")
Back to Journals » OncoTargets and Therapy » Volume 9
Spotlight on elotuzumab in the treatment of multiple myeloma: the evidence to date
Authors Weisel K
Received 26 May 2016
Accepted for publication 26 July 2016
Published 5 October 2016 Volume 2016:9 Pages 6037—6048
DOI https://doi.org/10.2147/OTT.S94531
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Katja Weisel
Department of Hematology/Oncology, University Hospital Tuebingen, Tuebingen, Germany
Abstract: Despite advances in the treatment of multiple myeloma, it remains an incurable disease, with relapses and resistances frequently observed. Recently, immunotherapies, in particular, monoclonal antibodies, have become important treatment options in anticancer therapies. Elotuzumab is a humanized monoclonal antibody to signaling lymphocytic activation molecule F7, which is highly expressed on myeloma cells and, to a lower extent, on selected leukocyte subsets such as natural killer cells. By directly activating natural killer cells and by antibody-dependent cell-mediated cytotoxicity, elotuzumab exhibits a dual mechanism of action leading to myeloma cell death with minimal effects on normal tissue. In several nonclinical models of multiple myeloma, elotuzumab was effective as a single agent and in combination with standard myeloma treatments, supporting the use of elotuzumab in patients. In combination with lenalidomide and dexamethasone, elotuzumab showed a significant increase in tumor response rates and progression-free survival in patients with relapsed and/or refractory multiple myeloma. This review summarizes the nonclinical and clinical development of elotuzumab as a single agent and in combination with established therapies for the treatment of multiple myeloma.
Keywords: multiple myeloma, elotuzumab, SLAMF7, CS1, antibody-based immunotherapy
Introduction
Multiple myeloma (MM) is a hematologic malignancy characterized by abnormal antibody-secreting B cells. In 2016, MM accounts for an estimated 30,330 new cases and 12,650 deaths in the US.1 In Europe, 40,570 new cases and 25,398 deaths were expected in 2015.2 During the last decade, patient outcome has significantly improved with the introduction of new, more effective, and less toxic therapies.3–6 Treatment regimens now include immunomodulatory drugs (IMiDs) such as thalidomide, lenalidomide, and pomalidomide and proteasome inhibitors (PIs) such as bortezomib, carfilzomib, and ixazomib. However, despite these therapeutic advances, patients with MM often relapse or develop refractory disease.7,8 MM remains an incurable disease.
Recently, treatment options for patients with relapsed and/or refractory MM (RRMM) have expanded to agents providing a novel mechanism of action: antibody-based immunotherapy. The potential targets of monoclonal antibodies (mAbs) in MM are diverse and may include tumor cell surface proteins involved in signaling, tumor growth, and survival, or cellular and noncellular components of natural killer (NK) cells, or the bone marrow microenvironment.4,9,10 By targeting the antigens present in tumor cells, mAbs activate the immune system against MM through direct cytotoxicity, antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity, or antibody-dependent cellular phagocytosis.11,12
The mAbs elotuzumab (anti-Signaling Lymphocyte Activation Molecule Family 7 [anti-SLAMF7]) and daratumumab (anti-CD38) successfully completed clinical development. Elotuzumab was approved in combination with lenalidomide and dexamethasone for the treatment of patients with RRMM who have received one to three prior therapies (US Food and Drug Administration [FDA]) or ≥1 prior therapy (European Medicines Agency [EMA]). Daratumumab was approved as monotherapy in patients with RRMM who have received ≥3 prior therapies (FDA) or whose previous treatment included a PI and an immunomodulatory agent and whose disease worsened after treatment (EMA conditional marketing authorization). This review summarizes published data on the development and clinical evaluation of elotuzumab, including the pharmacology and efficacy of elotuzumab in MM, and assesses the safety and tolerability of this therapy in patients with advanced disease.
The mAb elotuzumab
Elotuzumab (EMPLICITI™, Bristol-Myers Squibb Company, New York, NY, USA) is a fully humanized recombinant monoclonal immunoglobulin G1 (IgG1) antibody that binds human SLAMF7 (also CS1, CRACC). The unique epitope of elotuzumab is located within the membrane proximal C2 domain of SLAMF7 (Figure 1).
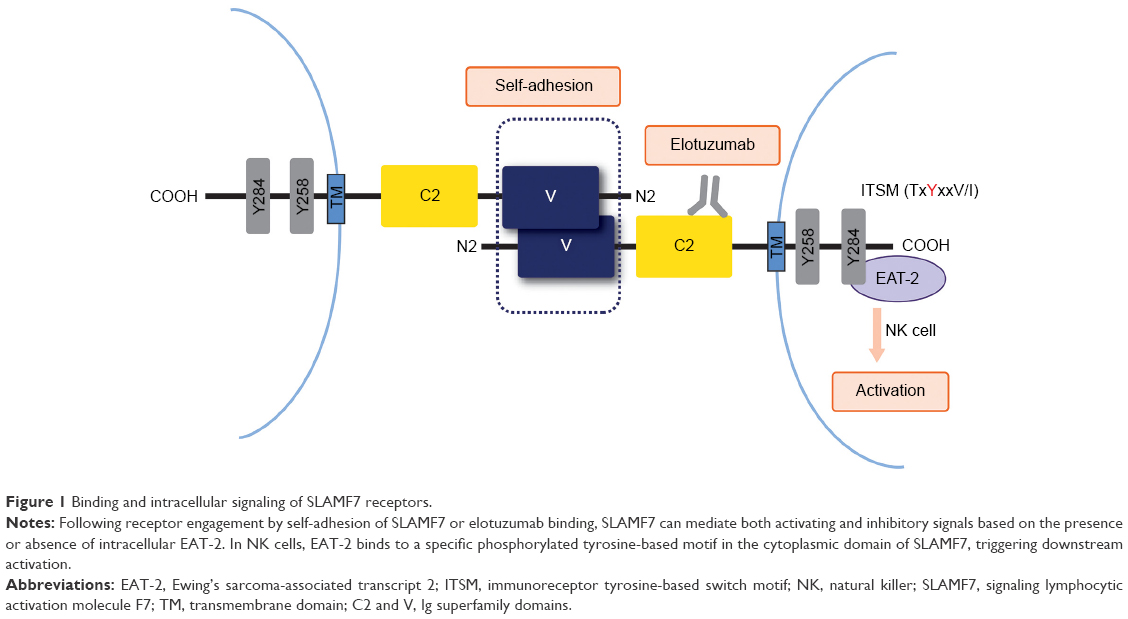 | Figure 1 Binding and intracellular signaling of SLAMF7 receptors. |
The SLAM family belongs to the Ig superfamily of cell surface receptors and is specifically expressed in hematopoietic cells.13 Most SLAM family receptors are self-ligands, that is, they bind to another molecule of the same receptor present on another cell, triggering interactions between identical or different types of hematopoietic cells. The SLAM family receptors play important roles in normal immune regulation and have been implicated in immunodeficiency, autoimmunity, and hematological malignancy.14,15
The cell surface glycoprotein SLAMF7 is universally and highly expressed on patient MM cells, irrespective of cytogenetic abnormalities and the degree of disease progression. To a lower extent, SLAMF7 is also expressed on lymphocytes such as NK cells, activated T cells, and most B cells. It is absent in other tissues, hematopoietic cells such as resting B cells, monocytes, CD4+ T cells or granulocytes, and hematopoietic stem cells.16–18 Self-adhesion of SLAMF7 receptors occurs at the membrane distal V domain.19–22
In the NK cells (Figure 1), SLAMF7 triggered activating or inhibitory effectors, depending on whether the cells expressed or did not express the intracellular adaptor protein Ewing’s sarcoma-associated transcript 2 (EAT-2). SLAMF7 bound to EAT-2 through one or two phosphorylated tyrosines in its cytoplasmic segment, thereby triggering signals of NK cell activation. In the absence of EAT-2, SLAMF7 potently inhibited NK cell function. Upon knockdown of SLAMF7 in vitro and in vivo, EAT-2-positive NK cells were functionally impaired and showed a reduced capacity to become activated. Myeloma cells lack EAT-2; therefore, SLAMF7 was unable to mediate activating signals. Of note, inhibitory signals by SLAMF7 signaling were also not inducible in these cells.14,16,23 Aside from failure of NK cells to show enhanced cytotoxicity, SLAMF7-deficient mice seemed healthy and showed no evidence of altered NK cell development.16
The exact role of SLAMF7 in myeloma cells is still unknown. Stable knockdown of SLAMF7 in MM cells resulted in decreased cell growth and colony formation in vitro,24 as well as reduced tumor burden and increased survival of animals in a xenograft mouse model.25 Homing of myeloma cells to the bone marrow, adhesion, and survival within the bone marrow microenvironment were impacted, indicating an involvement of SLAMF7 in the pathogenesis of MM.
