")
Back to Journals » OncoTargets and Therapy » Volume 10
Spotlight on bevacizumab and its potential in the treatment of malignant pleural mesothelioma: the evidence to date
Received 23 December 2016
Accepted for publication 11 March 2017
Published 7 April 2017 Volume 2017:10 Pages 2057—2066
DOI https://doi.org/10.2147/OTT.S113598
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yao Dai
Pavel A Levin,1 Jonathan E Dowell1,2
1Division of Hematology/Oncology, University of Texas Southwestern Medical Center, 2Section of Hematology/Oncology, Veteran Affairs North Texas Health Care System, Dallas, TX, USA
Abstract: Malignant pleural mesothelioma (MPM) is a rare, but aggressive cancer. Surgery and radiation offer limited benefit, and systemic chemotherapy remains the primary treatment modality for the majority of patients. Vascular endothelial growth factor (VEGF) and its receptor have been recognized as important players in the biology of this disease. Bevacizumab is a monoclonal antibody that binds VEGF and blocks its interaction with the VEGF receptor. Recent studies have shown benefit with the addition of bevacizumab to the combination of cisplatin and pemetrexed in MPM. This combination is now included in the National Comprehensive Cancer Network guidelines (with a category 2A recommendation) as a possible first-line treatment for unresectable MPM in appropriately selected patients. This review discusses the rationale behind the use of bevacizumab in MPM, as well as summarizes the pharmacology, efficacy, safety, and toxicity of bevacizumab across multiple trials. The use of small-molecule inhibitors of angiogenesis in the treatment of MPM is also discussed.
Keywords: angiogenesis, monoclonal antibody, VEGF
Introduction
Malignant pleural mesothelioma (MPM) is a rare, but aggressive cancer of the mesothelial surface of the pleural cavity, which has been linked to occupational exposure to asbestos. It accounts for the vast majority of cases of mesothelioma, with peritoneal and pericardial mesothelioma making up the remainder. Worldwide, the mortality from mesothelioma is reported as five people per million, with the largest number of reported deaths occurring in the USA.1 There are three recognized histologic subtypes, epithelioid, sarcomatoid, and mixed with epithelioid histology conferring a better prognosis. The prevalence is higher in males with a median age at diagnosis of 72 years. Patients typically present with signs and symptoms of a pleural effusion, notably chest pain and dyspnea. Systemic symptoms such as fatigue, weight loss, and fever are also common.
Management of MPM includes surgery, radiation, and chemotherapy. The role of surgery in MPM is controversial. Unlike non-small cell lung cancer (NSCLC), which has a well-defined anatomic site in the lung parenchyma, mesothelioma originates from the pleura, which surrounds the whole lung cavity and intrathoracic organs, therefore making a complete (R0) resection impossible. There are two techniques that have been traditionally used, pleurectomy/decortication, which includes removal of the involved pleura and all gross disease, but sparing the lung, and extrapleural pneumonectomy (EPP), which involves en bloc removal of the pleura, lung, ipsilateral diaphragm, and pericardium. Although EPP offers a better theoretical chance of complete disease resection, it is also associated with higher perioperative morbidity and mortality. Studies thus far have failed to demonstrate a survival advantage with EPP. Retrospective studies have shown seemingly improved survival after surgery, as compared to historical controls, but have been heavily skewed toward more fit patients with a better performance status and surgically more localized/favorable disease.2 The current guidelines agree that patients with higher risk disease, such as sarcomatoid or mixed histology, or mediastinal lymph node involvement, derive little benefit from a surgical approach and should be treated with systemic chemotherapy only.3 The decision on surgical intervention in fit patients with lower risk disease is usually made based on the institutional preference and surgical expertise. The role of radiation in pleural mesothelioma is limited as well. As a monotherapy, it does not offer a survival advantage and has been primarily used for palliative purposes with the goal of pain relief in areas of symptomatic disease. Although the rate of surgical intervention in MPM varies from 20% in the community setting to 40% in tertiary centers, the majority of patients receive systemic chemotherapy as their primary treatment modality.4,5
Single-agent chemotherapy has shown limited efficacy in MPM, with disappointing response rates (RRs) of <20%. The first chemotherapy combination of cisplatin and pemetrexed was approved in 2004, based on a randomized Phase III trial comparing the combination of cisplatin and pemetrexed to cisplatin monotherapy in untreated inoperable patients.6 The combination arm achieved an almost 3-month improvement in median overall survival (OS; 12.1 vs 9.3 months, P=0.02) as well as 1.8 months improvement in median time-to-progression (TTP; 5.7 vs 3.9 months, P=0.001) and a significantly superior tumor RR (41.3% vs 16.6%, P<0.0001). The combination remains the only National Comprehensive Center Network category 1 recommendation for patients with MPM who can tolerate both drugs. For patients who are unable to tolerate cisplatin, the combination of carboplatin and pemetrexed has shown similar outcomes in Phase II trials, with median OS of 12–14 months.7–9 The combination of gemcitabine and cisplatin has also been investigated in Phase II trials, and may be a reasonable option for patients who are unable to tolerate pemetrexed.
Angiogenesis
Angiogenesis is the process of formation and maintenance of new blood vessels. It is essential for tissue viability and growth, and plays an important role in wound healing as well as cancer proliferation. A variety of signaling proteins have been implicated in the regulation of this process. Among these, the interaction of vascular endothelial growth factor (VEGF) with its receptor (VEGFR) stands out as one of the main drivers of vasculogenesis, the formation of blood vessels from precursor cells during embryogenesis, and angiogenesis, the formation of blood vessels from the division of preexisting endothelial cells.
