeting inflammatory mediators with ferulic acid, a dietary polyphenol, for the suppression of monosodium urate crystal-induced inflammation in rats. Life Sci. 2016;148:201–210. doi:10.1016/j.lfs.2016.02.004
207. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. doi:10.1038/nature04516
208. Bauriedel G, Ganesh S, Uberfuhr P, Welsch U, Höfling B. [Growth-inhibiting effect of colchicine on cultured vascular wall myocytes from arteriosclerotic lesions]. Z Kardiol. 1992;81(2):92–98. German.
209. Cimmino G, Conte S, Morello A, et al. Colchicine inhibits the prothrombotic effects of oxLDL in human endothelial cells. Vascul Pharmacol. 2021;137:106822. doi:10.1016/j.vph.2020.106822
210. Robertson S, Martínez GJ, Payet CA, et al. Colchicine therapy in acute coronary syndrome patients acts on caspase-1 to suppress NLRP3 inflammasome monocyte activation. Clin Sci. 2016;130(14):1237–1246. doi:10.1042/CS20160090
211. Martínez GJ, Robertson S, Barraclough J, et al. Colchicine acutely suppresses local cardiac production of inflammatory cytokines in patients with an acute coronary syndrome. J Am Heart Assoc. 2015;4(8):e002128. doi:10.1161/JAHA.115.002128
212. Peng S, Xu LW, Che XY, et al. Atorvastatin inhibits inflammatory response, attenuates lipid deposition, and improves the stability of vulnerable atherosclerotic plaques by modulating autophagy. Front Pharmacol. 2018;9:438. doi:10.3389/fphar.2018.00438
213. Kong F, Ye B, Lin L, Cai X, Huang W, Huang Z. Atorvastatin suppresses NLRP3 inflammasome activation via TLR4/MyD88/NF-κB signaling in PMA-stimulated THP-1 monocytes. Biomed Pharmacother. 2016;82:167–172. doi:10.1016/j.biopha.2016.04.043
214. Altaf A, Qu P, Zhao Y, Wang H, Lou D, Niu N. NLRP3 inflammasome in peripheral blood monocytes of acute coronary syndrome patients and its relationship with statins. Coron Artery Dis. 2015;26(5):409–421. doi:10.1097/MCA.0000000000000255
215. Boland AJ, Gangadharan N, Kavanagh P, et al. Simvastatin suppresses interleukin Iβ release in human peripheral blood mononuclear cells stimulated with cholesterol crystals. J Cardiovasc Pharmacol Ther. 2018;23(6):509–517.
216. Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest. 2005;115(8):2047–2058.
217. Lamkanfi M, Mueller JL, Vitari AC, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187(1):61–70. doi:10.1083/jcb.200903124
218. Ling MY, Ma ZY, Wang YY, et al. Up-regulated ATP-sensitive potassium channels play a role in increased inflammation and plaque vulnerability in macrophages. Atherosclerosis. 2013;226(2):348–355. doi:10.1016/j.atherosclerosis.2012.11.016
219. Esposito K, Giugliano D, Nappo F, Marfella R. Regression of carotid atherosclerosis by control of postprandial hyperglycemia in type 2 diabetes mellitus. Circulation. 2004;110(2):214–219. doi:10.1161/01.CIR.0000134501.57864.66
220. Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet (London, England). 2006;368(9548):1696–1705. doi:10.1016/S0140-6736(06)69705-5
221. Scheen AJ. Cardiovascular effects of new oral glucose-lowering agents: DPP-4 and SGLT-2 Inhibitors. Circ Res. 2018;122(10):1439–1459. doi:10.1161/CIRCRESAHA.117.311588
222. Scheen AJ. Cardiovascular effects of gliptins. Nat Rev Cardiol. 2013;10(2):73–84. doi:10.1038/nrcardio.2012.183
223. Dai Y, Dai D, Wang X, Ding Z, Mehta JL. DPP-4 inhibitors repress NLRP3 inflammasome and interleukin-1beta via GLP-1 receptor in macrophages through protein kinase C pathway. Cardiovasc Drugs Ther. 2014;28(5):425–432. doi:10.1007/s10557-014-6539-4
224. Marx N, Libby P. Cardiovascular benefits of GLP-1 receptor agonism: is inflammation a key? JACC Basic Transl Sci. 2018;3(6):858–860. doi:10.1016/j.jacbts.2018.11.008
225. Luo X, Hu Y, He S, et al. Dulaglutide inhibits high glucose- induced endothelial dysfunction and NLRP3 inflammasome activation. Arch Biochem Biophys. 2019;671:203–209. doi:10.1016/j.abb.2019.07.008
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.