")
Back to Journals » Lung Cancer: Targets and Therapy » Volume 8
Spotlight on brigatinib and its potential in the treatment of patients with metastatic ALK-positive non-small cell lung cancer who are resistant or intolerant to crizotinib
Received 4 August 2017
Accepted for publication 13 September 2017
Published 13 October 2017 Volume 2017:8 Pages 169—177
DOI https://doi.org/10.2147/LCTT.S126507
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ignatius Ou
Rohit K Jain, Hongbin Chen
Department of Medicine, Roswell Park Cancer Institute, Buffalo, NY, USA
Abstract: In the last decade, there have been major therapeutic advances in the management of patients with anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer. Crizotinib was the first approved ALK inhibitor with significant benefits over chemotherapy. However, patients inevitably develop disease progression especially in central nervous system and acquire resistance to crizotinib. Several next-generation ALK inhibitors have been developed to overcome these resistance mechanisms and have demonstrated clinical benefits in crizotinib-refractory non-small cell lung cancer including in central nervous system. Brigatinib is a second-generation ALK inhibitor with high level of activity against ALK and several other targets. It is active in vitro against many ALK kinase domain mutations including L1196M, E1210K, and G1202R which may mediate acquired resistance to other ALK inhibitors. In Phase I/II and ALTA clinical studies, brigatinib has demonstrated substantial and durable responses and intracranial responses after progression on crizotinib. It has acceptable safety profile, but early pulmonary toxicity has been observed prompting to pursue daily dosing of 180 mg (with lead-in). Overall, 180 mg (with lead-in) has showed consistently better efficacy than 90 mg. In this review, we will discuss in detail these two pivotal trials that led to the accelerated approval for brigatinib and its future directions.
Keywords: non-small cell lung cancer, ALK rearrangement, crizotinib, ceritinib, alectinib, brigatinib
Introduction
Lung cancer is the leading cause of cancer death in USA with estimated 225,500 new cases and 155,870 deaths in 2017.1,2 Of all patients, only 18% are alive at 5 years.1 Before the turn of this century, the treatment for stage IV non-small cell lung cancer (NSCLC) was cytotoxic chemotherapy with platinum-based doublet backbone with limited overall survival (OS) benefit.3 However, there has been significant developments in screening, diagnosis, and treatment including targeted and immune therapy. Several biomarkers have emerged as predictive and prognostic markers for NSCLC.4 The increased understanding of the complex biology of NSCLC and identification of genetic and molecular subgroups have led to the development of specific inhibitors to target these oncogenic driver mutations.5 The presence of epidermal growth factor receptor (EGFR) exon 19 deletion or exon 21 L858R mutation has shown treatment benefit from tyrosine kinase inhibitors (TKIs) such as gefitinib, erlotinib, and afatinib with prolonged progression-free survival (PFS) and preserved quality of life when used as the first-line treatment in advanced NSCLC.6,7 Anaplastic lymphoma kinase (ALK) fusion oncogene and ROS1 rearrangement are also predictive biomarkers seen in small subset of patients that benefit from crizotinib.8 In patients with nonsquamous NSCLC and NSCLC not otherwise specified, testing for ALK gene rearrangement and EGFR mutation is recommended (category 1) so that they can receive effective treatment with targeted agents.9 This review will focus on ALK gene rearrangements and ALK inhibitors with emphasis on recently approved brigatinib.
ALK-positive NSCLC
Soda et al first described the fusion of kinase domain of ALK gene (exons 20→29) and the echinoderm microtubule-associated protein-like 4 (EML4) gene in NSCLC.10 The final product is the novel fusion oncogene EML4-ALK, which is a chimeric protein with oncogenic properties and defines a distinct clinicopathologic subset in NSCLC.10 This rearrangement of ALK gene is present in 2%–7% of patients with NSCLC.11 These patients are found to be younger, more likely men, never/light smokers with adenocarcinoma histology predominantly signet-ring cell subtype.12–14 It is estimated that 30% of patients in this selected population will have ALK rearrangement.14,15 ALK rearrangements are not present routinely in squamous cell carcinoma although positive patients can have mixed squamous cell histology.16 As per National Comprehensive Cancer Network guidelines, testing for ALK rearrangements is recommended in cases of small biopsy specimen used, mixed histology, or in patients with no smoking history.17 As NSCLC patients have multiple genetic alterations, various multiplex polymerase chain reactions (PCRs) have been developed to detect these point mutations.18 However, as ROS1 and ALK gene rearrangements are not point mutations, these can be detected using fluorescence in situ hybridization (FISH). Broad molecular profiling systems like next-generation sequencing (NGS) allow for comprehensive sequencing of entire genomes, exomes, and transcriptomes. NGS is able to detect EML4 and ALK genes that are separated by small rearrangements that prevent detection by FISH assay.19–24 The presence of ALK rearrangement is mutually exclusive from EGFR and KRAS mutations among other oncogenic drivers.25 Due to the constitutive activation of the ALK fusion oncogene, they become susceptible to ALK inhibitors.26 Studies have shown that these were effective in vitro and in vivo in the cell lines and mouse models of tumors harboring the EML4–ALK rearrangement.26,27 Four ALK inhibitors, crizotinib, ceritinib, alectinib, and now brigatinib, have established roles in the treatment for ALK-rearranged NSCLC, and additional agents are under development.