VEGF family
The VEGF family includes three receptors (VEGFR-1, VEGFR-2, and VEGFR-3) and five ligands (VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor). VEGFR-1, also known as FMS-related tyrosine kinase 1 (Flt1), has very weak tyrosine kinase activity, and although it binds to VEGF-A, VEGF-B, and placental growth factor tightly, it appears to serve more as a decoy receptor. VEGFR-2, also known as kinase insert domain receptor, is the main regulator of angiogenesis and is activated by VEGF-A. VEGFR-3 is primarily involved in the regulation of lymphogenesis and interacts with VEGF-C and VEGF-D.10
The importance of VEGF-A in angiogenesis is underscored by the fact that even one mutant allele for this protein is embryonically lethal in mice. Due to alternative splicing, VEGF-A has multiple isoforms, with peptide lengths of 121, 165, 189, and 206 amino acids. They differ by the basic stretch in the carboxyl terminus, which allows the peptides to have a different degree of binding to heparin/heparan sulfate and neuropilin-1, a coreceptor for VEGF-A. Among the above subtypes, the 165 amino acid peptide is quantitatively and qualitatively the most active. Reintroduction of VEGF-A165 is able to rescue the embryo of the VEGF-A knock-out mouse.10
Angiogenesis in pleural mesothelioma
Angiogenesis plays an important role in malignant mesothelioma. In preclinical studies, intraperitoneal injection of crocidolite asbestos fibers was found to cause development of peritoneal mesothelioma in mice after 30–50 weeks. However, formation of new blood vessels surrounding the implanted fibers was noted at 2 weeks after injection, implicating angiogenesis as one of the earliest events in mesothelioma.11 Moreover, several studies have shown that mesothelioma cell lines, as compared to the normal mesothelial cells and fibroblasts, produce high amounts of proangiogenic factors, including VEGF. Mesothelioma cells also express VEGFR-1 and VEGFR-2 and respond to VEGF in the environment by increasing cell proliferation. In addition, blocking the VEGFRs with a monoclonal antibody inhibits mesothelioma cell growth. This implicates VEGF as not only a promoter of angiogenesis, but also an autocrine growth factor for mesothelioma cells.
An analysis of mesothelioma tumor samples from patients as compared to non-neoplastic mesothelium revealed an increased expression of multiple proangiogenic cytokines, including VEGF (81% vs 20%) and fibrocyte growth factor (FGF)-1 (67% vs 50%) and FGF-2 (92% vs 40%).12 The expression of angiogenic cytokines also correlated with increased intratumoral microvessel density and worse patient survival.12,13 In addition, the expression of VEGF receptors has been confirmed in patient mesothelioma samples and varies between 20% and 70%, depending on the receptor subtype.14,15
Targeting angiogenesis
The idea to target angiogenesis as a means of controlling tumor growth was first proposed by Dr Judah Folkman in the 1970s. It came to fruition in 2004 with the United States Food and Drug Administration (FDA) approval of a monoclonal antibody against VEGF-A, bevacizumab. Since then, a variety of antiangiogenic molecules and peptides have been developed, including tyrosine kinase inhibitors, such as sorafenib, sunitinib, pazopanib, axitinib, vandetanib, regorafenib, and lenvatinib, all of which target VEGFR-2 among other tyrosine kinases with different degrees of selectivity.16 Other small-molecule inhibitors of VEGFR-2 are still in development and include promising molecules such as cediranib. An antibody against VEGFR-2, ramucirumab, has been developed and approved for use in metastatic gastric, colorectal, and NSCLCs. In addition, a decoy receptor, aflibercept, was made by fusion of VEGFR-1 and VEGFR-2 binding domains to the Fc portion of immunoglobulin and is approved for use in combination with chemotherapy in metastatic colorectal cancer. The different mechanisms of VEGF inhibition are depicted in Figure 1.
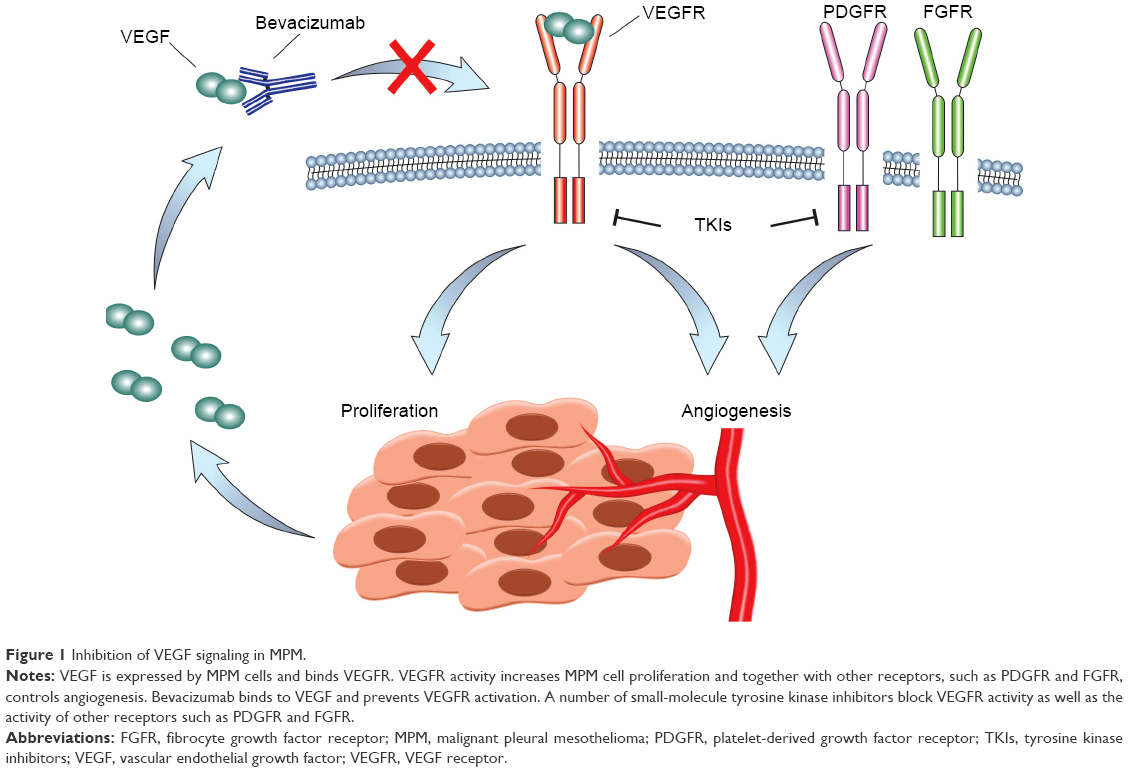 | Figure 1 Inhibition of VEGF signaling in MPM. |
Bevacizumab
Bevacizumab is a recombinant humanized monoclonal immunoglobulin antibody against VEGF-A ligand. It was created in 1997 when the complement determining region of murine anti-VEGF antibody (Mab A.4.6.1) was transferred to the human framework. This reduced antibody–VEGF binding 1,000-fold, compared to the original murine antibody. Further replacement of seven amino acids in the non-complement determining region was required to improve VEGF binding (Kd =1.1 nM), approximating binding of the original antibody. The final product contains 93% human and 7% murine sequences. Bevacizumab binds and neutralizes all biologically active isoforms of VEGF-A, including its bioactive proteolytic fragments. It sterically prevents binding and activation of VEGFR-1 and VEGFR-2 on the surface of the cells. However, it does not bind or inhibit other angiogenic cytokines, including VEGF-B, VEGF-C, and FGF.
