")
Back to Journals » Nature and Science of Sleep » Volume 13
Sporadic Creutzfeldt–Jakob Disease Appears to Be Sporadic Fatal Insomnia: A Case Report and Review of the Literature
Authors Sun Q, Shen P, Tang J, Meng H, Feng J, Wang Z, Cui L
Received 17 May 2021
Accepted for publication 3 August 2021
Published 23 August 2021 Volume 2021:13 Pages 1441—1448
DOI https://doi.org/10.2147/NSS.S319917
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ahmed BaHammam
Video abstract of "sFI in sCJD" [ID 319917].
Views: 1113
Qingqing Sun, Pingping Shen, Jiayi Tang, Hongmei Meng, Jiachun Feng, Zan Wang, Li Cui
Department of Neurology and Neuroscience Center, The First Hospital of Jilin University, Changchun, People’s Republic of China
Correspondence: Zan Wang; Li Cui Email [email protected]; [email protected]; [email protected]
Abstract: Creutzfeldt–Jakob disease (CJD) subtypes are difficult to identify due to the heterogeneity of the clinical phenotype, and early accurate identification of sporadic CJD (sCJD) subtypes aids prognosis prediction. Currently, the diagnosis of sCJD subtypes is mainly based on brain tissue biopsy or autopsy. In this report, we present a case of confirmed sCJD initially presenting as insomnia. We described detailed information including clinical, electroencephalographic, polysomnographic, positron emission tomography-computed tomographic and other neuroimaging findings, cerebrospinal fluid biomarkers, skin tissue biopsy and whole blood PRNP gene sequencing in this patient. An extensive literature search was performed in order to better understand the diagnosis of various sCJD subtypes, particularly the thalamic form, sCJDMM2 (also known as sporadic fatal insomnia). Our study highlights sporadic fatal insomnia as a differential diagnosis of sCJD.
Keywords: sporadic Creutzfeldt–Jakob disease, sporadic fatal insomnia, sCJDMM, polysomnography, PET
Introduction
Prion disease, also known as transmissible spongiform encephalopathy, comprises a group of rare and fatal neurodegenerative diseases caused by misfolded prion proteins (PrPSc). They may present as sporadic, genetic or acquired disorders.1,2 Sporadic Creutzfeldt–Jakob disease (sCJD), the most common type of human prion disease, accounts for 85–90% of prion disease cases.3 Based on the genotype of PRNP alleles at codon 129, different fragment sizes of PrPSc after proteinase K cleavage (type 1 and 2), the disease phenotypes of sCJD can be divided into five subtypes: sCJDMM(V)1, sCJDMM(V)2C, sCJDVV1, sCJDVV2, sCJDMV2K and sporadic fatal insomnia (sFI, or sCJDMM2T).4 sFI is considered as the thalamic form of sCJDMM2.5 Different phenotypes of sCJD are defined by the polymorphism (Met and Val) at codon 129 of the prion protein gene (PRNP) and by the molecular mass of PrPSc (type 1 and 2).5 sFI is a rare subtype, accounting for 0.9% of sporadic human prion disease cases.6 We herein present a confirmed sCJD case initially presenting as insomnia.