Crizotinib for ALK-positive advanced NSCLC
Crizotinib (Xalkori) is a multitargeted TKI active against ALK, ROS1, RON, and MET.28 In the Phase I study (PROFILE-1001), 149 patients with advanced ALK-positive NSCLC underwent treatment with crizotinib at a dose of 250 mg twice daily as part of an expansion cohort.29 Seventy-one percent of patients were non-smokers, 97% had adenocarcinoma histology, and most had received two lines of treatment. The overall response rate (ORR) was 60.8% (95% CI: 52.3–68.9), median PFS (mPFS) was 9.7 months (95% CI: 7.7–12.8), and median duration of response was 49 weeks (95% CI: 39.3–75.4). The estimated OS rates at 6 and 12 months were 87.9% (95% CI: 81.3–92.3) and 74.8% (95% CI: 66.4–81.5), respectively. Visual effects, nausea, and diarrhea were the most commonly reported adverse events (AEs). Neutropenia, lymphopenia, and raised alanine aminotransferase (ALT) were the most common grade 3 or 4 AEs. The multicenter, single-arm, Phase II study (PROFILE-1005) of crizotinib in advanced ALK-positive NSCLC after progression on at least one line of cytotoxic chemotherapy revealed an ORR of 53% (95% CI: 47–60) and an mPFS of 8.5 months (95% CI: 6.2–9.9).30 More than 250 heavily pretreated patients were evaluated, and this established that crizotinib is beneficial with good tolerance in this subgroup of population. Based on these results, in 2011 the US Food and Drug Administration (FDA) granted accelerated approval for crizotinib for use in advanced ALK-positive NSCLC.
A Phase III study (PROFILE-1007) compared crizotinib with second-line chemotherapy (pemetrexed or docetaxel) after failing one prior platinum-based chemotherapy.31 A total of 318 patients with advanced ALK-positive NSCLC were randomized between the two groups. The primary endpoint of mPFS was significantly longer in the crizotinib group (7.7 months, 95% CI: 6–8.8) compared with the chemotherapy group (3.0 months, 95% CI: 2.6–4.3; hazard ratio [HR]=0.49, 95% CI: 0.37–0.64, p<0.0001). The ORRs were 65% (95% CI: 58–72) in the crizotinib group and 20% (95% CI: 14–26) in the chemotherapy group (p<0.001). When comparing crizotinib versus pemetrexed, the mPFS was 7.7 versus 4.2 months (HR=0.59, 95% CI: 0.43–0.80, p<0.001); versus 2.6 months in the docetaxel arm (HR=0.3, 95% CI: 0.21–0.43, p<0.0001). No difference was observed in OS between the two groups (20.3 versus 22.8 months; HR=1.02, 95% CI: 0.67–1.54, p=0.54), likely owing to crossover of patients from chemotherapy to crizotinib. Patients in the crizotinib group experienced greater reduction of lung cancer-related symptoms with improvement in the overall quality of life compared with the chemotherapy group.31 The most common grade 3 or 4 AEs were elevated aminotransferase levels (16%) and neutropenia (13%) in patients who underwent crizotinib treatment.
Another Phase III study (PROFILE-1014) compared crizotinib monotherapy with pemetrexed-platinum chemotherapy as the first-line treatment for advanced ALK-positive nonsquamous NSCLC (determined by FISH).32 Patients with treated and stable brain metastases for 2 weeks were also included. A total of 343 patients were randomized and the primary endpoint of PFS was observed to be significantly longer in the crizotinib group (10.9 versus 7.0 months; HR=0.45, 95% CI: 0.35–0.60, p<0.001) than in the chemotherapy group. Response rate was 74% (95% CI: 67–81) in the crizotinib group and 45% (95% CI: 37–53) in the chemotherapy group with duration of response being 11.3 versus 5.3 months in the respective groups. In patients with brain metastases, the disease control rate at 12 weeks was 65% with crizotinib versus 46% with chemotherapy. OS was equivalent in both arms with better quality of life in the crizotinib arm (HR=0.60, 95% CI: 0.27–1.42). These results led to the FDA approval for crizotinib in 2013 as the first-line treatment for patients with advanced ALK-positive NSCLC.33
Mechanisms of crizotinib resistance
Despite the initial good response of crizotinib, ALK-rearranged NSCLC inevitably progresses after 8–12 months. Central nervous system (CNS) progression is observed despite systemic control on crizotinib.31 This has been largely due to inadequate levels of crizotinib in CNS, suggested by low CSF to serum ratio which is largely in the range of 0.06%–0.26%.34,35 Also, crizotinib is a substrate of PgP, a drug efflux membranous transporter, which limits its accumulation in CNS.28 Other cellular mechanisms studied extensively include secondary mutations in the kinase domain of ALK, like the “gatekeeper mutation” which is a substitution of leucine with methionine at position 1196 (L1196M), decreasing the affinity of crizotinib binding to ALK.36 Some other resistance mutations have also been reported including C1156Y, F1174L, G1269A, 1151Tins, L1152R, S1206Y, I1171T, V1180L, D1203N, and G1202R.37 In addition to the secondary mutations, activation of alternative pathways such as EGFR, Kras, and HER2 can also cause acquired resistance to crizotanib.38,39 ALK amplification, epithelial–mesenchymal transition, and insulin-like growth factor 1 receptor (IGF-1R) pathway activation have also resulted in crizotinib resistance.39–41 In some patients, the mechanism of acquired resistance remains unknown.42
Second-generation ALK-TKIs
Ceritinib
Ceritinib (LDK378, Zykadia) is a second-generation ALK inhibitor, derived from NVP-TAE684.43,44 It inhibits IGF-1R and ROS1 but not potent against MET.45 Preclinical studies have demonstrated that ceritinib has more significant antitumor activity than crizotinib, including in tumors with the most common L1196M and G1269A resistance mutations.46 In the dose expansion cohort of Phase I (ASCEND-1) clinical study, 246 patients with ALK-positive NSCLC were treated with ceritinib at a dose of 750 mg once daily.47 Due to the AEs, at least one dose reduction in nearly 60% of the patients was performed. The ORR was 56% (95% CI: 49–64) in patients with prior crizotinib exposure and 72% (95% CI: 61–82) in ALK inhibitor-naïve patients. The mPFS was 18.4 months (95% CI: 11.1–not estimated [NE]) in ALK inhibitor-naïve group and 6.9 months (95% CI: 5.6–8.7) in those previously treated with an ALK inhibitor. The most common grade 3 or 4 AEs were increased ALT level (30%), increased aspartate aminotransferase (AST) level (10%), and diarrhea (6%), which were reversible after discontinuation of ceritinib therapy. In regards to CNS activity, the ORRs were 65% (95% CI: 54–76) and 79% (95% CI: 54–94) in the ALK inhibitor-pretreated and ALK inhibitor-naïve groups, respectively. On the basis of these findings, ceritinib was approved by the FDA in April 2014 for patients who have progressed on, or are intolerant of, crizotinib.