Pharmacokinetics of bevacizumab
The pharmacokinetics of bevacizumab was studied in a Phase I trial, in which patients received bevacizumab at 0.1–10 mg/kg on days 0, 28, 35, and 42. Antibody clearance followed a fairly linear pattern with a half-life of 21 days, which is similar to other antibodies with the same backbone. The drug distribution was primarily intravascular, with undetectable serum VEGF achieved at doses as low as 0.3 mg/kg.17
Bevacizumab safety and toxicity
In a Phase I study, escalating doses of bevacizumab up to 10 mg/kg did not achieve dose-limiting toxicity, which was defined as grade 3 toxicity or higher (biologic response modifier-adjusted common toxicity criteria) occurring in two patients in a given cohort.17 The reported side effects were grade 1–2 headache, nausea, asthenia, and fever, which occurred in fewer than 20% of patients. No grade 3 or 4 liver, renal, or cardiac toxicity was noted. Grade 3–4 intracranial bleeding in a patient with central nervous system metastases and intratumoral hemorrhage in a patient with sarcoma were seen, both of which were attributed to the important role of VEGF in the maintenance of vascular integrity.
Subsequent Phase II and III trials continued to demonstrate a risk of bleeding with bevacizumab, varying from grade 1–2 epistaxis (the most common, ~35%) to clinically significant, sometimes fatal gastrointestinal, pulmonary, and intracranial bleeding with a cumulative incidence of grade 3–4 hemorrhage across trials of 1.2%–4.6%.18 In squamous cell carcinoma of the lung, use of bevacizumab is associated with 31% incidence of significant pulmonary hemorrhage and is currently contraindicated.19
Venous thromboembolic events (VTE) have also been reported in patients receiving bevacizumab and chemotherapy. The rates of VTE vary considerably, depending on the chemotherapy used and the malignancy being treated. A 2011 pooled analysis of Phase II and III trials of patients receiving chemotherapy and bevacizumab reported rates of VTE of 10.9% with bevacizumab versus 9.8% in controls (odds ratio, 1.14; 95% confidence interval: 0.96–1.35; P=0.13), suggesting that there is no increased risk of VTE in patients receiving bevacizumab.20
Other side effects include an increased incidence of arterial thrombotic events in patients over 65 years of age (5.7% grade ≥3 events as compared to 2.6% in the control arm) and hypertension (grade ≥3 in 15% as compared to 4% in the control arm).21 VEGF signaling utilizes the nitric oxide (NO) pathway, among others. Therefore, inhibition of VEGF-mediated signaling causes a decrease in NO-mediated signaling, resulting in arterial vasoconstriction and resultant hypertension. NO also plays a role in platelet aggregation, which is thought to be the mechanism behind the increased arterial thrombotic events associated with bevacizumab.
VEGF also regulates glomerular vascular permeability, which explains bevacizumab-associated proteinuria (grade ≥3 incidence is 0.7%–7.4%). The administration of bevacizumab warrants periodic monitoring of urine protein excretion and temporary suspension of bevacizumab if proteinuria exceeds 2 g in 24 h.22–25
Angiogenesis and neovascularization are important processes in wound healing and tissue repair. Consequently, there is a concern for bevacizumab-associated impairment of wound healing. The FDA approved drug label recommends suspension of bevacizumab treatment for 28 days before and after any surgical intervention.18 The incidence of wound healing complications following colorectal surgery was reported to be 15% in patients treated with bevacizumab, as compared to 4% in the control group.24
Other well-known adverse reactions to bevacizumab include gastrointestinal perforation (0.3%–2.4%), fistula formation (<0.3%), reversible posterior leukoencephalopathy (<0.1%), congestive heart failure (5% incidence; increased in patients concurrently treated with taxanes or with previous exposure to anthracyclines), ovarian failure (34% in bevacizumab arm as compared to 2% in the control arm; reversible with bevacizumab discontinuation in 22% of patients), and infusion reactions (severe reactions in <0.2% of patients).18
Bevacizumab in non-mesothelioma solid malignancies
At this time, bevacizumab is approved for use in the metastatic setting with chemotherapy for multiple tumor types, including cervical, colorectal, NSCLC (nonsquamous histology), ovarian, fallopian tube, primary peritoneal, and renal cell carcinomas. Bevacizumab as a monotherapy is also approved for the treatment of refractory glioblastoma.18
Bevacizumab in combination with chemotherapy in MPM
Bevacizumab was first evaluated in MPM in combination with erlotinib in a Phase II trial in MPM patients who were previously treated with systemic chemotherapy.26 Patients received erlotinib 150 mg daily and bevacizumab 15 mg/m2 every 21 days. Although there were no complete responses (CRs) or partial responses (PRs), 12 out of 24 patients achieved stable disease (SD) at least for two cycles. The median TTP was 2.2 months and the median OS was 5.8 months from the time of initiation of treatment. The trial did not achieve its primary endpoint (two objective responses) and was not continued further. The efficacy and toxicity of bevacizumab-based combinations across trials in MPM are summarized in Tables 1 and 2, respectively.