Case Presentation
A 47-year-old woman who had an unremarkable medical and family history (no previous blood transfusion and surgery) presented with a chief complaint of insomnia. One month after onset, she developed visual and auditory hallucinations, experienced psychotic and worsening hallucinations and agitation at night. A diagnosis of mania was originally considered by a psychiatrist. High blood pressure, excessive sweating, tachycardia, irregular breathing and other symptoms of autonomic hyperactivity were evident. Brain diffusion-weighted imaging (DWI) imaging showed hyperintense lesions in the bilateral basal ganglia and frontal cortical lobes (Figure 1A). Unfortunately, the patient’s symptoms progressively worsened. Repeated brain magnetic resonance imaging (MRI) revealed a wider distribution of abnormal high signals on DWI (Figure 1B). Electroencephalography (EEG) showed a slow activity background θ-δ with atypical periodic sharp-wave complexes (PSWCs) in the right temporal regions. Specifically, the first phase of typical PSWCs is a negative-phase sharp wave with a low amplitude, the second phase is a positive-phase sharp wave with a high amplitude and wide time frame, and the third phase is a slow wave with a high amplitude than the first phase. In our case, the whole PSWCs were approximately 1 Hz and the sleep staging was inconclusive (Figure 2A and B). To further understand the patient’s sleep structure, 24-hour polysomnography (PSG) was performed. The results demonstrated an evident abnormal sleep structure, with a significant reduction in sleep spindles and K-complexes (Figure 3). The total sleep time was 655.0 min, with a total sleep time in the N1 stage of being 91.9%, and N2 stage of being 8.1%, and the N3 stage and rapid eye movement stages of sleep being absent. The sleep efficiency was 48.8% and sleep apnea hypoventilation index (average number of occurrences per hour) was 5.7. The periodic leg movement index was 5.8 (Figure 3). As the symptoms worsened, the patient’s