In the Phase II trial (ASCEND-2) of 140 patients with two or more previous treatments, progressed on crizotinib, and had brain metastases, ceritinib showed an ORR of 38% (95% CI: 30.5–47.2) and a duration of response of 9.7 months (95% CI: 7.1–11.1).48 Of 100 patients with baseline brain metastases, the intracranial ORR was 45% (95% CI: 23.1–68.5). The common AEs included nausea (81.4%), diarrhea (80%), and vomiting (63%). In the Phase III (ASCEND-5) trial with two or less lines of cytotoxic treatment and crizotinib, ceritinib showed an ORR of 39% and a significantly superior mPFS versus chemotherapy (5.4 versus 1.6 months, HR=0.49, 95% CI: 0.36–0.47, p<0.001).49 Diarrhea, nausea, and vomiting were higher with ceritinib compared with chemotherapy. Recent Phase III trial (ASCEND-4) assessed ceritinib versus platinum-based chemotherapy as first-line therapy in ALK-positive advanced NSCLC.50 mPFS was twice as long with ceritinib (16.6 months, 95% CI: 12.6–27.2) as with standard chemotherapy (8.1 months, 95% CI: 5.8–11.1; HR 0.55, 95% CI: 0.42–0.73, p<0.001). Most common AEs (occurring in >50%) with ceritinib were diarrhea (85%), nausea (69%), vomiting (66%), ALT increase (60.3%), and AST increase (52.9%). In patients with measurable CNS lesions, intracranial ORR was 57% (95% CI: 37–76) with ceritinib and 22% (95% CI: 9–42) with chemotherapy. Based on these results, in May 2017, the FDA broadened ceritinib indication to previously untreated ALK-positive advanced NSCLC patients.
Alectinib
Alectinib (CH542802/RO542802, Alecensa) is a highly potent and selective ALK inhibitor, with activity against L1196M gatekeeper mutation as well as other secondary mutations such as G1269A.51–53 It does not inhibit MET kinase and has low inhibitory activity against ROS1 but exerts antiproliferative activity against RET kinase.54 It also showed potent activity against intracranial tumor in mouse models.52 In a Phase I/II study (AF-001JP) conducted in Japan, patients with ALK-positive and crizotinib-naïve NSCLC were treated with alectinib.55,56 As there were no dose-limiting toxicities (DLTs) or grade 4 AEs with highest dose, 300 mg twice daily was the recommended Phase II dose. In the Phase II portion, the ORR was 93.5% (95% CI: 82.1–98.6). Twenty-six percent patients developed grade 3 AEs including neutropenia and increased blood creatinine phosphokinase (CPK). This study led to alectinib approval in Japan in July 2014 for advanced ALK-positive NSCLC. In the Phase I/II study (AF-002JG) of alectinib, 600 mg twice daily was recommended for Phase II based on activity, tolerability, and pharmacokinetic data.57 In the USA, the activity of alectinib in crizotinib-refractory patients was tested in the global NP28763 and North American NP28761 Phase II studies.58,59 In NP28763, of the 138 patients pretreated with crizotinib with and without chemotherapy, alectinib 600 mg showed an ORR of 50% (95% CI: 41–59), with an mPFS of 8.9 months (95% CI: 5.6–11.3). In chemotherapy-naïve patients, the ORR was 69.2% (95% CI: 48–86) with an mPFS of 13 months (95% CI: 5.5 – not reached). A control rate of 83% (95% CI: 74–91) and a median duration of response of 10.3 months (95% CI: 7.6–11.2) were observed in CNS disease. The common AEs included constipation (33%), fatigue (26%), and peripheral edema (25%).58 In the North American phase NP28761 study, among 87 crizotinib-refractory patients the ORR was 48% (95% CI: 36–60) and the mPFS was 8.1 months (95% CI: 6.2–12.6).59 These results led to alectinib approval in December 2015 for ALK-positive advanced NSCLC patients who progressed on crizotinib. Alectinib is also known to be highly activated in CNS metastases including leptomeningeal diseases.57,60 In a pooled analysis of two Phase II studies, an intracranial ORR of 64% (22% complete response [CR]) was observed in 50 patients with measurable CNS disease.61 This is thought to be due to alectinib’s avoidance of efflux transporter PgP, thus achieving higher CNS therapeutic concentrations.62 In the recently reported open-label, Phase III (J-ALEX) trial, efficacy and safety of alectinib and crizotinib were compared in ALK inhibitor-naïve Japanese patients, with either no or one prior chemotherapy regimen.63 Patients were randomized in 1:1 fashion to receive alectinib 300 mg twice daily or crizotinib 250 mg twice daily. A total of 207 patients were enrolled; the mPFS was not reached for alectinib (95% CI: 20.3–NE) compared with crizotinib (10.2 months; 95% CI: 8.2–12; HR=0.34, 99.7% CI: 0.17–0.71). Similarly, the Phase III (ALEX) trial compared alectinib with crizotinib in previously untreated, advanced ALK-positive patients including those with asymptomatic CNS disease.64 A total of 303 patients were enrolled and randomized to receive either alectinib (600 mg twice daily) or crizotinib (250 mg twice daily). The investigator-assessed PFS rate was significantly higher with alectinib (68.4%, 95% CI: 61–75.9) than with crizotinib (48.7%, 95% CI: 40.4–56.9; HR 0.47, 95% CI: 0.34–0.65, p<0.001). Twelve percent of patients developed CNS disease progression on alectinib compared with 45% on crizotinib (HR 0.16, 95% CI: 0.1–0.28, p<0.001). Alectinib showed greater efficacy suppressing CNS disease than crizotinib. These Phase III data indicate the potential of alectinib being the first-line treatment for patients with ALK-positive NSCLC with and without CNS disease where it can be neuroprotective.