 | Table 1 Efficacy of bevacizumab-based combinations across MPM trials |
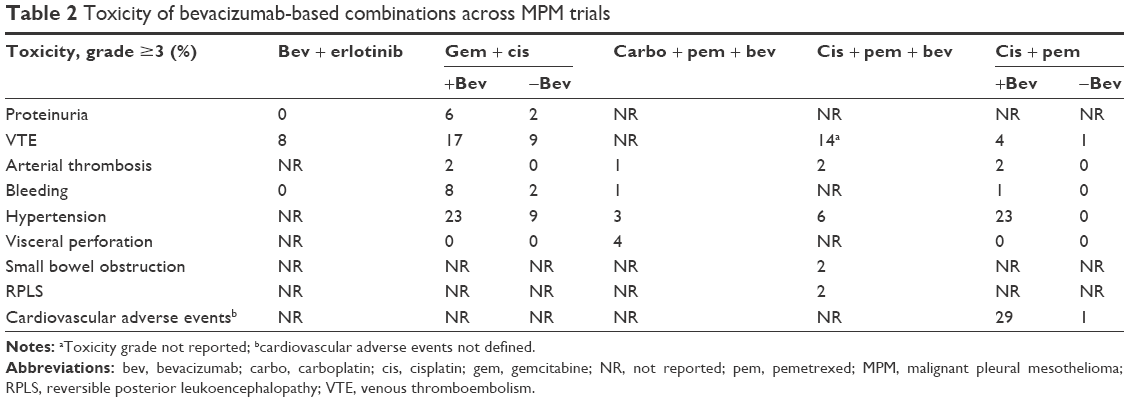 | Table 2 Toxicity of bevacizumab-based combinations across MPM trials |
The first randomized, double-blind, placebo-controlled Phase II trial evaluating the role of bevacizumab in combination with cytotoxic chemotherapy in MPM in the first-line setting was published in 2012.27 In this trial, patients were treated with gemcitabine (1,250 mg/m2 on days 1 and 8), cisplatin (75 mg/m2 on day 1), and bevacizumab (15 mg/m2 on day 1) or placebo every 21 days. One hundred fifteen patients were enrolled at 11 different sites. No difference in tumor RR (24.5% vs 21.8%), progression-free survival (PFS; 6.9 vs 6.0 months, P=0.88), or OS (15.6 vs 14.7 months, P=0.91) was noted between the bevacizumab and placebo arms, respectively. Pretreatment plasma VEGF levels were also analyzed, and although there was no difference between responders and nonresponders, a higher baseline plasma VEGF level was associated with a worse PFS and OS.
The observed lack of efficacy of bevacizumab in combination with gemcitabine/cisplatin backbone may, in part, be attributed to a negative interaction between gemcitabine and bevacizumab. As was pointed out by the authors of the study, preclinical studies have demonstrated that some of the chemotherapy drugs (such as the taxanes and 5-flurouracil) induce angiogenesis by mobilization of circulating endothelial cells.28 This effect counteracts the antitumor activity of chemotherapy and is blocked by anti-VEGF antibodies. Gemcitabine and cisplatin do not increase the number of circulating endothelial cells. Consistent with the above data, anti-VEGF therapy enhanced antitumor activity of taxanes, but had no effect when added to gemcitabine in a mouse model of lung cancer.
Bevacizumab was also evaluated in combination with carboplatin and pemetrexed in a Phase II trial in patients with unresectable MPM in the first-line setting.29 Patients were treated for a maximum of six cycles or until progressive disease (PD) and, in the absence of PD, were continued on maintenance bevacizumab for a maximum of 1 year. Out of 76 evaluable patients, response was seen in 34.2%, all of whom had either epithelioid or mixed histology MPM. SD was seen in 57.9% of patients, with an overall disease control rate of 92.1%. The median duration of response was 8 months. The median PFS was 6.9 months and the median OS was 15.3 months, with 1- and 2-year survivals of 62.6% and 25.9%, respectively. The study failed to achieve its primary endpoint of 50% improvement in PFS, compared to the standard pemetrexed/platinum combination.29
Dowell et al investigated the combination of cisplatin, pemetrexed, and bevacizumab in 53 patients with chemotherapy-naïve unresectable malignant pleural and peritoneal mesothelioma in a Phase II trial.30 The combination was relatively well tolerated, with the primary grade 3–4 toxicities being neutropenia (11%), grade 3 hypertension (6%), and venous thromboembolism (all grades) (13%). PR and SD were noted in 40% and 35% of patients, respectively. The 6-month PFS rate was 56%, and the median OS was 14.8 months. Unfortunately, the trial failed to meet its primary endpoint of a 33% improvement in the PFS rate at 6 months, as compared with the historical control of cisplatin and pemetrexed alone.30 In an unplanned post hoc analysis restricted to 44 patients with MPM, the PFS rate at 6 months was 52%, and an objective tumor RR of 35% and a median OS of 14.1 months were observed.