Elotuzumab nonclinical studies
Nonclinical pharmacology
Elotuzumab induced significant ADCC against various MM cell lines and patient MM cells in dose-dependent and SLAMF7-specific manners and regardless of sensitivity or resistance of these MM cells to conventional therapies (Figure 2A). Upon binding to SLAMF7 expressed on MM cells, the Fc region of elotuzumab recruited and engaged NK cells by their CD16 receptors (also known as FcγRIII). This crosslink of MM to NK cells resulted in NK cell activation, ADCC, and finally selective killing of myeloma cells.17,18 No signals of complement-dependent cytotoxicity were detected. Elotuzumab was unable to directly induce antiproliferation signals or cell death in MM cells. To mediate the antitumor activity of elotuzumab toward myeloma cells, the presence of functional NK cells was required.
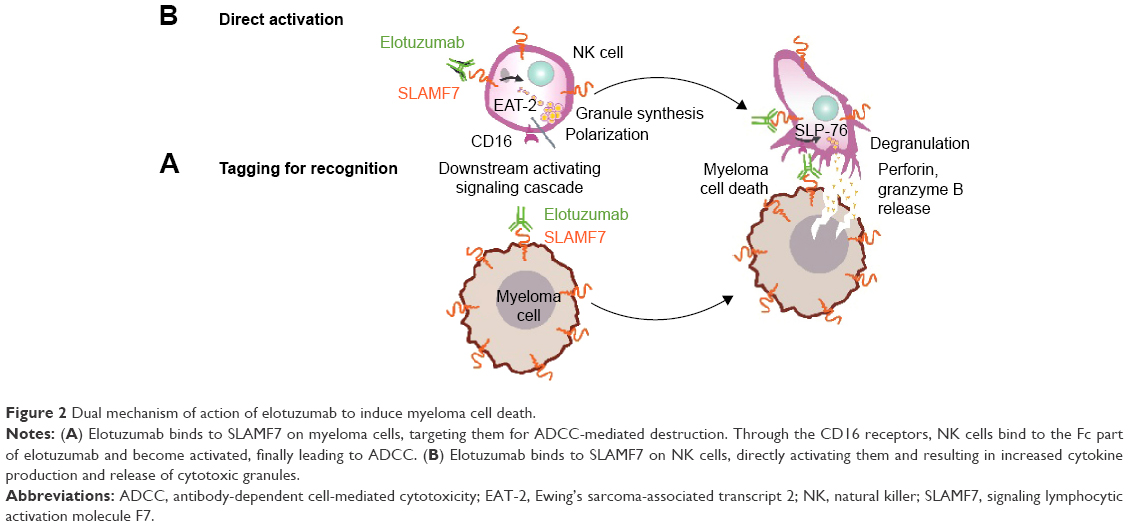 | Figure 2 Dual mechanism of action of elotuzumab to induce myeloma cell death. |
Elotuzumab also bound to SLAMF7 expressed on NK cells (Figure 2B). This was associated with increased cytokine production and degranulation, implicating direct NK cell activation and enhanced NK cell cytotoxicity as an additional mechanism of action of elotuzumab.17,18,26 Furthermore, elotuzumab inhibited SLAMF7-mediated MM cell adhesion to bone marrow stromal cells. Elotuzumab, but not control IgG1, specifically inhibited adhesion of SLAMF7-expressing MM lines and patient MM cells to bone marrow stromal cells in a dose-dependent manner.18
These in vitro findings were confirmed by in vivo pharmacological analyses. The in vivo antitumor activity of elotuzumab was studied in xenograft mouse models with tumors grown from SLAMF7-positive human MM cell lines L363, MM1S, and OPM2 and SLAMF7-negative H460 and PC3 cells.17,18,27 Treatment of animals with elotuzumab exhibited antitumor activity. In the OPM2 xenograft mouse model, maximal antitumor activity of elotuzumab was observed at the 10 mg/kg dose level, the highest dose level tested. The 10 mg/kg dose level correlated with elotuzumab serum concentrations of 70 and 430 μg/mL, respectively. In subsequent clinical studies, investigators sought an elotuzumab serum trough concentration of ≥70 μg/mL, which was achieved in patients at the proposed 10 mg/kg dose level.28
The antitumor activity of elotuzumab was also studied in combination with established therapies. In the OPM2 xenograft mouse model, elotuzumab, bortezomib, and lenalidomide each conferred similar antitumor activity; however, when elotuzumab was combined with either bortezomib29 or lenalidomide,27 enhanced antitumor activity was observed. Bortezomib is a reversible PI with significant activity in myeloma. Pretreatment of SLAMF7-positive myeloma cell lines and primary myeloma cells with bortezomib significantly enhanced elotuzumab-mediated ADCC.29 Lenalidomide is an IMiD known to alter NK cell function. The combination of elotuzumab and lenalidomide significantly decreased the number of myeloma cells, increased the activation of NK cells, and upregulated adhesion and activation markers including interleukin (IL)-2Rα, IL-2, and tumor necrosis factor-α.27 These nonclinical findings supported the use of combination regimens of elotuzumab and bortezomib or lenalidomide in patients.