Brigatinib
Brigatinib (AP26113, Alunbrig) is a pyrimidine-based molecule containing a C4 aniline with an ortho-dimethylphosphine oxide substituent.65 It is a synthetic oral TKI with a mean plasma elimination half-life of 25 hours and hepatic elimination as a major route of excretion. In the preclinical studies using ALK-positive cell lines, it inhibits ALK kinase with 12-fold greater potency than crizotinib. It showed a high degree of selectivity, inhibiting only 11 additional native or mutant kinases including ROS1, FLT3, and mutant variants of FLT3 (D835Y) and EGFR (L858R). It exhibited more modest activity against EGFR with a T790M resistance mutation (L858R/T790M), native EGFR, IGF1R, and INSR but did not inhibit MET. Compared with crizotinib, ceritinib, and alectinib, brigatinib displayed superior activity and inhibitory profile against all 17 secondary ALK mutations including the most recalcitrant G1202R.65 These promising preclinical results of brigatinib led to a Phase I/II study to assess safety, activity, and pharmacokinetic profile in advanced malignancies including ALK-rearranged NSCLC and the Phase II (ALTA) trial (Table 1).66,67 Based on results of the pivotal Phase II study, in April 2017, the FDA granted accelerated approval for brigatinib in patients with locally advanced or metastatic ALK-positive NSCLC who have progressed on or are intolerant to crizotinib.
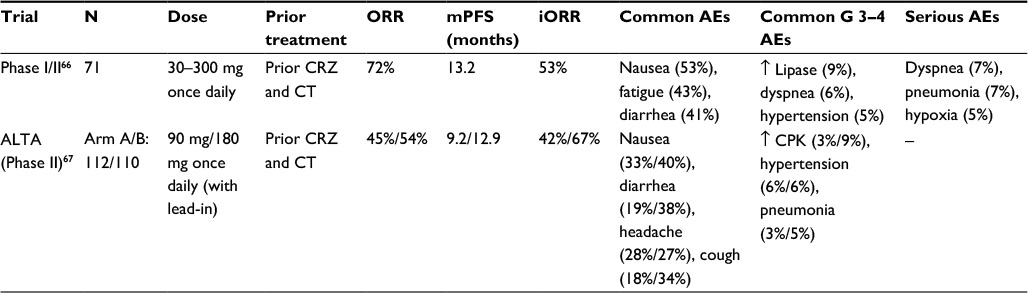 | Table 1 Summarizing the clinical outcomes and AEs of brigatinib in patients with advanced ALK-positive NSCLC Notes: ↑represented as increase; “–” represented as no data. Abbreviations: AEs, adverse events; ALK, anaplastic lymphoma kinase; CPK, creatinine phosphokinase; CRZ, crizotinib; CT, chemotherapy; mPFS, median progression-free survival; N, number of patients; NSCLC, non-small cell lung cancer; iORR, intracranial objective response rate; ORR, objective response rate. |
Phase I/II trial
This is a single-arm, open-label, Phase I/II trial performed at nine academic centers in USA and Spain.66 The Phase I dose escalation was conducted in histologically confirmed advanced malignancies except leukemia that were refractory to available therapies or no other treatment options were available. The primary objective of Phase I was to establish the recommended dose for Phase II. This was followed by expansion of Phase II in five cohorts defined by histological and molecular characteristics. These were as follows: cohort 1 – ALK inhibitor–naïve, ALK-rearranged NSCLC; cohort 2 – crizotinib-treated, ALK-rearranged NSCLC; cohort 3 – EGFRT790M-positive NSCLC with resistance to one prior EGFR-TKIs; cohort 4 – other cancers with alterations in brigatinib targets like ALK or ROS1; and cohort 5 – crizotinib-naïve or crizotinib-treated, ALK-rearranged NSCLC with active and measurable intracranial metastases. The primary outcome of Phase II was investigator-assessed ORR for cohorts 1–4 and CNS response for cohort 5. The secondary outcomes of Phase I included safety, tolerability, dose-limiting toxicities, and maximum tolerated dose of brigatinib. The Phase II secondary outcomes included PFS, OS, best target lesion response, and time to treatment failure. Inclusion criteria for both phases included patients ≥18 years old, with measurable disease by Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1); Eastern Cooperative Oncology Group Performance Status (ECOG PS) 0-1; adequate renal, hepatic, and bone marrow functions; normal QT interval; and those who were not pregnant. Patients with stable CNS metastases, not requiring anticonvulsants and higher doses of corticosteroids, were included. Patients with any number of treatments with previous systemic therapies were included. Patients with active or uncontrolled hypertension or cardiovascular diseases, history or active pulmonary interstitial disease, or drug-related pneumonitis were excluded.