The role of bevacizumab in MPM was further evaluated in the French Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS).31 The trial was started as a Phase II study that evaluated the disease control rate at 6 months (primary endpoint) and safety (secondary endpoint) with the addition of bevacizumab to cisplatin and pemetrexed. The Phase II portion of the trial met its goal with a 6-month disease control rate achieved in 27 out of 47 patients (57%) as was evaluated by an expert panel masked to the randomization. The trial was expanded to a multicenter, randomized, controlled, open-label Phase III study. Four hundred forty-eight chemotherapy-naïve patients with surgically inoperable MPM were randomized to receive cisplatin and pemetrexed (PC) vs PC with addition of bevacizumab (PCB). Eligible patients had biopsy-proven MPM that was not amenable to curative intent surgery, an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2, and adequate renal, hepatic, and bone marrow function. Patients were excluded based on the presence of central nervous system metastases, predisposition to bleeding (including hemoptysis, inherited coagulopathy, bleeding diathesis), use of antiplatelet agents (aspirin, nonsteroidal antiinflammatory drugs, clopidogrel, ticlopidine, or dipyridamole), or patients on therapeutic doses of anticoagulation (low-molecular-weight heparin or a vitamin K antagonist). Patients were also excluded for uncontrolled hypertension, major surgery within 28 days of enrollment, recent myocardial infarction or cerebral vascular accident (within 6 months), uncontrolled ischemic cardiomyopathy, congestive heart failure, cardiac arrhythmia, recent gastrointestinal perforation (within 6 months), or fistula. Patients completed a maximum of six cycles of cisplatin and pemetrexed in both PC and PCB arms. After six cycles of chemotherapy, the PCB group was allowed to continue with bevacizumab maintenance every 3 weeks until PD or limiting toxicity. Patients experiencing ≥ grade 2 renal toxicity from cisplatin could be changed to carboplatin (area under the curve 5). Tumor response assessments were performed after every three cycles of treatment. The primary outcome was median OS, and patients were stratified by mesothelioma histology, performance status, and smoking history. The study was designed to have an 80% power to detect an improvement in median OS from 13 months in the control arm to 17.3 months in the experimental arm.
Seventy-five percent of patients enrolled were male, 97% had an ECOG performance status of 0 or 1, and 81% had epithelioid histology. After a median follow-up of 39.4 months, patients who received PCB were noted to have a statistically significant improvement in median PFS (9.2 vs 7.3 months; adjusted hazard ratio [HR] =0.61; P<0.0001) and median OS (18.8 vs 16.1 months; adjusted HR =0.75; P=0.0167), as compared to patients who received PC. The PFS and OS observed in the control arm of this trial were superior to those seen in the pivotal trial of cisplatin and pemetrexed by Vogelzang et al.6 This was likely due to the strict inclusion criteria required for enrollment, such as the exclusion of patients with cardiovascular comorbidities. A preplanned exploratory analysis noted that the baseline serum concentration of VEGF was prognostic of worse outcome, although not predictive of response to bevacizumab. As expected, the incidence of VEGF-related adverse events was significantly higher in PCB group as compared to PC group, specifically grade 3 and 4 hypertension (23% vs 0%), cardiovascular events (28.8% vs 0.9%), and arterial and venous thromboembolic events (5.8% vs 0.9%), respectively. Bevacizumab-treated patients also experienced a higher incidence of grade 1–2 hemorrhage (epistaxis) and serum creatinine elevation. There was no significant difference in other hematologic and nonhematologic toxicities, including the incidence of febrile neutropenia, fatigue, nausea, and vomiting. More patients in the PCB arm stopped first-line treatment due to toxicity than in PC arm (24.3% vs 13%, respectively). Nevertheless, the reported quality of life scores worsened for activity-related items (PC 52% vs PCB 37%) and general condition items (PC 48% vs PCB 36%) more commonly in the group that did not receive bevacizumab. The quality-of-life data need to be interpreted with caution due to low rate of questionnaire completion (50%–70%) and possible reporting bias in this open-label study.
Small-molecule inhibitors of VEGFR in pleural mesothelioma
In addition to antibody-mediated interference with the VEGF–VEGFR interaction, as seen with bevacizumab, small-molecule inhibitors of VEGFR signaling have also been explored in MPM.
Sunitinib
Sunitinib is an oral tyrosine kinase inhibitor of VEGFR, platelet-derived growth factor receptor (PDGFR), FMS-related tyrosine kinase 3 (Flt3), and c-Kit tyrosine kinase (c-Kit). It is currently FDA approved for use in gastrointestinal stroma tumors and renal cell carcinoma. It was studied in a Phase II trial in patients with unresectable MPM, whose disease had shown progression on a platinum-based, antimetabolite (pemetrexed or gemcitabine)-containing regimen.32 Out of 53 enrolled patients, 6 patients (12%) had a PR and 34 patients (65%) had SD. There were no CRs. The median TTP was 3.5 months, and the median OS from treatment initiation was 6.1 months. The most common grade 3–4 toxicities were fatigue (30%), thrombocytopenia (9%), and neutropenia (7%). One patient experienced grade 3 cardiac toxicity, which was attributed to sunitinib.
A combination of sunitinib together with cytotoxic chemotherapy (cisplatin and pemetrexed) was attempted in a Phase I trial in patients with NSCLC and one patient with mesothelioma. Although the patient with mesothelioma achieved a PR, the combination was not well tolerated due to prohibitive myelosuppression.33
Sorafenib
Sorafenib is a potent tyrosine kinase inhibitor of the Raf tyrosine kinase/mitogen-activated protein kinase signaling pathway with additional activity against VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-β, FGF receptor 1 (FGFR-1), Flt3, and c-Kit. It is FDA approved for use in renal cell and hepatocellular carcinomas. In 2010, a Phase II trial examined the activity of sorafenib in patients with MPM. Sixty percent of enrolled patients had been previously treated with one line of pemetrexed-based chemotherapy.34 Among 50 patients, only 3 (6%) achieved a PR, 27 patients (54%) had SD, with the remaining patients showing disease progression (PD). The median duration of response/SD was 7.9 months, with some patients achieving ongoing long-term stability for 40 months at the time of study completion. In the chemotherapy-naïve population, a 10% PR was observed.
