")
Back to Journals » Infection and Drug Resistance » Volume 12
Spoligotyping analysis of Mycobacterium tuberculosis in Khyber Pakhtunkhwa area, Pakistan
Authors Ali S , Khan MT, Anwar Sheed K , Khan MM, Hasan F
Received 14 December 2018
Accepted for publication 5 April 2019
Published 20 May 2019 Volume 2019:12 Pages 1363—1369
DOI https://doi.org/10.2147/IDR.S198314
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eric Nulens
Sajid Ali,1 Muhammad Tahir Khan,2 Khan Anwar Sheed,3 Muhammad Mumtaz Khan,4 Fariha Hasan1
1Department of Microbiology, Quaid-i-Azam University, Islamabad, Pakistan; 2Department of Bioinformatics and Biosciences, Capital University of Science and Technology, Islamabad, Pakistan; 3Provincial TB Reference Laboratory, Provincial TB Control Program, Khyber Pakhtunkhwa, Pakistan; 4Department of Microbiology, University of Haripur, Haripur, Pakistan
Background: Spoligotyping is a reproducible, reverse hybridization approach for genotyping of Mycobacterium tuberculosis complex (MTBC). Molecular typing of MTBC is helpful for understanding and controlling tuberculosis epidemics.
Methods: Spoligotyping was performed on 166 clinical isolates of Mycobacterium tuberculosis (MTB) collected from 25 districts of Khyber Pakhtunkhwa, Pakistan. Results were compared to SITVIT2, an online database developed by the Institut Pasteur de la Guadeloupe, France.
Results: Spoligotyping results showed that 145 strains (88%) displayed known patterns while 21 (12%) were new. Lineage 3/Central Asian strain (L3/CAS) was the predominant family (73%, χ2=19.9, P=0.001), followed by L2/Beijing (5.4%) and L4 (4.2%). L3/CAS1-Delhi was the major sublineage (82%) among the L3/CAS family (χ2=664, P=0.0001). Analysis showed that the majority of the clinical isolates with an unknown pattern had an evolutionary link with the L3/CAS strain, and nine (5.4%) of the unknown strains were epidemiologically linked and were tentatively named L3/CAS-KP (Khyber Pakhtunkhwa).
Conclusion: The present study demonstrated that L3/CAS is the predominant lineage of MTB, widely distributed in different areas of the Khyber Pakhtunkhwa province of Pakistan. Spoligotyping patterns of some clinical isolates could not be matched to other reported patterns in an international database. Other tools, such as mycobacterial interspersed repetitive unit–variable number tandem repeat (MIRU-VNTR), will be helpful in future investigations into the epidemiological characteristics of clinical isolates in the Khyber Pakhtunkhwa province.
Keywords: spoligotyping, VNTR, L3/CAS, genotyping, molecular characterization, genetic diversity
Introduction
Tuberculosis (TB) has become a major public health problem in Pakistan over the past few decades, and was declared a global emergency by the WHO in 1993. Various national as well as international authorities are highly concerned and are contributing their efforts to abolish TB. Pakistan is among 22 countries identified by the WHO for its high burden of TB cases reported annually. Drug resistance is a major obstacle to TB control in Pakistan.1,2 Millions of Afghan refugees have seasonal traffic between the two countries, which suggests an epidemiological linkage and evolutionary genomic characteristics of Mycobacterium tuberculosis (MTB) strains in Pakistan. The genome of MTB contains variable copies of 36 bp direct repeats (DRs) in a specific locus, the CRISPR (clustered regularly interspaced short palindromic repeats) locus. These DR loci are interspersed with 34–41 bp conserved sequences called spacers. More than 100 spacers have been identified so far; however, only 43 spacers are used for genotyping while the others contribute only a small amount of information.3 This set of 43 unique spacer sequences has been used to classify MTB strains based on the pattern of the presence or absence of DRs.4 Variation in the number of spacers contributes to combinational or individual effects of the genomic process, such as homologous recombination, DNA replication or transposition of the IS6110 element in the DR locus. The function of CRISPR in bacterial metabolism and its relationship with the spacers have not yet been well defined in MTB;5 however, it is evident from the literature that certain patterns of DRs are linked to evolutionary processes.3
Spoligotyping is one of the most highly adopted PCR-based methods for genotyping analysis and is considered both reliable and cost-effective as a first-line genotyping method. Various new techniques have been proposed for spoligotyping analysis, including the DNA microarray,6 PixSysn QUAD 4500 Microarrayer,7 TB-SPRINT8 and hydrogel microarray.9 TB-SPRINT is a microbead assay that provides a spoligotyping profile and simultaneous detection of common mutations in the hot-spot regions of rpoB, katG and inhA genes to evaluate drug resistance to rifampicin and isoniazid in MTB. TB-SPRINT is a useful tool for epidemiological studies of TB in resource-limited countries because of its high throughput and acceptable cost.8 The aim of the current study was to investigate the epidemiological link between MTB strains and their genotypic families found in Pakistan using the spoligotyping method.