EEG voltage was gradually decreased, suggesting a reduced whole-brain function. The PSWCs remained atypical with the disappearance of sleep staging and periodic synchronous discharge (Figure 2B–D). Three months after disease onset, the patient developed cognitive impairment, aphasia and intermittent bilateral lower limb myoclonus. Four months later, she developed laryngeal stridor. To further clarify the diagnosis, the positron emission tomography-computed tomography (PET-CT) was performed (Figure 1C), showing heterogeneous hypometabolism in the whole brain parenchyma with pronounced hypometabolism being noted in bilateral frontal, temporal lobes, basal ganglia and thalamus. Her routine cerebrospinal fluid (CSF) tests including color, clarity, cell count, protein, and other routine biochemical markers were normal. Tests were also negative for autoimmune encephalitis antibodies (NMDAR, AMPA1, AMPA2, LGI1, CASPR2, GABA, and GAD), and negative for paraneoplastic antibodies (Hu, Yo, Ri, MA2, CV2 and amphiphysin). For further detection of CSF 14-3-3 protein, real-time quaking-induced conversion (RT-QuIC) assay of CSF, skin biopsy and whole blood PRNP gene sequencing were performed, and the skin tissue biopsy, whole blood samples, and remaining CSF samples were sent to the China Center for Disease Control and Prevention. The CSF 14-3-3 protein was negative using Western blot. The whole blood PRNP gene sequence was compared to the standard sequence (NCBI: NM-183079.1) and no homozygous or heterozygous mutations were found. The biopsied skin and CSF samples were positive according to RT-QuIC (Figure 4A and B). Therefore, this patient was diagnosed with sCJD. Six months after disease onset, repeated brain MRI was performed. However, due to patient’s tolerance, only an apparent diffusion coefficient sequence was completed, and the results showed slight hypointensities in the left temporal and parietal lobes, and ventricular enlargement compared to the previous brain MRI (Figure 1D).