Toxicology
Due to the lack of a pharmacologically relevant animal species to conduct toxicology studies with a humanized antibody, the toxicological evaluation for elotuzumab was limited to a tissue cross-reactivity study and in vivo studies that assessed the potential for off-target toxicity and local tolerance. Across 332 normal adult tissue samples representing 86 different organs as well as 148 leukocyte samples representing 12 different cell types, SLAMF7 expression was found to be restricted to leukocytes and was not expressed in any major body organs.17 In human tissues examined, elotuzumab only stained the membrane and/or cytoplasm of plasma cells and/or immunoblasts present in these tissues, but not the tissues themselves.30 Consequently, the risk of elotuzumab for off-target toxicity or a significant local adverse reaction was considered low.
The impact of elotuzumab on reproduction and fetal development is unknown. As an IgG1 mAb, elotuzumab has the potential to cross the placental barrier, permitting direct fetal exposure.31 However, SLAMF7-deficient mice appear healthy, suggesting SLAMF7 does not play a role in embryo–fetal development.16
A single dose of intravenously infused elotuzumab (0, 30, or 100 mg/kg) was well tolerated in rhesus monkeys, with no clinical signs at any dose. There were no local adverse reactions when 5 mg of elotuzumab was intravenously injected into the ears of New Zealand white rabbits, and a solution of 10 mg/mL elotuzumab did not cause hemolysis of human whole blood. An in vitro cytokine release experiment identified elevated levels of ten cytokines in response to elotuzumab treatment.30
Clinical pharmacology
Pharmacodynamics
Saturation of SLAMF7 by elotuzumab on bone marrow target cells increased as the dose of elotuzumab increased. At doses of 10–20 mg/kg elotuzumab, SLAMF7 receptors on bone marrow-derived myeloma cells (CD45dim/CD38+ cells) were consistently saturated (≥95% of SLAMF7 receptors) (Figure 3).28 The addition of bortezomib (Study 1702) or lenalidomide/dexamethasone (Study 1703) did not affect elotuzumab serum trough levels or SLAMF7 saturation.32,33
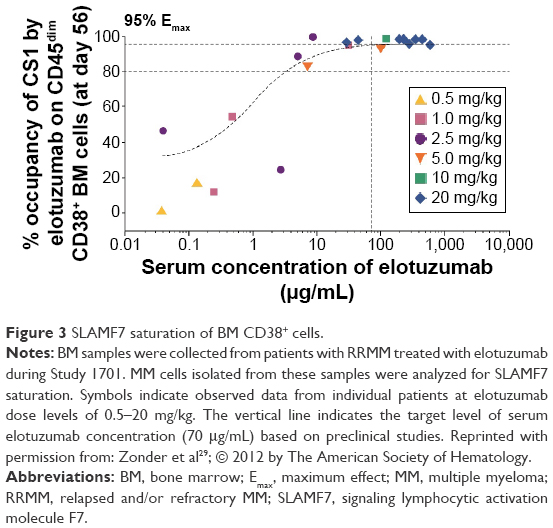 | Figure 3 SLAMF7 saturation of BM CD38+ cells. |
In MM patients, soluble SLAMF7 (sSLAMF7) serum levels may be associated with disease stage and could be used as a surrogate marker for disease burden. Upon treatment with elotuzumab alone or in combination with bortezomib/dexamethasone (E–B/d) or lenalidomide/dexamethasone (E–L/d), a significant dose- and time-dependent increase in total sSLAMF7 (both free and elotuzumab-bound sSLAMF7) was observed, likely reflecting stabilization of the protein in serum by elotuzumab. Early changes in sSLAMF7 levels following elotuzumab treatment may indicate the likelihood of a response.34
Pharmacokinetics (PK)
The PK parameters of elotuzumab were analyzed in 34 patients enrolled in the first-in-man Phase I study28 and in 375 patients enrolled in four Phase I–III clinical trials.35 Based on noncompartmental analysis, elotuzumab across the dose range of 0.5–20 mg/kg exhibited a nonlinear PK behavior. Elotuzumab plasma levels and terminal half-life increased proportionally with dose, while exposure (area under the curve [AUC] and AUCinf) increased greater than proportionally. The clearance of elotuzumab decreased with increasing doses, indicating a saturation of target-mediated elimination.28 Clearance of elotuzumab correlated with body weight, thus justifying a weight-based dosing of elotuzumab.35 Elimination of elotuzumab increased with rising baseline serum monoclonal (M) protein concentrations, the abnormal immunoglobulin fragment produced in excess by myeloma cells. Accordingly, patients with high baseline serum M protein had a lower exposure to elotuzumab.35
Coadministration of lenalidomide/dexamethasone (E–L/d) decreased elotuzumab nonspecific clearance by 35%,35 which was shown to be predominantly due to coadministration of dexamethasone.36 Impaired renal function did not impact elotuzumab serum concentrations. In MM patients with various levels of renal dysfunction, mean elotuzumab serum concentrations were comparable across all treatment groups and support the use of elotuzumab (in combination with lenalidomide and dexamethasone) without dose adjustments.37
Immunogenicity
The development of antidrug antibodies (ADA) to elotuzumab was observed in about 40% of patients on elotuzumab monotherapy (Study 1701) and in about 15% of patients on combination therapy (E–L/d; Studies 1703 and ELOQUENT-2).28,38,39 The presence of these ADA varied across dose groups and was usually transient, resolving by 2–4 months. During this time, an increase in nonspecific elotuzumab clearance was noted.35 Immunogenicity did not correlate with any efficacy or safety parameter. Immunosuppressants such as dexamethasone may have contributed to the reduced ADA titers observed on combination therapy compared with elotuzumab monotherapy.36
Clinical efficacy
Elotuzumab has been evaluated for the treatment of MM in a series of clinical trials (Table 1). The first-in-human study (Study 1701) assessed safety and tolerability as well as PK of elotuzumab as a single agent. However, the antitumor effects of elotuzumab were largely determined in combination with established myeloma therapies.