Another single-arm Phase II trial of sorafenib was conducted in patients previously treated with pemetrexed-based chemotherapy and demonstrated similar efficacy. Fifty-three patients were enrolled, with 3 patients (6%) achieving a PR and 30 patients (56%) having SD at 8 months. The median PFS and OS were 5.1 and 9 months, respectively, and 9% of patients were still receiving the drug after 1 year of treatment.35
Cediranib
Cediranib is a potent small-molecule tyrosine kinase inhibitor of VEGFR-1, VEGFR-2, and VEGFR-3, as well as c-Kit and PDGFR-β, that is currently being evaluated in multiple cancer types. Garland et al evaluated the efficacy of single-agent cediranib (45 mg) in a Phase II trial in MPM patients who were previously treated with platinum-based chemotherapy (SWOG S0509).36 In 43 evaluable patients, there were no CRs and only 4 patients (9%) achieved a PR. However, in responding patients, as much as 91% tumor shrinkage was observed. Sixteen patients (34%) achieved SD with a median TTP of 4.9 months. Nine percent of patients had ≥ grade 3 toxicity, including fatigue, proteinuria, and gastrointestinal symptoms (diarrhea, colitis, ileal perforation, and esophageal necrosis).
A similar outcome was achieved in another Phase II trial of cediranib in patients with MPM who were chemotherapy naïve (12%) or whose disease progressed on one line of previous chemotherapy (88%). An almost identical number of responders were seen, with 10% having a PR and 34% having SD. The 45 mg dose of the drug was more effective than the 30 mg dose (disease control rate 67% vs 34%, respectively, P=0.04), but also had a higher incidence of grade ≥3 toxicity (87% vs 43%, respectively, P=0.002).37
There is an ongoing Phase I/II randomized clinical trial examining the efficacy of low-dose cediranib (20 mg daily) in MPM when given concurrently with cisplatin and pemetrexed chemotherapy as first-line treatment for chemotherapy-naïve patients. Enrollment in this trial has been completed and results are expected in the next year (NCT01064648).
Nintedanib
Nintedanib is a tyrosine kinase inhibitor of VEGFR, PDGFR, and FGFR, in addition to the Src and Abl tyrosine kinases. Currently, nintedanib is approved in the European Union and other countries to be used in combination with docetaxel in the treatment of NSCLC. Nintedanib is being evaluated in a randomized, Phase II/III trial in combination with cisplatin–pemetrexed chemotherapy in chemotherapy-naïve patients with unresectable MPM.38 The trial has completed the Phase II component, which demonstrated that nintedanib is well tolerated in combination with cisplatin–pemetrexed chemotherapy and significantly improved the median PFS survival in this population.39 The Phase III portion of the trial is currently recruiting patients (NCT01907100). Nintedanib is also being studied in a Phase II trial in the second-line setting in patients with unresectable MPM whose cancer has progressed or recurred after prior platinum-based therapy (NCT02568449).
Vatalanib (PTK787)
Vatalanib is an oral tyrosine kinase inhibitor of VEGFR-1, VEGFR-2, and PDGFR-β. It was investigated by the Cancer and Leukemia Group B in a multicenter Phase II trial as a monotherapy in unresectable MPM in the first-line setting.18 Although the toxicity of the drug was very mild with few grade 3–4 adverse events reported, the drug showed limited efficacy. Out of 47 enrolled patients, only 6% achieved PR and 72% had SD. The median PFS was 4.1 months with a median OS of 10 months. The trial failed to achieve its primary endpoint of a 75% PFS at 3 months (a 55% PFS was observed), and therefore, further investigation of this drug as a monotherapy will not be continued in this disease.
Semaxanib (SU5416)
Semaxanib is a selective tyrosine kinase inhibitor of VEGFR-2. It was evaluated in a Phase II trial in 23 previously untreated patients with MPM. Eleven percent of patients showed a PR with a median OS of 12.4 months. No correlation between pretreatment serum VEGF, vascular cell adhesion molecule-1 and response or survival was identified. No further investigation of this drug in MPM is planned.40
Pazopanib
Pazopanib is a small-molecule inhibitor of VEGFR, PDGFR, FGFR, and c-Kit. It was studied in a single-arm Phase II trial in patients with MPM who received less than one prior systemic therapy. Patients were given pazopanib daily until toxicity or PD for up to 2 years. The primary endpoint was PFS at 6 months. A total of 34 patients were analyzed, with 16 patients (47.8%) being progression free at 6 months of treatment. The median OS was 11.5 months (95% confidence interval: 6.2–18.2 months).41
Axitinib
Axitinib is another oral small-molecular inhibitor of VEGFR-1, VEGFR-2, VEGFR-3, PDGFR, and c-Kit. In a Phase II trial, chemotherapy-naïve MPM patients, who were medically suitable for limited surgical intervention, were randomized to receive pemetrexed and cisplatin with or without daily axitinib.42 Patients underwent thoracoscopy with biopsies before the treatment and after three cycles of chemotherapy. The second biopsy revealed that the number of new blood vessels in the tumor (microvessel density and immature blood vessels) increased after treatment with cisplatin and pemetrexed. This increase was blocked with axitinib. However, patients who were treated with axitinib showed increased expression of mRNA for a number of proangiogenic growth factors. Since axitinib had to be stopped 5 days before second thoracoscopy, rebound overexpression of proangiogenic factors could have confounded the results. There was no difference in PFS or OS between the treatment groups. Of note, in the axitinib group, one patient developed a transient cerebrovascular accident on postoperative day 2 and another developed a pulmonary embolus during treatment.