A total of 137 patients were enrolled in the Phase I dose escalation and Phase II dose expansion cohorts. In the drug escalation Phase I, the dosing ranged from 30 to 300 mg once daily. One grade 3 dose-limiting toxicity with increased ALT was observed at 240 mg daily and one grade 4 dyspnea was observed at 300 mg daily. Although, as per protocol, no formal establishment of maximum tolerated dose was made, a dose of 180 mg once daily was recommended as Phase II dose based on no DLTs, pharmacokinetics, and preliminary data. Due to early pulmonary events at 180 mg once daily, two additional regimens were explored which were later recommended for Phase II dose expansion cohorts including 90 mg once daily and 180 mg once daily with a 7-day lead-in at 90 mg.
In the Phase II dose expansion cohorts, the ORRs were as follows: cohort 1 – ALK inhibitor–naïve, ALK-rearranged NSCLC patients, the ORR was seen in 4/4 (100%, 95% CI: 40–100); cohort 2 – crizotinib-treated, ALK-rearranged NSCLC patients, ORR was seen in 31/42 (74%, 95% CI: 58–86); cohort 3 – none of EGFRT790M-positive NSCLC patients with resistance to one prior EGFR-TKI had ORR; cohort 4 – 3/18 (17%, 95% CI: 4–41) patients with other cancers with alterations in brigatinib targets like ALK or ROS1 had an objective response; and cohort 5 – crizotinib-naïve or crizotinib-treated, ALK-rearranged NSCLC with active, measurable, intracranial metastases, 5/6 (83%, 95% CI: 36–100) patients had ORR. In patients with ALK-rearranged NSCLC treated with crizotinib, 51/71 had an objective response (72%, 95% CI: 60–82). These included 47 patients with partial response (PR) and 4 patients with CR. In crizotinib-naïve patients, 8/8 (100%, 95% CI: 63–100) achieved confirmed objective response including 3 (38%) who displayed CR. In patients with previous crizotinib treatment, when treated with 90 mg brigatinib once daily, 10/13 (77%, 95% CI: 46–95) had objective response; with 180 mg once daily with 7-day lead-in at 90 mg, 20/25 (80%, 95% CI: 59–93) had objective response; and of those who received 180 mg once daily, 15/23 (65%, 95% CI: 43–84) had objective response. The mPFS in crizotinib-pretreated, ALK-rearranged NSCLC was 13.2 months (95% CI: 9.1–18.7) and was not reached in crizotinib-naïve patients (95% CI: 7.4–not reached). The probability of OS at 1 year in crizotinib-pretreated patients was 78% (95% CI: 67–86), and in crizotinib-naïve patients, it was 100% (95% CI: 100–100).
The treatment-emergent AEs (TEAEs) were predominantly grade 1–2 including nausea (53%), fatigue (43%), and diarrhea (41%). Among grade 3–4 AEs were increased lipase concentration (9%), dyspnea (6%), and hypertension (5%). Serious AEs were dyspnea (7%), pneumonia (7%), and hypoxia (5%). Sixteen patients died during treatment with brigatinib, which included eight deaths due to disease progression. Of the remaining eight patients, seven deaths were related to treatment (sudden death, hypoxia, and unknown cause) and one due to pneumonia.
During Phase I and the initial Phase II expansion, a subset of pulmonary events was observed within 7 days of treatment initiation or reinitiation following a period of dose interruption. These events included dyspnea, hypoxia, cough, pneumonia, and pneumonitis. Most events happened within 24–48 hours of dosing, requiring dose interruptions with steroids and antibiotics. These events were seen across all doses; however, incidence increased with higher starting doses. Only 2% of the events occurred in patients starting at 90 mg daily while 14% of the events occurred in patients starting on 180 mg daily dosing. Due to these TEAEs, two additional dosing regimens at 90 mg daily and 180 mg daily with a 7-day lead-in at 90 mg daily were explored. None of the 32 patients treated with loading dose of 90 mg daily for 7 days followed by 180 mg daily reported such events.
Sixty-three percent (50/79) of patients with ALK-rearranged NSCLC had brain metastases at baseline. Forty-six percent (23/50) had no prior brain radiation treatment. Forty-six patients had baseline MRI, of which 15 patients had measurable CNS metastases and 31 had nonmeasurable disease. The intracranial ORR seen was 53% (95% CI: 27–79) in the measurable disease group and 35% (95% CI: 19–55) in the nonmeasurable disease group. The median duration of response for all 19 assessable patients was 18.9 months (95% CI: 5.5 – not reached). The intracranial mPFS for all 46 patients was 15.6 months (95% CI: 13 – not reached), and for patients with no prior radiation treatment, it was 22.3 months (95% CI: 8–22.3).