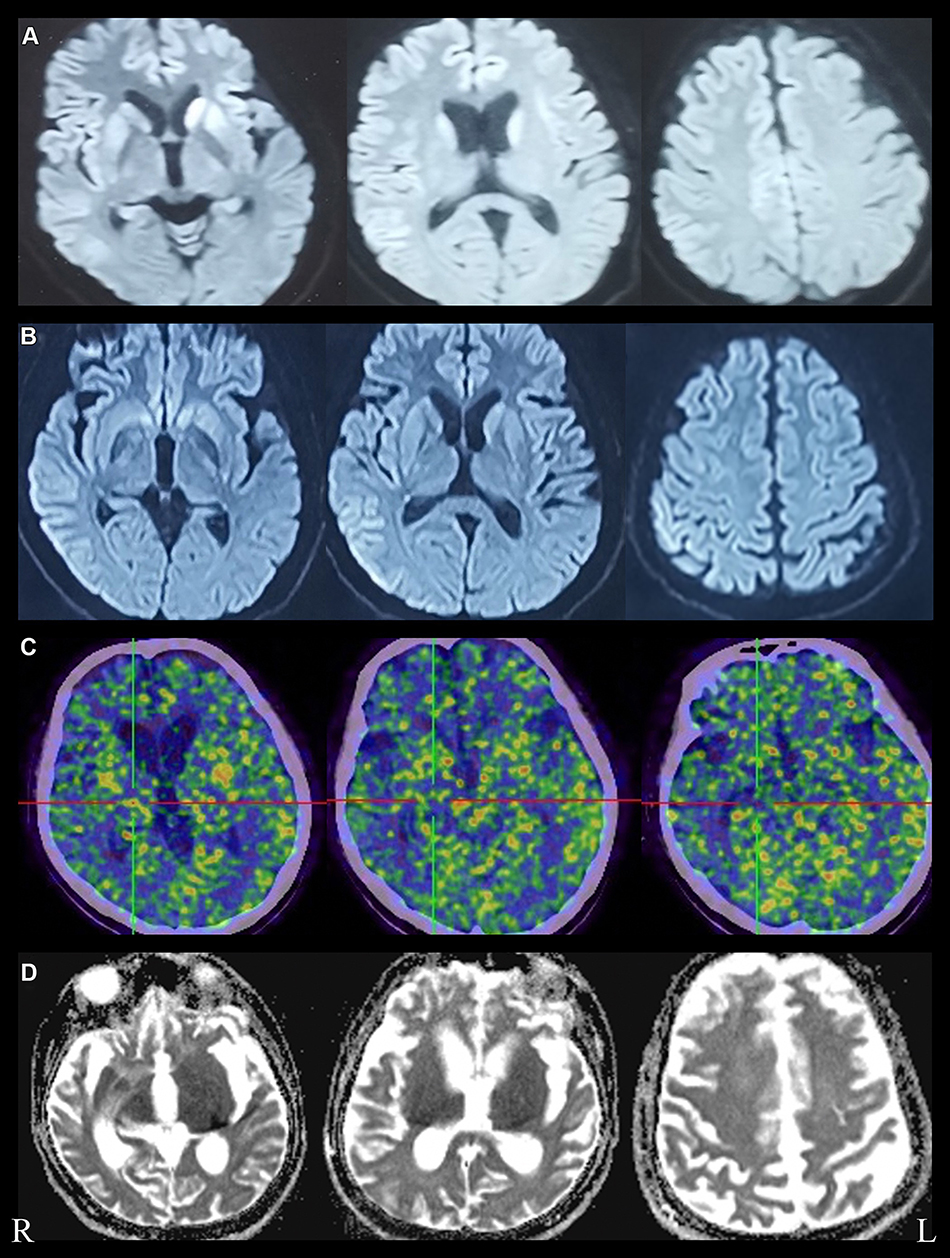 |
Figure 1 Brain imaging features of this patient. (A) Restricted diffusion in bilateral caudate nucleus, putamen nucleus and frontal cortical areas on DWI, obtained at one month, (B) wider distribution of abnormal high signal on DWI, “cortical ribboning” of frontal, temporal, parietal and cingulate cortex, obtained at one and a half months, (C) Brain PET-CT images showed uneven hypometabolism of the whole brain parenchyma most evident in the bilateral frontotemporal lobes, bilateral basal ganglia and bilateral thalamus, obtained at four months. (D) Brain ADC images showed significant ventricular enlargement, obtained at six months after disease onset. |
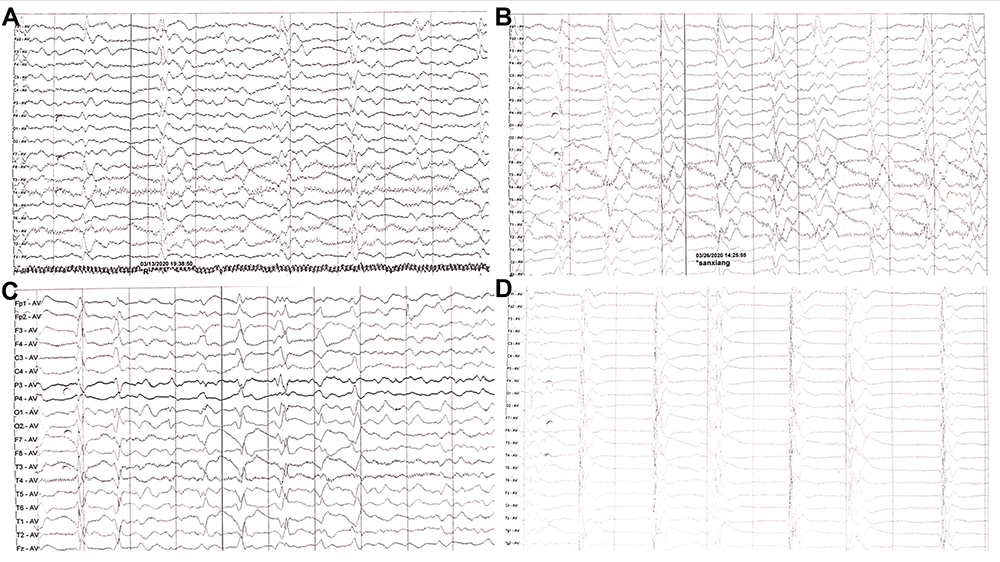 |
Figure 2 EEG showed generalized slowing without typical PSWCs, obtained at (A) three months, (B) three and a half months, (C) four months (D) four and a half months after disease onset. |
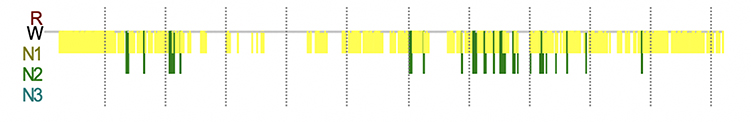 |
Figure 3 PSG trend graph of abnormal sleep structures obtained three months after onset. Recording Duration (min) 1342.1, from 11:01:45 to 09:23:52 the next day. |
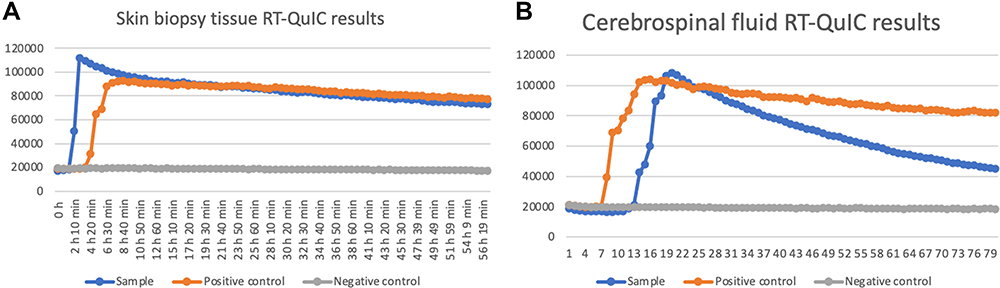 |
Figure 4 Positive skin biopsy tissue (A) and CSF samples (B) RT-QuIC results at four months after onset (Updated diagnostic guidelines for sCJD have incorporated RT-QuIC for early diagnosis of disease, surveillance, evaluation of PrPsc inoculation in different tissues, and in trial monitoring).18 |
Discussion
In this report, we presented a rare case of sCJD with persistent insomnia. Sporadic prion disease with codon 129 polymorphism MM was confirmed by skin, CSF RT-QuIC, and genetic testing 4 months after disease onset. As the patient’s family members refused brain tissue biopsy or autopsy, we could not determine whether the patient’s PrPSc was type 1, type 2 or both. MM2 can be divided into cortical and thalamic types. Clinical features of the thalamic type overlap with those of fatal familial insomnia (FFI). Thus, it is also referred to