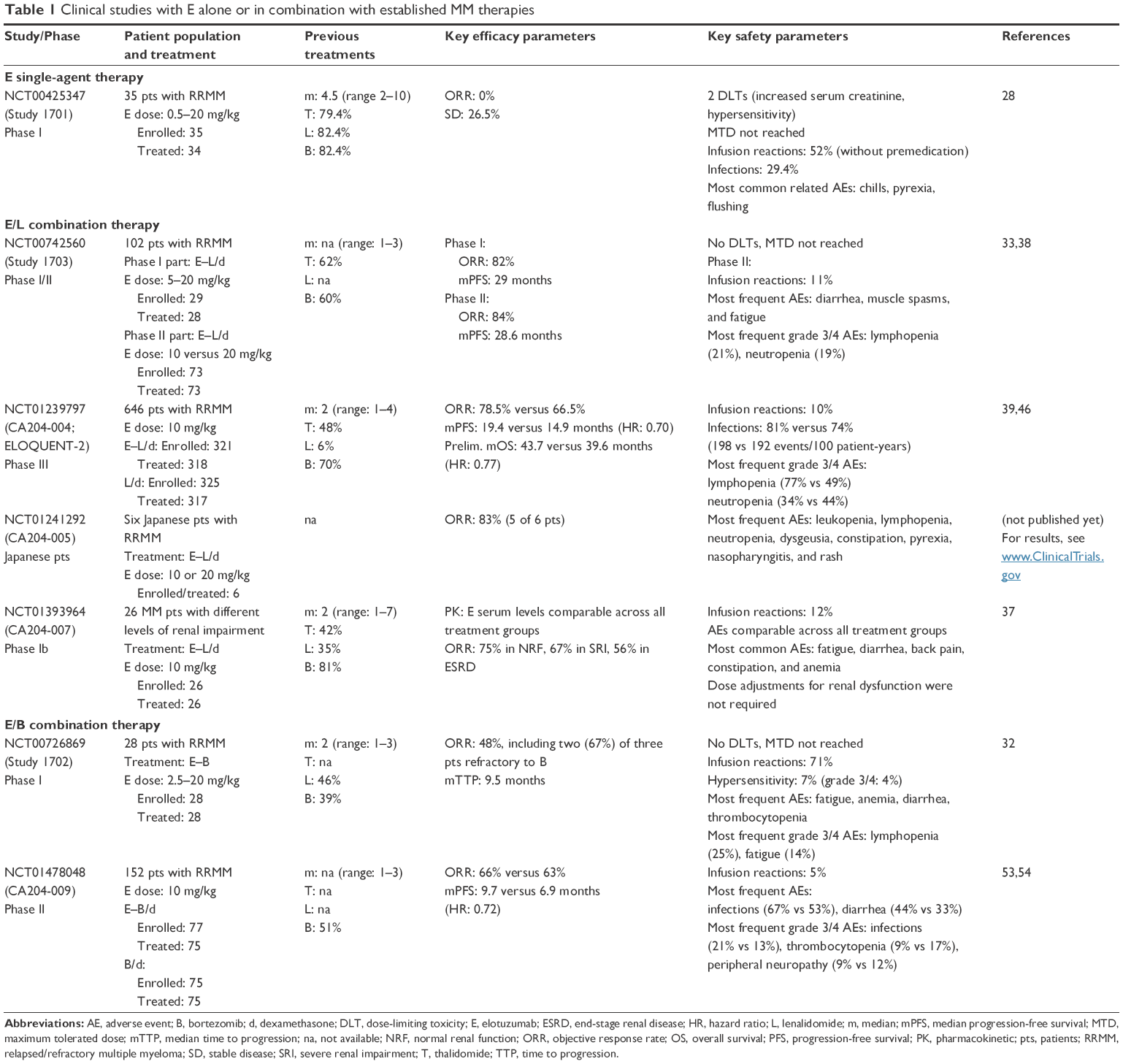 | Table 1 Clinical studies with E alone or in combination with established MM therapies |
Elotuzumab monotherapy
In Study 1701 (Table 1), safety and tolerability as well as PK and pharmacodynamics of different doses of elotuzumab (dose cohorts from 0.5 to 20 mg/kg body weight) were analyzed in 35 patients with RRMM. Saturation of SLAMF7 receptors was observed at elotuzumab doses of 10–20 mg/kg (Figure 3). Despite plasma cell target saturation, no objective myeloma responses were observed.28 Based on the European Society of Blood and Marrow Transplantation myeloma response criteria, 25 patients (73.5%) had progressive disease and nine patients (26.5%) were classified as having stable disease, demonstrating minimal single-agent activity of elotuzumab. These findings substantiate the challenge to effectively activate and direct cells of the immune system to kill myeloma cells in patients with compromised immune function, in particular, with compromised NK cell function. In Study 1701, patients were heavily pretreated and had received a median of 4.5 prior therapies including lenalidomide, thalidomide, and/or bortezomib.28 To further evaluate the association between NK cell status and the efficacy of elotuzumab monotherapy, a Phase II biomarker study (Study CA204-011) is currently ongoing in patients with high-risk smoldering MM.
Combination therapy of elotuzumab and lenalidomide
Lenalidomide is an oral IMiD that has been shown to directly kill MM cells, stimulate T cells to produce IL-2, increase NK cell proliferation, and lower the threshold required for NK cell activation.40,41 Based on data of several Phase III trials, lenalidomide in combination with low-dose dexamethasone (L/d) was approved for treatment of newly diagnosed MM patients ineligible for stem cell transplantation42 and of patients with relapsed MM who have received at least one prior therapy.43,44 A meta-analysis of randomized controlled trials confirmed the survival advantage of treatment with L/d compared with other first-line therapies in patients with previously untreated MM.45
The triple combination therapy of elotuzumab, lenalidomide, and dexamethasone (E–L/d) was evaluated in a Phase I/II dose escalation study (Study 1703) and two large Phase III studies (ELOQUENT-1 and -2) in patients with newly diagnosed MM and RRMM, respectively. While ELOQUENT-1 is still ongoing with first results to be expected in 2017, data of ELOQUENT-2 are already available showing substantial evidence of effectiveness for E–L/d in the treatment of patients with RRMM who have received one to three prior therapies (Table 1).
In this study, a total of 646 patients were enrolled (321 in the E–L/d arm and 325 in the L/d arm). Efficacy results of 635 treated patients showed a significant increase in tumor response of 78.5% versus 65.5% and median progression-free survival (mPFS) of 19.4 versus 14.9 months of E–L/d over L/d, respectively (Table 2).39 Based on data of a 3-year follow-up analysis, the progression-free survival (PFS) benefit of patients on E–L/d was maintained during this period. Exploratory analysis revealed a median delay of 1 year in the time to next treatment in patients treated with E–L/d compared with L/d-treated patients (33 months on E–L/d vs 21 months on L/d). Preliminary analysis of overall survival (OS) also indicated an advantage for patients treated with E–L/d.46 Of note, the benefit in PFS and OS of patients on E–L/d was observed for all patient subgroups independent of age, disease stage, response to most recent line of therapy, number and nature of prior therapies, as well as high-risk cytogenetic profile.39,46
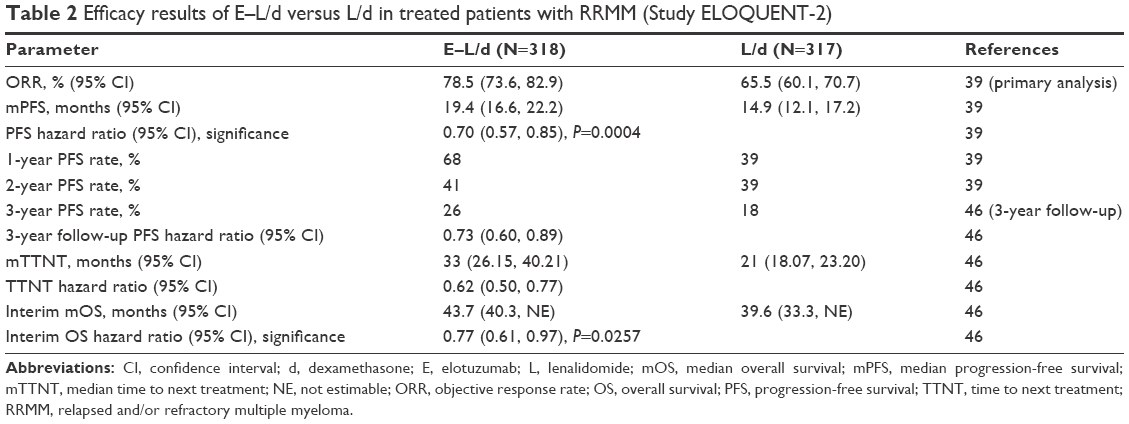 | Table 2 Efficacy results of E–L/d versus L/d in treated patients with RRMM (Study ELOQUENT-2) |
Based on the Phase III efficacy data and the observed adverse events (AEs) (see “Safety of elotuzumab treatment”), both the FDA and the EMA approved elotuzumab in combination with lenalidomide and dexamethasone for the treatment of patients with RRMM.