Dovitinib (CHIR258, TKI258)
Dovitinib is a small-molecule multikinase inhibitor that inhibits several receptors, including VEGFRs, FGFRs, PDGFR-b, c-Kit, rearranged during transfection (RET), and Flt3.43 It was used in a Phase II trial as a single agent in patients with advanced pleural mesothelioma that had progressed after a first-line platinum–antifolate combination. The study was aborted after stage I accrual due to preliminary data showing lack of efficacy.44
Conclusion
VEGF-targeted therapies, including bevacizumab, have been extensively evaluated in MPM. Although initial Phase II trials in MPM failed to demonstrate a clear benefit with the addition of bevacizumab to standard chemotherapy in this disease, these studies were limited by small numbers of patients. The recently reported French MAPS demonstrated a statistically significant improvement in median OS with the combination of cisplatin, pemetrexed, and bevacizumab in MPM. The toxicity from the regimen was similar to that seen with the addition of bevacizumab to other chemotherapies in other malignancies, but significant rates of hypertension, cardiovascular events, and arterial and venous thromboembolic events were noted. The study included primarily patients with an ECOG performance status of 0 or 1 and employed extensive exclusion criteria to ensure that those patients at risk for serious side effects from bevacizumab were not included. Therefore, the regimen is not appropriate for all patients with MPM. In addition, 81% of patients enrolled had epithelioid histology, and, as a result, the utility of this regimen in MPM patients with nonepithelioid histology is unclear. The MAPS study represents a significant step forward in the treatment of unresectable MPM and has established the combination of cisplatin, pemetrexed, and bevacizumab as a reasonable option for appropriately selected MPM patients who are candidates to receive bevacizumab. Previous attempts to identify potential biomarkers as predictors of response to antiangiogenic treatment (including microvessel density of the tumor, serum/plasma VEGF levels, and tissue VEGF/VEGFR expression), across a variety of malignancies and with numerous antiangiogenic agents, have failed to elucidate an ideal candidate. Hopefully, future trials will identify molecular correlates that can better elucidate the subset of MPM patients who benefit the most from bevacizumab. In addition, immunotherapy agents (including programmed cell death 1 [PD-1], programmed death ligand 1 [PD-L1], and cytotoxic T lymphocyte-associated 4 [CTLA-4] inhibitors) are currently being investigated in MPM and preliminary studies show encouraging activity with these drugs. Emerging evidence also suggests that VEGF may suppress T-cell-mediated immune response, and therefore, anti-VEGF therapies may augment the effect of immunotherapy in cancer.45 This provides a strong preclinical rationale for future trials targeting VEGF in combination with immunotherapy in MPM. In fact, a trial of pembrolizumab and nintedanib is currently recruiting patients across different malignancies, including MPM (NCT02856425). In addition, the combination of bevacizumab with immunotherapy is being studied in renal and ovarian cancers, melanoma, and glioblastoma multiforme. Further clinical studies with bevacizumab-based chemotherapy regimens in combination with immunotherapy in patients with MPM are also anticipated.
Disclosure
Dr JE Dowell receives research funding from Astex, Medimmune, and Taiho. PA Levin reports no conflicts of interest in this work.
References
Delgermaa V, Takahashi K, Park EK, Le GV, Hara T, Sorahan T. Global mesothelioma deaths reported to the World Health Organization between 1994 and 2008. Bull World Health Organ. 2011;89(10):716–724, 724A–724C. | ||
Kaufman AJ, Flores RM. Surgical treatment of malignant pleural mesothelioma. Curr Treat Options Oncol. 2011;12(2):201–216. | ||
Ettinger DS, Wood DE, Akerley W, et al. NCCN Guidelines Insights: malignant pleural mesothelioma, Version 3. 2016. J Natl Compr Canc Netw. 2016;14(7):825–836. | ||
Flores RM, Riedel E, Donington JS, et al. Frequency of use and predictors of cancer-directed surgery in the management of malignant pleural mesothelioma in a community-based (Surveillance, Epidemiology, and End Results [SEER]) population. J Thorac Oncol. 2010;5(10):1649–1654. | ||
Flores RM, Zakowski M, Venkatraman E, et al. Prognostic factors in the treatment of malignant pleural mesothelioma at a large tertiary referral center. J Thorac Oncol. 2007;2(10):957–965. | ||
Vogelzang NJ, Rusthoven JJ, Symanowski J, et al. Phase III study of pemetrexed in combination with cisplatin vs cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21(14):2636–2644. | ||
Katirtzoglou N, Gkiozos I, Makrilia N, et al. Carboplatin plus pemetrexed as first-line treatment of patients with malignant pleural mesothelioma: a phase II study. Clin Lung Cancer. 2010;11(1):30–35. | ||
Ceresoli GL, Zucali PA, Favaretto AG, et al. Phase II study of pemetrexed plus carboplatin in malignant pleural mesothelioma. J Clin Oncol. 2006;24(9):1443–1448. | ||
Castagneto B, Botta M, Aitini E, et al. Phase II study of pemetrexed in combination with carboplatin in patients with malignant pleural mesothelioma (MPM). Ann Oncol. 2008;19(2):370–373. | ||
Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer. 2011;2(12):1097–1105. | ||
Branchaud RM, MacDonald JL, Kane AB. Induction of angiogenesis by intraperitoneal injection of asbestos fibers. FASEB J. 1989;3(6):1747–1752. | ||
Kumar-Singh S, Weyler J, Martin MJ, Vermeulen PB, Van Marck E. Angiogenic cytokines in mesothelioma: a study of VEGF, FGF-1 and −2, and TGF beta expression. J Pathol. 1999;189(1):72–78. | ||
Edwards JG, Cox G, Andi A, et al. Angiogenesis is an independent prognostic factor in malignant mesothelioma. Br J Cancer. 2001;85(6):863–868. | ||
Ohta Y, Shridhar V, Bright RK, et al. VEGF and VEGF type C play an important role in angiogenesis and lymphangiogenesis in human malignant mesothelioma tumours. Br J Cancer. 1999;81(1):54–61. | ||