sFI by some researchers.7 Clinical findings of our case were consistent with the sFI subtype. Therefore, this case can be used to expand our understanding of the characteristics of the sCJDMM2T/sFI, sCJDMM2C, and sCJDMM1 subtypes (Table 1). Sleep disorders are not uncommon in sCJD, while have not received extensive attention in other disease subtypes. This patient’s PSG is different from our previous study8 of PSG in patients with sCJD. We propose that PSG in patients may be useful in the diagnosis of clinical subtypes of sCJD. The various subtypes are discussed as below:
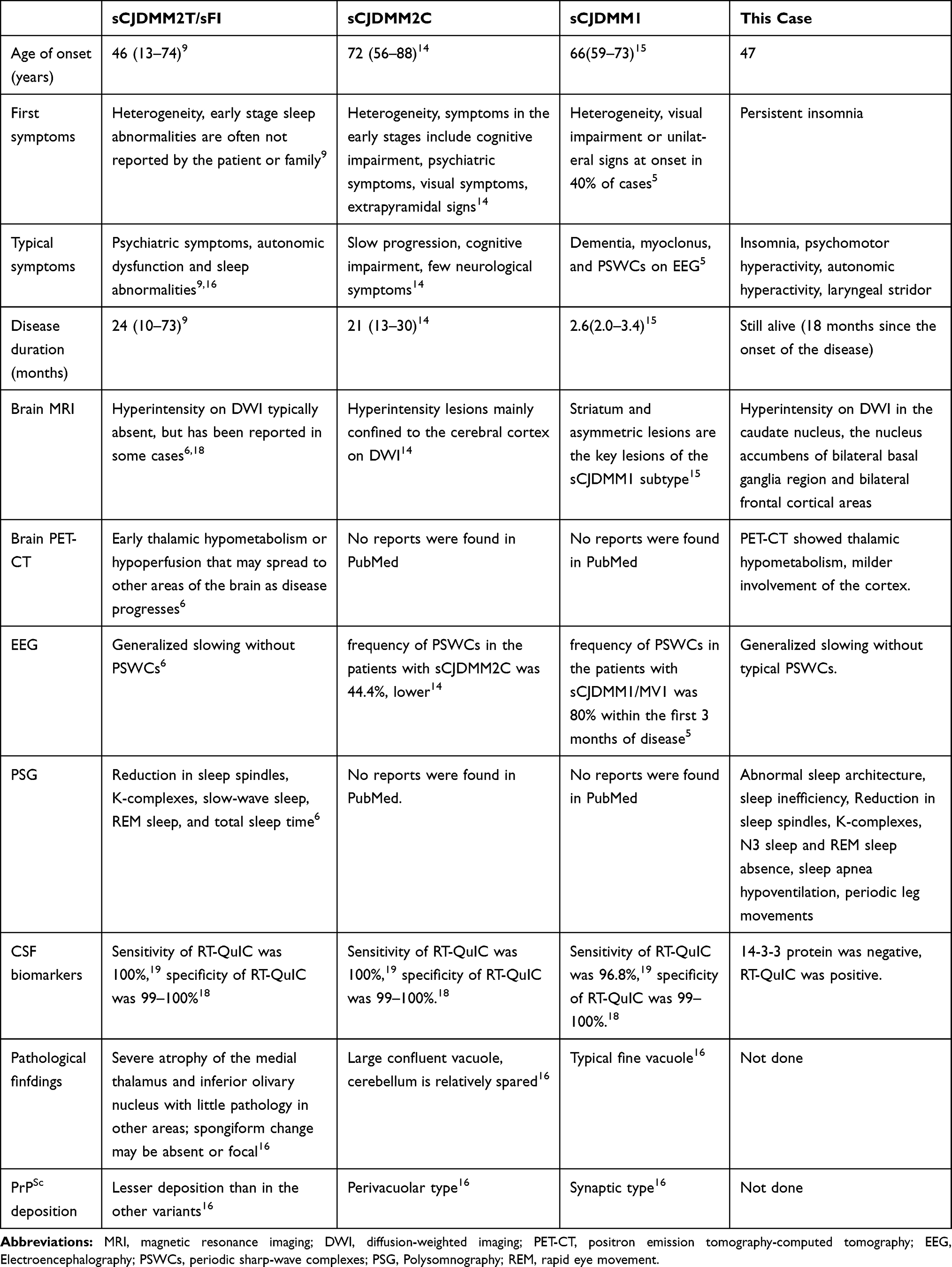 |
Table 1 Comparison of sCJDMM2T/sFI, sCJDMM2C, sCJDMM1 and Our Case |
sCJDMM2T/sFI
sFI (the thalamic form of CJD) is the most rare type of sporadic prion disease.9 The symptoms of sFI overlap with those of FFI.6 However, insomnia is not evident in most sFI cases, unless the patients and family members are surveyed for sleep disorders10 (Table 1). Neuroimaging findings of sFI are similar to FFI, while most sFI cases do not exhibit typical MRI features of sCJD,11 with DWI showing hyperintensity in approximately 8% cases.9 A previous study revealed that 71.4% of sFI cases had significant bilateral thalamic hypometabolism and scattered cortical hypometabolic areas using fluorodeoxyglucose-PET.12 The PSG and EEG features of sFI are similar to FFI. The characteristic manifestations of PSG include the reduction of sleep-related EEG activities, such as K complexes and sleep spindles.13 EEG recordings had a slow background activity (or normal), dominated by the θ-δ rhythm. PSWCs were noted in only 7.7% reported cases,12 and elevated level of 14-3-3 protein was rarely found in the CSF of patients with sFI.11