The combination of elotuzumab and lenalidomide is further being evaluated in various ongoing clinical studies of Phase I–III (www.ClinicalTrials.gov). Analyses include the treatment of earlier stages of MM, such as patients with newly diagnosed, untreated MM (ELOQUENT-1) or high-risk smoldering MM (NCT01668719), treatment of Japanese patients with MM (NCT02272803), or maintenance therapy after autologous stem cell transplantation (NCT02420860).
Combination therapy of elotuzumab and bortezomib
Bortezomib is a potent, selective, and reversible PI that exhibits direct antitumor effects on myeloma cells by cell cycle arrest and apoptosis, but is also able to sensitize the tumor cells to cellular effector pathways including NK cell-mediated killing.47 Based on Phase II data, bortezomib was approved for the treatment of patients with RRMM.48,49 Following generation of Phase III data, approval was extended to MM patients who have received at least one prior therapy50 and, in combination with melphalan and prednisone, to patients with newly diagnosed MM.51 The beneficial effects of bortezomib in patients with MM were corroborated lately by a Cochrane systematic review and meta-analysis.52
To evaluate the clinical effectiveness of elotuzumab in combination with bortezomib, a Phase I dose escalation study (Study 1702)32 administering elotuzumab and bortezomib (E–B) and a Phase II proof-of-concept study (Study CA204-009)53 administering elotuzumab, bortezomib, and dexamethasone (E–B/d) were performed (Table 1). The Phase II study enrolled a total of 152 patients (77 in the E–B/d arm and 75 in the B/d arm). Despite ongoing follow-up of patients, all results of efficacy parameters were in favor of the triple combination therapy. The objective response rate was 66% in the E–B/d arm versus 63% in the B/d arm. The mPFS was 9.7 months (95% confidence interval [CI]: 7.4, 12.2) on E–B/d compared with 6.9 months (95% CI: 5.1, 10.2) on B/d, accounting for an estimated hazard ratio for PFS of 0.72 for E–B/d over B/d (70% CI: 0.59, 0.88; P=0.09).53 The PFS benefit for E–B/d was observed in all patient subgroups analyzed. At the time of data analysis, median OS was not estimable in patients of the E–B/d arm and 34.7 months in the B/d arm, corresponding to an OS hazard ratio of 0.75 for E–B/d over B/d (95% CI: 0.45, 1.24).54
In Study CA204-009, patients were also analyzed for genetic polymorphisms of their Fc receptor FcγRIIIa (CD16) expressed on NK cells.53 Binding of the Fc part of elotuzumab to the FcγRIIIa receptor is mandatory to induce ADCC. Genetic polymorphisms of this receptor, particularly, the V158F polymorphism, were described to influence activation of NK cells and effectiveness of ADCC. The high-affinity “VV” genotype of the FcγRIIIa receptor cells has been linked with enhanced ADCC.55,56 Indeed, in Study CA204-009, a strong trend toward longer PFS was reported for patients homozygous for the high-affinity FcγRIIIa V allele (Figure 4). In the E–B/d treatment group, patients with “VV” genotype had an mPFS of 22.3 months versus 9.8 months observed in patients with the low-affinity “FF” genotype.53
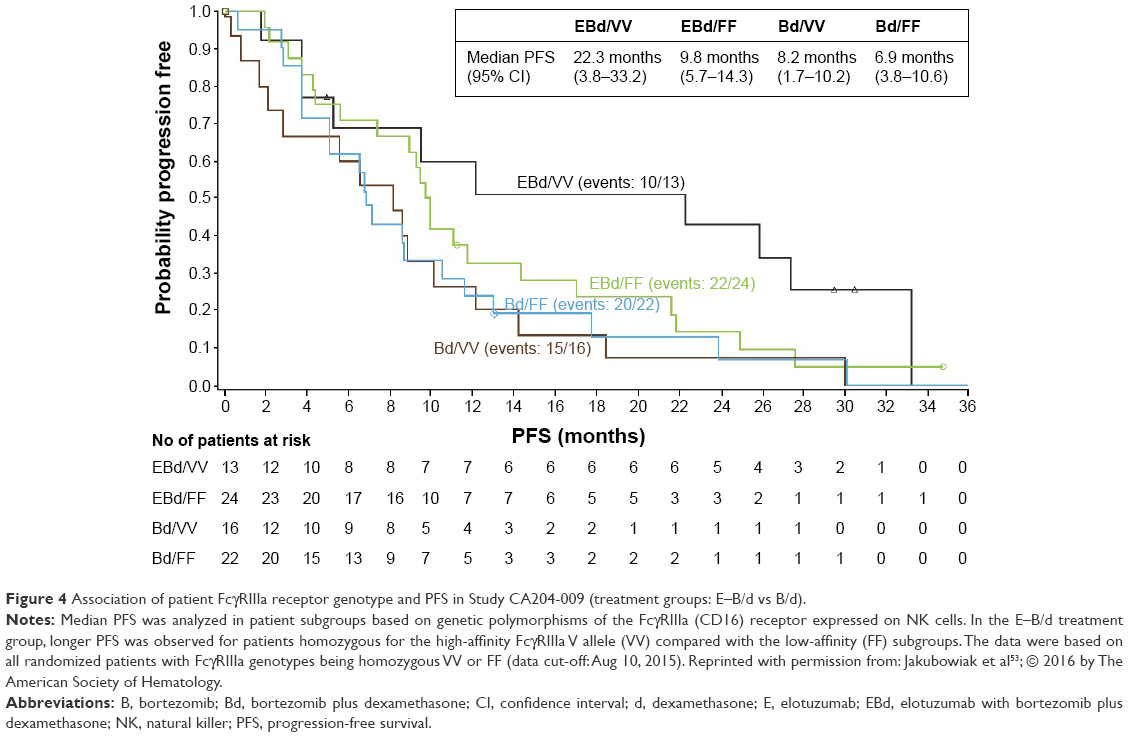 | Figure 4 Association of patient FcγRIIIa receptor genotype and PFS in Study CA204-009 (treatment groups: E–B/d vs B/d). |
In several Phase I and II studies, different treatment regimens of elotuzumab and bortezomib/dexamethasone are currently being evaluated in combination with IMiDs such as lenalidomide (studies NCT01668719 and NCT02375555) in patients with newly diagnosed MM or pomalidomide (Study NCT02718833) in patients with RRMM. A Phase III study is ongoing evaluating E–BL/d versus BL/d consolidation therapy followed by E–L/d versus L/d maintenance therapy after BL/d induction in patients with newly diagnosed myeloma (Study NCT02495922).