ALTA (Phase II)
The ALK in Lung Cancer Trial of AP26113/brigatinib (ALTA) is an open-label, randomized, multicenter, international Phase II study.67 It was designed to evaluate safety and efficacy of two different regimens (90 mg daily and 180 mg daily with 7-day lead-in of 90 mg) of brigatinib in locally advanced or metastatic ALK-rearranged NSCLC patients with prior exposure to crizotinib. The inclusion criteria included adequate organ and hematologic function, at least one measurable lesion per RECIST v1.1, ECOG PS ≤2, and should not have received any ALK inhibitor other than crizotinib. Patients with history of or active interstitial lung disease, drug-related pneumonitis, neurologically unstable/symptomatic CNS metastases, or requiring increasing doses of corticosteroids were excluded. Patient stratification was based on the presence of brain metastases at baseline (present versus absent) and best investigator-assessed response (CR, PR, or unknown) to prior crizotinib. These patients were randomized in 1:1 fashion to receive 90 mg once daily (arm A) or 180 mg once daily with a 7-day lead-in at 90 mg (arm B). The primary endpoint was investigator-assessed ORR per RECIST v1.1. The secondary endpoints included PFS and independent review committee-assessed confirmed ORR and CNS response (intracranial confirmed ORR and PFS) along with duration of response, OS, safety, and tolerability. Although statistical design did not include comparisons between arms, post hoc hazard ratios were estimated for PFS and OS to support dose selection.
A total of 222 patients were randomly assigned to arm A (112) and arm B (110). Of these, 69% had brain metastases at baseline, 74% had prior chemotherapy, 65% had a best response (CR or PR) to prior crizotinib, and 31% were Asians. The investigator-assessed ORR in arm A was 45% (97.5% CI: 34–56) including one CR. The confirmed ORR in arm B was 54% (97.5% CI: 43–65) including four CRs. In patients with prior chemotherapy, ORR was 42% in arm A and 54% in arm B, while without chemotherapy it was 52% in each arm. One patient with G1202R mutation at baseline in arm B was confirmed to have PR. The median time to response was 1.8 months (range 1.7–9.1 months) and 1.9 months (range 1–11 months) in arms A and B, respectively. The mPFS was 9.2 months (95% CI: 7.4–15.6) and 12.9 months (95% CI: 11.1–not reached) in arms A and B, respectively (HR=0.55, 95% CI: 0.35–0.86). The 1-year OS probability was 71% in arm A (95% CI: 60%–79%) and 80% (95% CI: 67%–80%) in arm B. In patients with measurable baseline brain metastasis, the independent review committee-assessed ORR was 42% (95% CI: 23–63) and 67% (95% CI: 41–87) in arms A and B, respectively. It was noted that in patients with measurable and active brain metastases, the response rate was similar within each arm. Among patients with nonmeasurable baseline brain metastases, 7% (95% CI: 2–18) and 18% (95% CI: 9–31) had complete resolution of intracranial lesions in arms A and B, respectively. The median intracranial PFS was 15.6 months (95% CI: 7.3–15.7) in arm A and 12.8 months (95% CI: 11–not reached) in arm B.
The AEs (any grade) in both arms A and B included nausea (33%/40%), diarrhea (19%/38%), headache (28%/27%), fatigue (20%/27%), vomiting (24%/23%), and dyspnea (21%). Visual disturbances including blurred vision, diplopia, and reduced visual acuity also occurred in patients (7.3%/10%) receiving brigatinib. The most common grade ≥3 AEs in arms A and B included hypertension (6%/6%), increased blood CPK (3%/9%), pneumonia (3%/5%), and increased lipase (4%/3%). Six percent of the patients developed early onset pulmonary events which included dyspnea, hypoxia, cough, pneumonia, or pneumonitis. Seven patients (3%) developed grade ≥3 toxicity. These pulmonary AEs were noted at 90 mg in both arms, and no further events occurred after dose escalation to 180 mg. Seven patients discontinued the treatment including one death due to lymphangitic carcinomatosis, widespread lung scarring, and diffuse alveolar damage. In a multivariable analysis, older age and shorter interval between last crizotinib dose and first brigatinib dose were significantly associated with increased pulmonary toxicity.
Future trials
ALTA-1L (NCT 02737501) is an ongoing open-label, multicenter, randomized Phase III trial designed to assess the efficacy and safety of brigatinib versus crizotinib in patients with advanced ALK-rearranged NSCLC who are naive to TKI therapy (including ALK inhibitors). Approximately 270 patients are planned to be enrolled from April 2016 with an estimated primary completion date of April 2019. These patients are randomized in 1:1 fashion to receive either brigatinib 180 mg (with 7-day lead-in of 90 mg) or crizotinib and stratified by presence of brain metastasis at baseline and prior chemotherapy. The primary endpoint is PFS and secondary endpoints are ORR, duration of response, OS, and intracranial ORR/PFS.68 Another ongoing Phase II trial (NCT 02706626) is studying brigatinib after treatment with second-generation ALK inhibitors. This is to assess safety and effectiveness of brigatinib in patients who have progressed on second-generation ALK inhibitors including ceritinib and alectinib. Approximately 40 patients will be enrolled with primary completion date in June 2018 and primary endpoint will be ORR. This will help to identify patient population in which brigatinib can be used as a salvage treatment option to overcome resistance to other ALK inhibitors.