In patients with rapidly progressing cognitive impairment, behavioral or mood changes associated with ataxic signs, and sleep abnormalities, sFI should be considered as a differential diagnosis. PSG examination, cerebral blood flow single-photon emission CT and/or PET can be used if sFI is suspected. While standard EEG, CSF tests, and MRI are not helpful for diagnosing this disorder, PSG findings including reductions in sleep-related EEG activities (K-complexes and spindles) can be beneficial for a final diagnosis even during an early disease stage. PET examinations have consistently shown thalamic hypometabolism, associated with variable participation in cerebral cortical regions.9 Cracco et al6 proposed clinical diagnostic criteria for sFI by summarizing 32 cases of pathologically confirmed sFI (Table 2). It is thus recommended to complete the PSG examination as early as possible. Additional PSG and PET-CT examinations can also be performed.
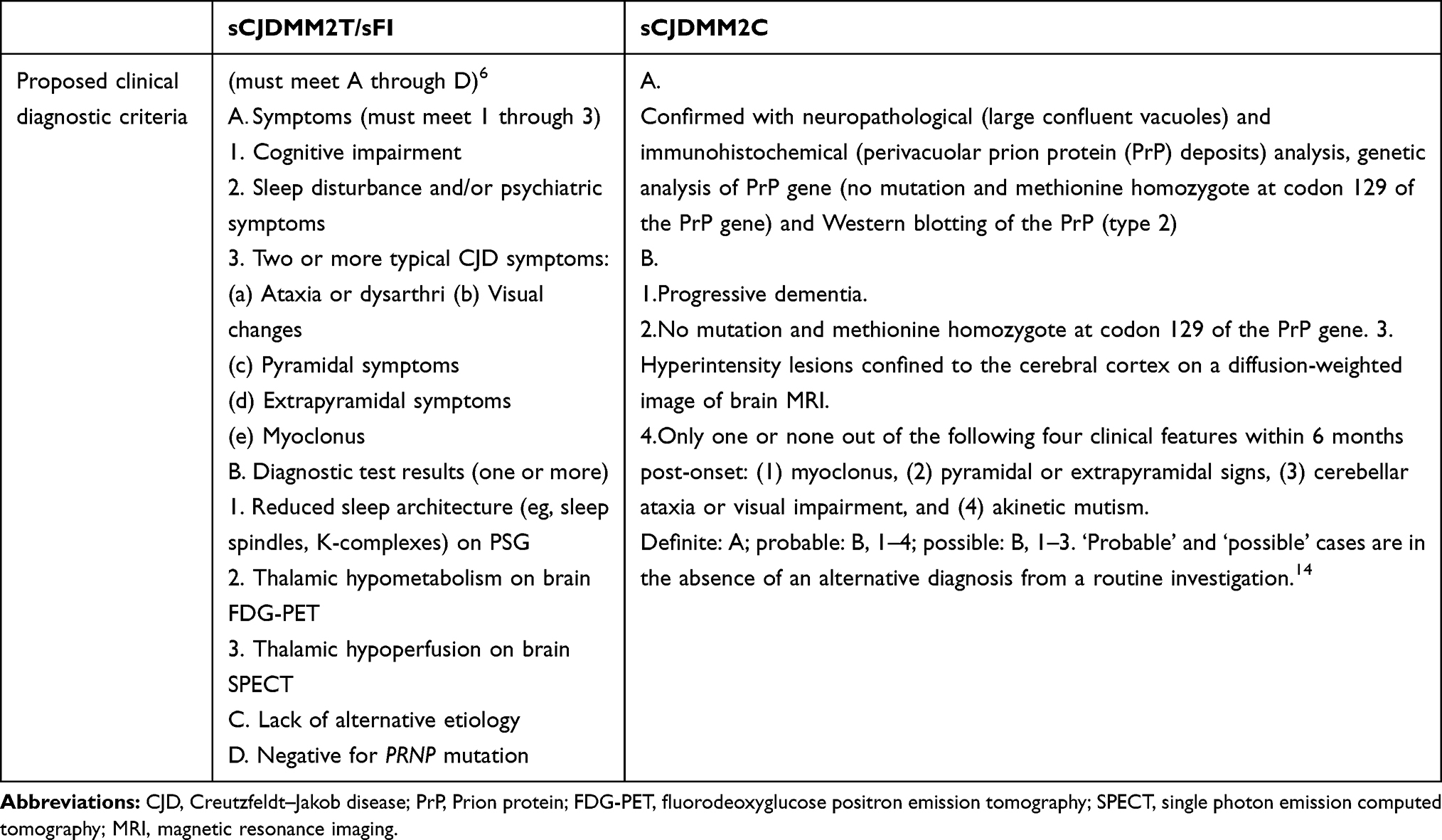 |
Table 2 Proposed Clinical Diagnostic Criteria of sCJDMM2T/sFI, sCJDMM2C |
sCJDMM2C
Compared to other sCJD subtypes, patients with sCJDMM2C experience slower disease progression and a higher frequency of high-intensity lesions on DWI.14 Of note, parietal lobes were abnormal in 96% of patients with sCJDMM2C.15 The frequency of PSWCs in patients with sCJDMM2C was 44.4%, and the CSF 14-3-3 protein was positive in 62.5% patients.14 Pathological features of sCJDMM2C include large confluent vacuoles, cerebellar sparing, and PrPSc perivacuolar deposition.16 Hamaguchi et al14 reviewed 254 patients with pathologically confirmed prion disease in Japan and proposed the clinical diagnostic criteria for sCJDMM2C (Table 2). sCJDMM2C should be included in the differential diagnosis if a patient has a relatively long survival period, few neurological symptoms, and slow progression and the hyperintensity lesions are mainly confined to the cerebral cortex on DWI.
sCJDMM1
sCJDMM1 is the most common subtype of sCJD, accounting for 60–70% of sporadic cases. Previously classified as typical CJD, this subtype has a shorter duration of symptoms than other subtypes.5 Striatal and asymmetric lesions are frequently noted in sCJDMM1.15 PSWCs were detected in 80% cases within 3 months of disease onset.5 Elevated