Assessment of other combination regimens
To further evaluate the potential clinical benefit of elotuzumab in myeloma, clinical studies are being conducted administering different combination regimens. In two Phase II studies, elotuzumab plus pomalidomide and low-dose dexamethasone are being assessed in patients with RRMM (Study CA204-125, NCT02654132) and in patients with MM relapsed or refractory to prior treatment with lenalidomide (Study CA204-142, NCT02612779). Furthermore, in an exploratory treatment arm of a Phase III study (NCT02726581), elotuzumab is administered in combination with nivolumab (humanized IgG4 anti-PD-1 mAb), pomalidomide, and dexamethasone to patients with RRMM.
Safety of elotuzumab treatment
The safety dataset of elotuzumab is primarily based on the randomized controlled trials ELOQUENT-2 and CA204-009 evaluating elotuzumab in combination with lenalidomide and bortezomib, respectively, in patients with RRMM. Supportive data have been generated in a similar patient population from studies 1701, 1702, and 1703, in Japanese RRMM patients (Study CA204-005), and in RRMM patients with different levels of renal function (Study CA204-007). An overview of these studies is presented in Table 1.
The rates of treatment-emergent AEs and AEs of grade 3 or 4 were similar between treatment groups, indicating limited added toxicity when combining elotuzumab with L/d or B/d. The AEs observed on elotuzumab alone or in combination with L/d or B/d included gastrointestinal, hematological, and respiratory disorders. Common AEs were fatigue, diarrhea, pyrexia, constipation, cough, and muscle spasms.57,58
Study ELOQUENT-2 evaluating E–L/d
In Study ELOQUENT-2, patient baseline characteristics were well balanced between the treatment groups.39 At data cut-off for a recent follow-up analysis (May 16, 2015), 29% of patients treated with E–L/d remained on study therapy compared with 15% of patients treated with L/d.59 Treatment discontinuation was mainly due to disease progression (46% on E–L/d vs 51% on L/d).
Safety and tolerability data showed that minimal incremental toxicity was associated with addition of elotuzumab to L/d. Significant differences in grade 3/4 AEs were observed for lymphopenia (78% on E–L/d vs 49% on L/d) and neutropenia (35% on E–L/d vs 44% on L/d). The rates for other grade 3/4 AEs including anemia, thrombocytopenia, cardiac or renal disorders were similar between the treatment groups. Infections (any grade) were reported in 83% of patients in the elotuzumab group versus 75% in the control group. After adjustment for drug exposure, infection rates were equal in the two groups (196 and 193 events per 100 patient-years on E–L/d and L/d, respectively).59 The incidence of herpes zoster infections was 4.1 versus 2.2 per 100 patient-years, respectively. Despite the finding of increased lymphopenia which may reflect alterations in lymphocyte trafficking, there was no evidence of increased autoimmunity or other sequelae of immune dysregulation.39 This confirmed previous results in whole blood samples from healthy individuals tested for the effects of elotuzumab on the numbers of T, NK, and B cells. Elotuzumab treatment did not result in substantial depletion of major lymphocyte subsets.17
Infusion reactions (mostly grade 1/2) occurred in only 11% of patients treated with E–L/d59 and usually presented as pyrexia, chills, headache, flushing, and/or hypertension. Most infusion reactions were observed during the first dose of elotuzumab. They were usually mild to moderate and resolved spontaneously within 24 hours or after management as clinically indicated. No patient experienced grade 4–5 reactions, and all but two patients could continue elotuzumab treatment.39
Furthermore, second primary malignancies were observed in 7% of patients on E–L/d compared with 4% on L/d. However, after adjustment for exposure to study therapy, the incidence rates were similar at 3.5 and 2.8 second primary cancers per 100 patient-years, respectively.39
In Study CA204-112 (NCT02159365), treatment of elotuzumab administered over ~1 hour in combination with L/d was evaluated in 70 patients with newly diagnosed or relapsed/refractory MM.60,61 All patients were required to have a premedication regimen before each dose of elotuzumab. The infusion rate was accelerated from 0.5 to 5 mL/min at the start of the third dose in treatment cycle 1. At data cut-off, infusion reactions were observed in only two patients. The reactions were of grade 1/2 intensity and both patients continued treatment with elotuzumab. No grade 3/4 infusion reactions had occurred by the end of treatment cycle 2. The administration of elotuzumab at a faster rate may provide a safe option to reduce treatment time for patients.
Study CA204-009 evaluating E–B/d
At data cut-off (August 10, 2015) for a recent analysis of Study CA204-009, 8% of patients treated with E–B/d remained on therapy compared with 1% treated with B/d.53 The median number of treatment cycles was 12 on E–B/d and seven on B/d. Treatment discontinuation was mainly due to disease progression (57% across treatment groups). Grade 3/4 AEs were reported in 53 (71%) and 45 (60%) patients in the E–B/d and B/d arms, respectively. Differences in grade 3/4 AEs were observed for infections (21% on E–B/d vs 13% on B/d) and thrombocytopenia (9% on E–B/d vs 17% on B/d). Furthermore, the incidence of grade 3/4 peripheral neuropathy was higher on B/d alone, whereas grade 3/4 diarrhea was more often observed on E–B/d. Infusion reactions (all grade 1/2) occurred in 4/75 patients (5%) in the elotuzumab treatment arm and included pyrexia in two patients and bone pain, chills, flushing, nausea, and ear pruritus in one patient each. In this study, 27 patients received elotuzumab as an infusion over a 1-hour period, leading to a reaction in only one patient. Overall, the safety profile was consistent over time, with minimal incremental toxicity of E–B/d compared with B/d alone.53
Conclusion
According to the International Myeloma Working Group, management of relapsed MM requires a systematic approach based on several characteristic features of the patients, including prior treatment, degree and depth of response, treatment-related toxicities, and genetic risk stratification.7 Furthermore, the selection of MM therapies demands careful consideration of the balance between maximizing efficacy and ensuring acceptable tolerability.62 Today, first-line treatment for MM usually includes PIs and/or IMiDs, which have improved the patients’ survival. However, there is still an unmet need for drugs achieving deep and sustained myeloma responses, even during relapse of the disease. In particular, drugs with novel modes of action are required exhibiting effects that are additive or synergistic to the established backbone regimens.