Conclusion
With the discovery of ALK translocation in NSCLC patients, there have been several therapeutic developments targeting this genetic alteration. Brigatinib is a next-generation ALK inhibitor with broader coverage against several clinically relevant ALK mutations including acquired resistance mutations which emerge after treatment with other TKIs. The Phase I and II trials have shown that it is very potent in ALK-rearranged NSCLC patients with previous crizotinib exposure in terms of overall (54%–72%) as well as intracranial (53%–67%) response rates. The mPFS between 12.9 and 13.2 months in Phase I and II trials is substantial relative to ceritinib and alectinib.69 In patients with measurable brain metastases, the ORR of 67% with 180 mg (with lead-in) brigatinib is favorable compared with other second-generation ALK inhibitors.69 The treatment landscape of ALK-rearranged NSCLC is changing dramatically especially after the results of J-ALEX and ALEX trials showing improved PFS with alectinib over crizotinib.63,64 Based on these results, alectinib would likely be approved in the front-line setting and brigatinib could be used as a potential second-line agent after progression on alectinib. The ongoing clinical trial of brigatinib in ALK-rearranged NSCLC with prior exposure to other second-generation ALK inhibitors will further elucidate its role in this setting. In treatment-naïve patients, like alectinib, brigatinib may also improve duration of disease control and intracranial disease control along with delaying resistance.69 The pivotal Phase III study (ALTA-1L) is ongoing and will help to further identify its role in the first-line setting. In terms of safety profile, alectinib has been observed to be more tolerable and ceritinib is noticed to have high drug-related AEs. Brigatinib, in general, is well tolerated but the development of early onset pulmonary toxicity is concerning prompting the dose to start at 90 mg for 7 days followed by 180 mg daily. Physicians need to closely monitor these patients and may require dose interruptions with early clinical intervention.
In summary, brigatinib as a novel ALK inhibitor is promising and may play an important role in the treatment for ALK-positive, NSCLC-targeting, ALK-resistant mutations which are not covered by other ALK inhibitors. Further clinical trials are required to better understand the resistance mechanisms and establish appropriate sequence of drugs for optimal management.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1–19. | ||
Scagliotti GV, De Marinis F, Rinaldi M, et al; Italian Lung Cancer Project. Phase III randomized trial comparing three platinum-based doublets in advanced non-small-cell lung cancer. J Clin Oncol. 2002;20(21):4285–4291. | ||
Johnson DH, Schiller JH, Bunn PA Jr. Recent clinical advances in lung cancer management. J Clin Oncol. 2014;32(10):973–982. | ||
Metro G, Crino L. Novel molecular trends in the management of advanced non-small-cell lung cancer. Expert Rev Anticancer Ther. 2012;12(6):729–732. | ||
Miller VA, Riely GJ, Zakowski MF, et al. Molecular characteristics of bronchioloalveolar carcinoma and adenocarcinoma, bronchioloalveolar carcinoma subtype, predict response to erlotinib. J Clin Oncol. 2008;26(9):1472–1478. | ||
Sequist LV, Martins RG, Spigel D, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26(15):2442–2449. | ||
Mazieres J, Zalcman G, Crino L, et al. Crizotinib therapy for advanced lung adenocarcinoma and a ROS1 rearrangement: results from the EUROS1 cohort. J Clin Oncol. 2015;33(9):992–999. | ||
Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol. 2013;8(7):823–859. | ||
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. | ||
Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. | ||
Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol. 2009;22(4):508–515. | ||
Rodig SJ, Mino-Kenudson M, Dacic S, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res. 2009;15(16):5216–5223. | ||
Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253. | ||
Sun JM, Lira M, Pandya K, et al. Clinical characteristics associated with ALK rearrangements in never-smokers with pulmonary adenocarcinoma. Lung Cancer. 2014;83(2):259–264. | ||
Wong DW, Leung EL, So KK, et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009;115(8):1723–1733. | ||
Ettinger DS, Wood DE, Aisner DL, et al. Non-small cell lung cancer, version 5.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2017;15(4):504–535. | ||
Lovly CM, Horn L, Pao W. Molecular profiling of lung cancer. My Cancer Genome. 2016;(updated March 28, 2016). Available from: https://www.mycancergenome.org/content/disease/lung-cancer/. Accessed October 03, 2017. | ||
Aziz N, Zhao Q, Bry L, et al. College of American Pathologists’ laboratory standards for next-generation sequencing clinical tests. Arch Pathol Lab Med. 2015;139(4):481–493. | ||
Drilon A, Wang L, Arcila ME, et al. Broad, hybrid capture-based next-generation sequencing identifies actionable genomic alterations in lung adenocarcinomas otherwise negative for such alterations by other genomic testing approaches. Clin Cancer Res. 2015;21(16):3631–3639. | ||
Robson ME, Bradbury AR, Arun B, et al. American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol. 2015;33(31):3660–3667. | ||
Yu PP, Vose JM, Hayes DF. Genetic cancer susceptibility testing: increased technology, increased complexity. J Clin Oncol. 2015;33(31):3533–3534. | ||
Cardarella S, Ortiz TM, Joshi VA, et al. The introduction of systematic genomic testing for patients with non-small-cell lung cancer. J Thorac Oncol. 2012;7(12):1767–1774. | ||
Li T, Kung HJ, Mack PC, Gandara DR. Genotyping and genomic profiling of non-small-cell lung cancer: implications for current and future therapies. J Clin Oncol. 2013;31(8):1039–1049. | ||
Takahashi T, Sonobe M, Kobayashi M, et al. Clinicopathologic features of non-small-cell lung cancer with EML4-ALK fusion gene. Ann Surg Oncol. 2010;17(3):889–897. | ||
Soda M, Takada S, Takeuchi K, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A. 2008;105(50):19893–19897. | ||
Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14(13):4275–4283. | ||