Soini Y, Puhakka A, Kahlos K, et al. Endothelial nitric oxide synthase is strongly expressed in malignant mesothelioma but does not associate with vascular density or the expression of VEGF, FLK1 or FLT1. Histopathology. 2001;39(2):179–186. | ||
Al-Husein B, Abdalla M, Trepte M, Deremer DL, Somanath PR. Antiangiogenic therapy for cancer: an update. Pharmacotherapy. 2012;32(12):1095–1111. | ||
Gordon MS, Margolin K, Talpaz M, et al. Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J Clin Oncol. 2001;19(3):843–850. | ||
Jahan T, Gu L, Kratzke R, et al. Vatalanib in malignant mesothelioma: a phase II trial by the Cancer and Leukemia Group B (CALGB 30107). Lung Cancer. 2012;76(3):393–396. | ||
Johnson DH, Fehrenbacher L, Novotny WF, et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2004;22(11):2184–2191. | ||
Hurwitz HI, Saltz LB, Van Cutsem E, et al. Venous thromboembolic events with chemotherapy plus bevacizumab: a pooled analysis of patients in randomized phase II and III studies. J Clin Oncol. 2011;29(13):1757–1764. | ||
Cassidy J, Saltz LB, Giantonio BJ, Kabbinavar FF, Hurwitz HI, Rohr UP. Effect of bevacizumab in older patients with metastatic colorectal cancer: pooled analysis of four randomized studies. J Cancer Res Clin Oncol. 2010;136(5):737–743. | ||
Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–2550. | ||
Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370(9605):2103–2111. | ||
Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–2342. | ||
Saltz LB, Clarke S, Diaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26(12):2013–2019. | ||
Jackman DM, Kindler HL, Yeap BY, et al. Erlotinib plus bevacizumab in previously treated patients with malignant pleural mesothelioma. Cancer. 2008;113(4):808–814. | ||
Kindler HL, Karrison TG, Gandara DR, et al. Multicenter, double-blind, placebo-controlled, randomized phase II trial of gemcitabine/cisplatin plus bevacizumab or placebo in patients with malignant mesothelioma. J Clin Oncol. 2012;30(20):2509–2515. | ||
Shaked Y, Henke E, Roodhart JM, et al. Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell. 2008;14(3):263–273. | ||
Ceresoli GL, Zucali PA, Mencoboni M, et al. Phase II study of pemetrexed and carboplatin plus bevacizumab as first-line therapy in malignant pleural mesothelioma. Br J Cancer. 2013;109(3):552–558. | ||
Dowell JE, Dunphy FR, Taub RN, et al. A multicenter phase II study of cisplatin, pemetrexed, and bevacizumab in patients with advanced malignant mesothelioma. Lung Cancer. 2012;77(3):567–571. | ||
Zalcman G, Mazieres J, Margery J, et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet. 2016;387(10026):1405–1414. | ||
Nowak AK, Millward MJ, Creaney J, et al. A phase II study of intermittent sunitinib malate as second-line therapy in progressive malignant pleural mesothelioma. J Thorac Oncol. 2012;7(9):1449–1456. | ||
Camidge DR, Blais N, Jonker DJ, et al. Sunitinib combined with pemetrexed and cisplatin: results of a phase I dose-escalation and pharmacokinetic study in patients with advanced solid malignancies, with an expanded cohort in non-small cell lung cancer and mesothelioma. Cancer Chemother Pharmacol. 2013;71(2):307–319. | ||
Dubey S, Janne PA, Krug L, et al. A phase II study of sorafenib in malignant mesothelioma: results of Cancer and Leukemia Group B 30307. J Thorac Oncol. 2010;5(10):1655–1661. | ||
Papa S, Popat S, Shah R, et al. Phase 2 study of sorafenib in malignant mesothelioma previously treated with platinum-containing chemotherapy. J Thorac Oncol. 2013;8(6):783–787. | ||
Garland LL, Chansky K, Wozniak AJ, et al. Phase II study of cediranib in patients with malignant pleural mesothelioma: SWOG S0509. J Thorac Oncol. 2011;6(11):1938–1945. | ||
Campbell NP, Kunnavakkam R, Leighl N, et al. Cediranib in patients with malignant mesothelioma: a phase II trial of the University of Chicago Phase II Consortium. Lung Cancer. 2012;78(1):76–80. | ||
Scagliotti GV, Gaafar RM, Nowak AK, et al. Lume-meso: a double-blind, randomized, phase II/III study of nintedanib (N) + pemetrexed (P)/cisplatin (C) followed by maintenance N vs placebo + P/C followed by maintenance placebo for patients with unresectable malignant pleural mesothelioma (MPM). J Clin Oncol. 2016;34. | ||
Grosso F. Nintedanib plus Pemetrexed/Cisplatin in Patients with MPM: Phase II Findings from the Placebo-Controlled LUME-Meso Trial. Paper presented at: International Association for the Study of Lung Cancer – World Conference on Lung Cancer; July 12; 2016; Vienna, Austria. | ||
Kindler HL, Vogelzang NJ, Chien K, et al. SU5416 in Malignant Mesothelioma: a University of Chicago Phase II Consortium Study [abstract]. Proceedings of ASCO, 2001.20:341:a1359. | ||
Molina J. Phase II Study of GW786034 in Patients With Malignant Pleural Mesothelioma. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US); 2000. Available from: https://clinicaltrials.gov/ct2/show/study/NCT00459862. Accessed November 23, 2016. | ||
Buikhuisen WA, Scharpfenecker M, Griffioen AW, Korse CM, van Tinteren H, Baas P. A randomized phase II study adding axitinib to pemetrexed-cisplatin in patients with malignant pleural mesothelioma: a single-center trial combining clinical and translational outcomes. J Thorac Oncol. 2016;11(5):758–768. | ||
Porta C, Giglione P, Liguigli W, Paglino C. Dovitinib (CHIR258, TKI258): structure, development and preclinical and clinical activity. Future Oncol. 2015;11(1):39–50. | ||
Laurie S. A Phase II Study of Single-agent DOVitinib in Advanced Malignant PlEural Mesothelioma Which Has Progressed Following Prior Platinum-Antifolate Chemotherapy; 2015. Available from: https://clinicaltrials.gov/ct2/show/record/NCT01769547. Accessed November 26, 2016. | ||
Kaur S, Chang T, Singh SP, et al. CD47 signaling regulates the immunosuppressive activity of VEGF in T cells. J Immunol. 2014;193(8):3914–3924. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.