Recently, promising clinical results have been observed for mAb therapies targeting plasma cell antigens such as SLAMF7, CD38, and CD138.8 Elotuzumab showed minimal added toxicity when combined with established double-agent therapies such as L/d or B/d, while increased antitumor activity in terms of tumor response and PFS has been observed. The overall clinical benefit of elotuzumab remains to be determined based on final OS data of the patients. However, elotuzumab demonstrated, for the first time, the antitumor efficiency of antibody-based immunotherapy targeting myeloma cells in a large, randomized controlled Phase III study. Based on these data, E–L/d has now been approved by FDA and EMA for treatment of patients with RRMM. For these patients, the treatment spectrum has been further expanded by the recently approved double or triple combinations of carfilzomib/d and carfilzomib–L/d. In the US, treatment options also include the combination of ixazomib–L/d. Patients with more advanced disease may be treated with pomalidomide/d, panobinostat–B/d, or daratumumab. The possibility to select the most suitable treatment(s) from an increasing variety of available therapies may give rise to cautious optimism to be able to transform MM into a chronic disease in the future.
Clinical data on elotuzumab acknowledge the high threshold of immune-mediated killing of myeloma cells that needs to be overcome: elotuzumab as a single agent is unable to reduce this threshold and requires additional stimuli for effective immune activation to kill myeloma cells. Possibly, patients at earlier disease stages and bearing less-compromised immune systems may be more susceptible to elotuzumab-containing treatment regimens. Therefore, data on first-line treatment regimens including elotuzumab will be of high interest. In advanced stages, the combination of elotuzumab with other agents or combinations such as E–LB/d may compensate for these impairments and lower the threshold needed for elotuzumab-mediated killing of myeloma cells. Clinical trials are currently ongoing to evaluate all these treatment options.
In conclusion, elotuzumab is the first mAb introduced to the treatment of MM that acts by a dual, immuno-oncologic mechanism. Addition of elotuzumab to lenalidomide and dexamethasone did not only lead to a significant improvement in PFS, but also resulted in a promising course of the PFS curve, suggesting a potential for long-term immunological control of MM in a subset of patients. The generation of additional clinical data on biomarkers, treatment of earlier disease stages, and studies on selected combination regimens are ongoing to further optimize the use of elotuzumab in patients with MM.
Acknowledgments
Assistance in preparation and writing of the manuscript was provided by Birgit Glasschroeder, PhD, and funded by Bristol-Myers Squibb.
Disclosure
The author has received travel grants, honoraria, and/or research support from AMGEN, Bristol-Myers Squibb, Celgene, Janssen, Novartis, Onyx, and Takeda. The author reports no other conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
World Health Organization/International Agency for Research on Cancer (WHO/IARC). GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence worldwide in 2012. Online analysis on multiple myeloma. Available from: globocan.iarc.fr. Accessed April 29, 2016. | ||
Mina R, Cerrato C, Bernardini A, Aghemo E, Palumbo A. New pharmacotherapy options for multiple myeloma. Expert Opin Pharmacother. 2016;17(2):181–192. | ||
Ayed AO, Chang LJ, Moreb JS. Immunotherapy for multiple myeloma: current status and future directions. Crit Rev Oncol Hematol. 2015;96(3):399–412. | ||
Broijl A, Sonneveld P. An update in treatment options for multiple myeloma in nontransplant eligible patients. Expert Opin Pharmacother. 2015;16(13):1945–1957. | ||
Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28(5):1122–1128. | ||
Laubach J, Garderet L, Mahindra A, et al. Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia. 2016;30(5):1005–1017. | ||
Ocio EM, Richardson PG, Rajkumar SV, et al. New drugs and novel mechanisms of action in multiple myeloma in 2013: a report from the International Myeloma Working Group (IMWG). Leukemia. 2014;28(3):525–542. | ||
Sondergeld P, van de Donk NW, Richardson PG, Plesner T. Monoclonal antibodies in myeloma. Clin Adv Hematol Oncol. 2015;13(9):599–609. | ||
Van de Donk NW, Kamps S, Mutis T, Lokhorst HM. Monoclonal antibody-based therapy as a new treatment strategy in multiple myeloma. Leukemia. 2012;26(2):199–213. | ||
Magarotto V, Salvini M, Bonello F, Bringhen S, Palumbo A. Strategy for the treatment of multiple myeloma utilizing monoclonal antibodies: a new era begins. Leuk Lymphoma. 2016;57(3):537–556. | ||
Van de Donk NW, Moreau P, Plesner T, et al. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood. 2016;127(6):681–695. | ||
Veillette A. SLAM-family receptors: immune regulators with or without SAP-family adaptors. Cold Spring Harb Perspect Biol. 2010;2(3):a002469. | ||
Guo H, Cruz-Munoz ME, Wu N, Robbins M, Veillette A. Immune cell inhibition by SLAMF7 is mediated by a mechanism requiring src kinases, CD45, and SHIP-1 that is defective in multiple myeloma cells. Mol Cell Biol. 2015;35(1):41–51. | ||
Veillette A, Guo H. CS1, a SLAM family receptor involved in immune regulation, is a therapeutic target in multiple myeloma. Crit Rev Oncol Hematol. 2013;88(1):168–177. | ||
Cruz-Munoz ME, Dong Z, Shi X, Zhang S, Veillette A. Influence of CRACC, a SLAM family receptor coupled to the adaptor EAT-2, on natural killer cell function. Nat Immunol. 2009;10(3):297–305. | ||
Hsi ED, Steinle R, Balasa B, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res. 2008;14(9):2775–2784. | ||
Tai YT, Dillon M, Song W, et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood. 2008;112(4):1329–1337. | ||
Kumaresan PR, Lai WC, Chuang SSS, Bennett M, Mathew PA. CS1, a novel member of the CD2 family, is homophilic and regulates NK cell function. Mol Immunol. 2002;39(1–2):1–8. | ||
Boles KS, Stepp SE, Bennett M, Kumar V, Mathew PA. 2B4 (CD244) and CS1: novel members of the CD2 subset of the immunoglobulin superfamily molecules expressed on natural killer cells and other leukocytes. Immunol Rev. 2001;181:234–249. | ||
Boles KS, Mathew PA. Molecular cloning of CS1, a novel human natural killer cell receptor belonging to the CD2 subset of the immunoglobulin superfamily. Immunogenetics. 2001;52(3–4):302–307. | ||
Bouchon A, Cella M, Grierson HL, Cohen JI, Colonna M. Activation of NK cell-mediated cytotoxicity by a SAP-independent receptor of the CD2 family. J Immunol. 2001;167(10):5517–5521. | ||
Pérez-Quintero LA, Roncagalli R, Guo H, Latour S, Davidson D, Veillette A. EAT-2, a SAP-like adaptor, controls NK cell activation through phospholipase Cγ, Ca++, and Erk, leading to granule polarization. J Exp Med. 2014;211(4):727–742. | ||