Leprieur EG, Fallet V, Cadranel J, Wislez M. Spotlight on crizotinib in the first-line treatment of ALK-positive advanced non-small-cell lung cancer: patients selection and perspectives. Lung Cancer (Auckl). 2016;7:83–90. | ||
Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13(10):1011–1019. | ||
Kim D, Ahn M, Shi Y, et al. Results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC). J Clin Oncol. 2012;30(Suppl 15):7533. | ||
Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. | ||
Solomon BJ, Mok T, Kim DW, et al; PROFILE 1014 Investigators. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. | ||
Gandhi S, Chen H, Zhao Y, Dy GK. First-line treatment of advanced ALK-positive non-small-cell lung cancer. Lung Cancer (Auckl). 2015;6:71–82. | ||
Kaneda H, Okamoto I, Nakagawa K. Rapid response of brain metastasis to crizotinib in a patient with ALK rearrangement-positive non-small-cell lung cancer. J Thorac Oncol. 2013;8(4):e32–e33. | ||
Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol. 2011;29(15):e443–e445. | ||
Choi YL, Soda M, Yamashita Y, et al; ALK Lung Cancer Study Group. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–1739. | ||
Toyokawa G, Seto T. Updated evidence on the mechanisms of resistance to ALK inhibitors and strategies to overcome such resistance: clinical and preclinical data. Oncol Res Treat. 2015;38(6):291–298. | ||
Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–1482. | ||
Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4(120):120ra17. | ||
Tanizaki J, Okamoto I, Okabe T, et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin Cancer Res. 2012;18(22):6219–6226. | ||
Yamaguchi N, Lucena-Araujo AR, Nakayama S, et al. Dual ALK and EGFR inhibition targets a mechanism of acquired resistance to the tyrosine kinase inhibitor crizotinib in ALK rearranged lung cancer. Lung Cancer. 2014;83(1):37–43. | ||
Costa DB, Kobayashi S. Acquired resistance to the ALK inhibitor crizotinib in the absence of an ALK mutation. J Thorac Oncol. 2012;7(3):623–625. | ||
Galkin AV, Melnick JS, Kim S, et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci U S A. 2007;104(1):270–275. | ||
Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem. 2013;56(14):5675–5690. | ||
Shaw AT, Mehra R, Kim DW, et al. Clinical activity of the ALK inhibitor LDK378 in advanced, ALK-positive NSCLC. J Clin Oncol. 2013;31(Suppl 15):8010. | ||
Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4(6):662–673. | ||
Kim DW, Mehra R, Tan DS, et al. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol. 2016;17(4):452–463. | ||
Crino L, Ahn MJ, De Marinis F, et al. Multicenter phase II study of whole-body and intracranial activity with ceritinib in patients with ALK-rearranged non-small-cell lung cancer previously treated with chemotherapy and crizotinib: results from ASCEND-2. J Clin Oncol. 2016;34(24):2866–2873. | ||
Shaw AT, Kim TM, Crino L, et al. Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017;18(7):874–886. | ||
Soria JC, Tan DSW, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet. 2017;389(10072):917–929. | ||
Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem. 2012;20(3):1271–1280. | ||
Kodama T, Tsukaguchi T, Yoshida M, Kondoh O, Sakamoto H. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014;351(2):215–221. | ||
Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19(5):679–690. | ||
Ou SH, Janne PA, Bartlett CH, et al. Clinical benefit of continuing ALK inhibition with crizotinib beyond initial disease progression in patients with advanced ALK-positive NSCLC. Ann Oncol. 2014;25(2):415–422. | ||
Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1–2 study. Lancet Oncol. 2013;14(7):590–598. | ||
Yang JC. A selective ALK inhibitor in ALK-rearranged patients. Lancet Oncol. 2013;14(7):564–565. | ||
Gadgeel SM, Gandhi L, Riely GJ, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014;15(10):1119–1128. | ||
Ou SH, Ahn JS, De Petris L, et al. Alectinib in crizotinib-refractory ALK-rearranged non-small-cell lung cancer: a phase II global study. J Clin Oncol. 2016;34(7):661–668. | ||
Shaw AT, Gandhi L, Gadgeel S, et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: a single-group, multicentre, phase 2 trial. Lancet Oncol. 2016;17(2):234–242. | ||
Ou SH, Sommers KR, Azada MC, Garon EB. Alectinib induces a durable (>15 months) complete response in an ALK-positive non-small cell lung cancer patient who progressed on crizotinib with diffuse leptomeningeal carcinomatosis. Oncologist. 2015;20(2):224–226. | ||
Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small-cell lung cancer. J Clin Oncol. 2016;34(34):4079–4085. | ||
Kodama T, Hasegawa M, Takanashi K, Sakurai Y, Kondoh O, Sakamoto H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother Pharmacol. 2014;74(5):1023–1028. | ||
Hida T, Nokihara H, Kondo M, et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): an open-label, randomised phase 3 trial. Lancet. 2017;390(10089):29–39. | ||
Peters S, Camidge DR, Shaw AT, et al; ALEX Trial Investigators. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med. 2017;377(9):829–838. | ||
Zhang S, Anjum R, Squillace R, et al. The potent ALK inhibitor brigatinib (AP26,113) overcomes mechanisms of resistance to first- and second-generation ALK inhibitors in preclinical models. Clin Cancer Res. 2016;22(22):5527–5538. | ||
Gettinger SN, Bazhenova LA, Langer CJ, et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: a single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016;17(12):1683–1696. | ||
Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490–2498. | ||
Tiseo M, Popat S, Gettinger SN, et al. Design of ALTA-1L (ALK in lung cancer trial of brigatinib in first-line), a randomized phase 3 trial of brigatinib (BRG) versus crizotinib (CRZ) in tyrosine kinase inhibitor (TKI)-naive patients (pts) with advanced anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC). J Clin Oncol. 2017;35(Suppl 15):TPS9098. | ||
Sabari JK, Santini FC, Schram AM, et al. The activity, safety, and evolving role of brigatinib in patients with ALK-rearranged non-small cell lung cancers. Onco Targets Ther. 2017;10:1983–1992. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.