level of 14-3-3 protein is common in the CSF with 95% being sensitive.17 To our knowledge, PSG manifestations are rarely noted in patients with sCJDMM1 subtype. Our previous studies found that sCJD can present with multiple sleep disorders including hypersomnia, insomnia, sleep apnea, and periodic limb movement disorder.8 Pathological findings include typical fine vacuoles and PrPSc synaptic deposition.16 The clinical diagnostic criteria has not been established for sCJDMM1 yet. Although sCJD can present with a variety of sleep disorders including insomnia, the PSG of sCJD patients with insomnia symptoms differs from those of patients with sFI.
Conclusions
Although our patient’s clinical phenotype was consistent with fatal insomnia, the case fulfilled the proposed clinical diagnostic criteria for sFI. Phenotypic variability is one of remarkable features in sCJD. In the absence of brain tissue biopsy or autopsy, the subtype is only highly suspected. sFI is a rare subtype and it should be distinguished from other sCJD subtypes. In individuals with rapidly progressive cognitive impairment, behavioral abnormalities and sleep abnormalities, differential diagnosis of sFI should be considered. Once the sFI subtype is considered. Apart from brain MRI and EEG, PSG, PET and RT-QuIC are also recommended.
Ethical Approval
The present study was approved by ethics committees of the First Hospital of Jilin University.
Informed Consent
Written informed consent was obtained from the patient’s relatives for publication of this manuscript and any accompanying images.
Acknowledgments
We are grateful to Prof. Xiao Kang at China Center for Disease Control and Prevention for his technical help regarding RT-QuIC analysis of skin biopsy tissue and CSF. We thank Prof. Zhou Wei at China Center for Disease Control and Prevention for the detection of 14-3-3 protein in CSF and prion gene in blood. We are grateful to our hospital neurophysiology unit for providing help regarding dynamic EEG examinations and 24-hour PSG.