Xie Z, Gunaratne J, Cheong LL, et al. Plasma membrane proteomics identifies biomarkers associated with MMSET overexpression in T(4;14) multiple myeloma. Oncotarget. 2013;4(7):1008–1018. | ||
Glavey S, Reagyn M, Manier S, et al. Dissecting the mechanisms of activity of SLAMF7 and the targeting antibody elotuzumab in multiple myeloma. Blood. 2014;124(21):3431. | ||
Collins SM, Bakan CE, Swartzel GD, et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother. 2013;62(12):1841–1849. | ||
Balasa B, Yun R, Belmar NA, et al. Elotuzumab enhances natural killer cell activation and myeloma cell killing through interleukin-2 and TNF-α pathways. Cancer Immunol Immunother. 2015;64(1):61–73. | ||
Zonder JA, Mohrbacher AF, Singhal S, et al. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood. 2012;120(3):552–559. | ||
Van Rhee F, Szmania SM, Dillon M, et al. Combinatorial efficacy of anti-CS1 monoclonal antibody elotuzumab (HuLuc63) and bortezomib against multiple myeloma. Mol Cancer Ther. 2009;8(9):2616–2624. | ||
Food and Drug Administration (U.S.), FDA-approved drug products. EMPLICITI Center for Drug Evaluation and Research (CDER) Summary Review 2015. Available from: http://www.fda.gov. Accessed April 29, 2016. | ||
Malek A, Sager R, Kuhn P, Nicolaides KH, Schneider H. Evolution of maternofetal transport of immunoglobulins during human pregnancy. Am J Reprod Immunol. 1996;36(5):248–255. | ||
Jakubowiak AJ, Benson DM, Bensinger W, et al. Phase I trial of anti-CS1 monoclonal antibody elotuzumab in combination with bortezomib in the treatment of relapsed/refractory multiple myeloma. J Clin Oncol. 2012;30(16):1960–1965. | ||
Lonial S, Vij R, Harousseau JL, et al. Elotuzumab in combination with lenalidomide and low-dose dexamethasone in relapsed or refractory multiple myeloma. J Clin Oncol. 2012;30(16):1953–1959. | ||
Postelnek J, Sheridan J, Keller S, et al. Effects of elotuzumab on soluble SLAMF7 levels in multiple myeloma. American Society of Hematology Annual Meeting 2015. Blood. 2015;126(23):abstract 2964. | ||
Gibiansky L, Passey C, Roy A, Bello A, Gupta M. Model-based pharmacokinetic analysis of elotuzumab in patients with relapsed/refractory multiple myeloma. J Pharmacokinet Pharmacodyn. 2016;43(3):243–257. | ||
Passey C, Gibiansky L, Mora J, Roy A, Bello A, Gupta M. Effect of dexamethasone co-administration on the pharmacokinetics and immunogenicity of elotuzumab. Clin Pharmacol Ther. 2016;99:S86. | ||
Berdeja J, Jagannath S, Zonder J, et al. Pharmacokinetics and safety of elotuzumab combined with lenalidomide and dexamethasone in patients with multiple myeloma and various levels of renal impairment: results of a phase Ib study. Clin Lymphoma Myeloma Leuk. 2016;16(3):129–138. | ||
Richardson PG, Jagannath S, Moreau P, et al. Elotuzumab in combination with lenalidomide and dexamethasone in patients with relapsed multiple myeloma: final phase 2 results from the randomised, open-label, phase 1b–2 dose-escalation study. Lancet Haematol. 2015;2(12):e516–e527. | ||
Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373(7):621–631. | ||
Lagrue K, Carisey A, Morgan DJ, Chopra R, Davis DM. Lenalidomide augments actin remodeling and lowers NK-cell activation thresholds. Blood. 2015;126(1):50–60. | ||
Kotla V, Goel S, Nischal S, et al. Mechanism of action of lenalidomide in hematological malignancies. J Hematol Oncol. 2009;2:36. | ||
Benboubker L, Dimopoulos MA, Dispenzieri A, et al. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2014;371(10):906–917. | ||
Dimopoulos M, Spencer A, Attal M, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357(21):2123–2132. | ||
Weber DM, Chen C, Niesvizky R, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357(21):2133–2142. | ||
Weisel K, Doyen C, Dimopoulos M, et al. A systematic literature review and network meta-analysis of treatments for patients with untreated multiple myeloma not eligible for stem cell transplantation. Leuk Lymphoma. 2016;1–9. | ||
Dimopoulos MA. Eloquent-2 update: A phase 3, randomized, open-label study of elotuzumab in combination with lenalidomide/dexamethasone in patients with relapsed/refractory multiple myeloma – 3-year safety and efficacy follow-up. Blood. 2015;126(23):28. | ||
Hallett WH, Ames E, Motarjemi M, et al. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol. 2008;180(1):163–170. | ||
Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–2617. | ||
Jagannath S, Barlogie B, Berenson J, et al. A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br J Haematol. 2004;127(2):165–172. | ||
Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352(24):2487–2498. | ||
San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359(9):906–917. | ||
Scott K, Hayden PJ, Will A, Wheatley K, Coyne I. Bortezomib for the treatment of multiple myeloma. Cochrane Database Syst Rev. 2016;4:CD010816. | ||
Jakubowiak A, Offidani M, Pégourie B, et al. Randomized phase 2 study of elotuzumab plus bortezomib/dexamethasone (Bd) versus Bd for relapsed/refractory multiple myeloma. Blood. 2016;127(23):2833–2840. | ||
Palumbo A, Offidani M, Pégourie B, et al. Elotuzumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in patients with relapsed/refractory multiple myeloma: 2-year follow-up. Blood. 2015;126(23):510. | ||
Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21(21):3940–3947. | ||
Cartron G, Dacheux L, Salles G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99(3):754–758. | ||
Mateos MV. Elotuzumab for treating multiple myeloma. Exp Opin Orphan Drugs. 2016;4(2):215–222. | ||
Palumbo A, Sonneveld P. Preclinical and clinical evaluation of elotuzumab, a SLAMF7-targeted humanized monoclonal antibody in development for multiple myeloma. Expert Rev Hematol. 2015;8(4):481–491. | ||
Dimopoulos MA, Lonial S, White D, et al. Eloquent-2 update: A phase 3, randomized, open-label study of elotuzumab in combination with lenalidomide/dexamethasone in patients with relapsed/refractory multiple myeloma – 3-year safety and efficacy follow-up. Blood. 2015;126(23):28. | ||
Berenson J, Swift R, Hobson S, Rifkin R. Elotuzumab administered over approximately 60 minutes in combination with lenalidomide and dexamethasone in patients with multiple myeloma: a phase 2 safety study. In: Hematology/Oncology Pharmacy Association (HOPA) 12th Annual Conference; March 16–19, 2016; Atlanta, GA. Poster No. 31; 2016. | ||
Swift R, Hobson S, Rifkin R, Berenson J. Elotuzumab administered over approximately 60 minutes in combination with lenalidomide and dexamethasone in patients with multiple myeloma: a phase 2 safety study. In: Oncology Nursing Society 41st Annual Congress, April 28–May 1, 2016; San Antonio, TX. Poster No. E59. | ||
Dimopoulos MA, Terpos E, Niesvizky R, Palumbo A. Clinical characteristics of patients with relapsed multiple myeloma. Cancer Treat Rev. 2015;41(10):827–835. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.