Disclosure
The authors have no conflicts of interest for this work to declare.
References
1. Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi:10.1073/pnas.95.23.13363
2. Prusiner SB, Hsiao KK. Human prion diseases. Ann Neurol. 1994;35:385–395. doi:10.1002/ana.410350404
3. Zerr I, Parchi P. Sporadic Creutzfeldt-Jakob disease. Handb Clin Neurol. 2018;153:155–174. doi:10.1016/b978-0-444-63945-5.00009-x
4. Nemani SK, Xiao X, Cali I, et al. A novel mechanism of phenotypic heterogeneity in Creutzfeldt-Jakob disease. Acta Neuropathol Commun. 2020;8:85. doi:10.1186/s40478-020-00966-x
5. Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. doi:10.1002/1531-8249(199908)46:2<224::AID-ANA12>3.0.CO;2-W
6. Cracco L, Appleby BS, Gambetti P. Fatal familial insomnia and sporadic fatal insomnia. Handb Clin Neurol. 2018;153:271–299. doi:10.1016/b978-0-444-63945-5.00015-5
7. Geschwind MD. Prion diseases. Continuum (Minneap Minn). 2015;21:1612–1638. doi:10.1212/con.0000000000000251
8. Dai Y, Shao J, Lang Y, Lv Y, Cui L. Clinical manifestations and polysomnography-based analysis in nine cases of probable sporadic Creutzfeldt-Jakob disease. Neurol Sci. 2021;Online ahead of print. doi:10.1007/s10072-021-05102-8
9. Puoti G, Bizzi A, Forloni G, et al. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11:618–628. doi:10.1016/s1474-4422(12)70063-7
10. Moda F, Suardi S, Di Fede G, et al. MM2-thalamic Creutzfeldt-Jakob disease: neuropathological, biochemical and transmission studies identify a distinctive prion strain. Brain Pathol. 2012;22:662–669. doi:10.1111/j.1750-3639.2012.00572.x
11. Hamaguchi T, Kitamoto T, Sato T, et al. Clinical diagnosis of MM2-type sporadic Creutzfeldt-Jakob disease. Neurology. 2005;64:643–648. doi:10.1212/01.Wnl.0000151847.57956.Fa
12. Abu-Rumeileh S, Redaelli V, Baiardi S, et al. Sporadic fatal insomnia in Europe: phenotypic features and diagnostic challenges. Ann Neurol. 2018;84:347–360. doi:10.1002/ana.25300
13. Gambetti P, Parchi P, Chen SG. Hereditary Creutzfeldt-Jakob disease and fatal familial insomnia. Clin Lab Med. 2003;23:43–64. doi:10.1016/s0272-2712(02)00065-3
14. Hamaguchi T, Sanjo N, Ae R, et al. MM2-type sporadic Creutzfeldt-Jakob disease: new diagnostic criteria for MM2-cortical type. J Neurol Neurosurg Psychiatry. 2020;91:1158–1165. doi:10.1136/jnnp-2020-323231
15. Bizzi A, Pascuzzo R, Blevins J, et al. Subtype diagnosis of sporadic Creutzfeldt–Jakob disease with diffusion magnetic resonance imaging. Ann Neurol. 2021;89:560–572. doi:10.1002/ana.25983
16. Iwasaki Y. Creutzfeldt-Jakob disease. Neuropathology. 2017;37:174–188. doi:10.1111/neup.12355
17. Otto M, Wiltfang J, Cepek L, et al. Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology. 2002;58:192–197. doi:10.1212/wnl.58.2.192
18. Hermann P, Appleby B, Brandel J-P, et al. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Lancet Neurol. 2021;20:235–246. doi:10.1016/s1474-4422(20)30477-4
19. Abu-Rumeileh S, Baiardi S, Polischi B, et al. Diagnostic value of surrogate CSF biomarkers for Creutzfeldt-Jakob disease in the era of RT-QuIC. J Neurol. 2019;266:3136–3143. doi:10.1007/s00415-019-09537-0
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.