Methods
This study was approved by the research and ethics committee of the Provincial TB Control Program Khyber Pakhtunkhwa (TB/KP/R&D-25-17). Samples for the current study were collected at the Provincial TB Reference Laboratory, Peshawar, which is equipped with a biosafety level-III (BSL-III) facility. Samples were digested and decontaminated using a standard N-acetyl-L-cysteine sodium hydroxide (NALC-NaOH) method. Decontaminated samples were inoculated on Lowenstein–Jensen (LJ) medium and a mycobacterium growth indicator tube (MGIT). Positive growth in the tubes was confirmed using a TBc Identification Test device (ref. 245159; Becton Dickinson, Franklin Lakes, NJ, USA). All confirmed mycobacterial isolates were subsequently processed for genotyping analysis targeting the DR locus in MTB, as described by Kamerbeek et al.4 DNA was extracted from the culture using the cetyl–trimethyl-ammonium-bromide (CTAB) method.4
Spoligotyping assay was performed on all samples using a Luminex 200 (Luminex Corp., Austin, TX, USA) instrument.8 Spoligotype patterns were entered in binary format into the SITVIT2, a proprietary database of the Institut Pasteur de la Guadeloupe, which is an updated version of the SpolDB4 and SITVIT-WEB databases.10,11 Clinical isolates were initially grouped in one of nine lineage and sublineages: L1/EAI, L2/Beijing, L3/CAS, L4.1.1/X, L4.1.2/Haarlem, L4.3/LAM, L4.4/S, L4/T-others, L5 and L6 (Mycobacterium africanum WA1 and WAS2).11 The selected clinical isolates that matched with a published spoligo-international type (SIT) were assigned with the corresponding SIT label, while strains with an unknown pattern were declared “orphans”. Lineages were deduced based on the latest next-generation sequencing-based taxonomy when correspondence between whole-genome-based and spoligotyping-based taxonomy was possible.12,13 All strains whose pattern did not fall within the above-mentioned lineages were declared new patterns. Furthermore, the patterns of all strains originating from Pakistan were collected from the SITVIT2 database. Evolutionary relationships between the unclustered spoligotype patterns and strains reported from Pakistan were determined using MEGA7 software.14
The genetic diversity was laid out as a maximum parsimony (MP) tree construct using default settings in MEGA7. The phylogenetic tree was constructed using the subtree–pruning–regrafting (SPR) algorithm, taking the 43 DR pattern as a sequence.14
Results
In total, 200 isolates of MTB were analyzed by the spoligotyping method. Of these isolates, 166 produced valid and interpretable results. Patterns of 145 isolates were found in the international database (SITVIT2) and 21 unique patterns (13%) were declared new types (Table 1). The results showed that lineage 3/Central Asian strain (L3/CAS) was the predominant genotype lineage with 72.8% (n=121), followed by L2/Beijing with 5.4% (n=9) and L4/T with 4.2% (n=7), and 4.8% (n=8) were other unknown types (Figure 1).
 | Table 1 Spoligotypes detected in Mycobacterium tuberculosis isolates from KP, Pakistan |
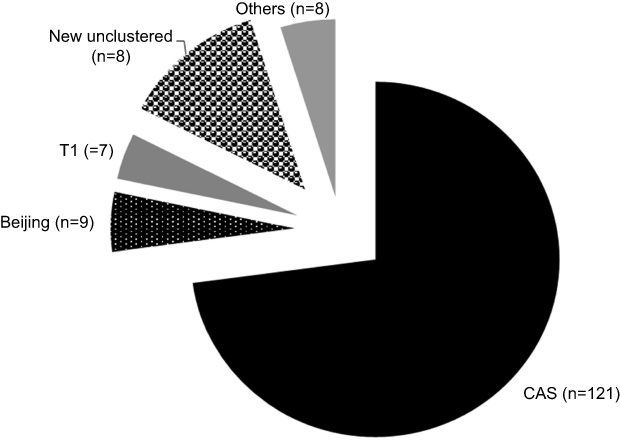 | Figure 1 Graphical analysis of 166 Mycobacterium tuberculosis isolates collected from different areas of Pakistan. Note: Lineage 3/Central Asian strain (L3/CAS), shown in black, is the dominant spoligotype family. New unclustered strains are shown in the black dotted segment. Less frequent spoligotypes are grouped in the “Others” category in the gray segment. |
Cluster analysis of widely distributed lineages
Detailed analysis of L3/CAS resulted in 11 different clusters or sub-families. Among the sub-families, L3/CAS1-Delhi (SIT26) was the predominant genotype (n=78), followed by L3/SIT1401 (n=5) and L3/SIT794 (n=4). The pattern of one member of the L3/CAS family that did not find a match in the SITVIT2 database was declared as L3/CAS-Orphan. All strains belonging to the L2/Beijing family were grouped in two clusters, SIT1 (n=8) and SIT941 (n=1). Members of L4/T-other spoligotypes were grouped in five different clusters (SIT51, SIT53, SIT273, SIT732 and SIT189) and one other T orphan. It was also found that EAI1-SOM, EAI5, EAI6-BGD1, H1, LAM9-ZWE, Manu, EAI3-IND and X1 were rarely distributed in this region.
All 21 strains with unique spoligotyping patterns were grouped into 13 clusters based on the similarity index of the MP test. The new clusters were further analyzed using different methods to determine any potential evolutionary relationship to previously published genotypes. The results indicated that the majority of the unique patterns were sublineages of the L3/CAS family, except for some orphans (KP1–KP3) that were more closely related to L4. The detailed phylogenetic tree is represented in Figure 2. These spoligotyping patterns were unique not only to Pakistan but also to the international database. Therefore, no SIT has been assigned so far (Table 1).
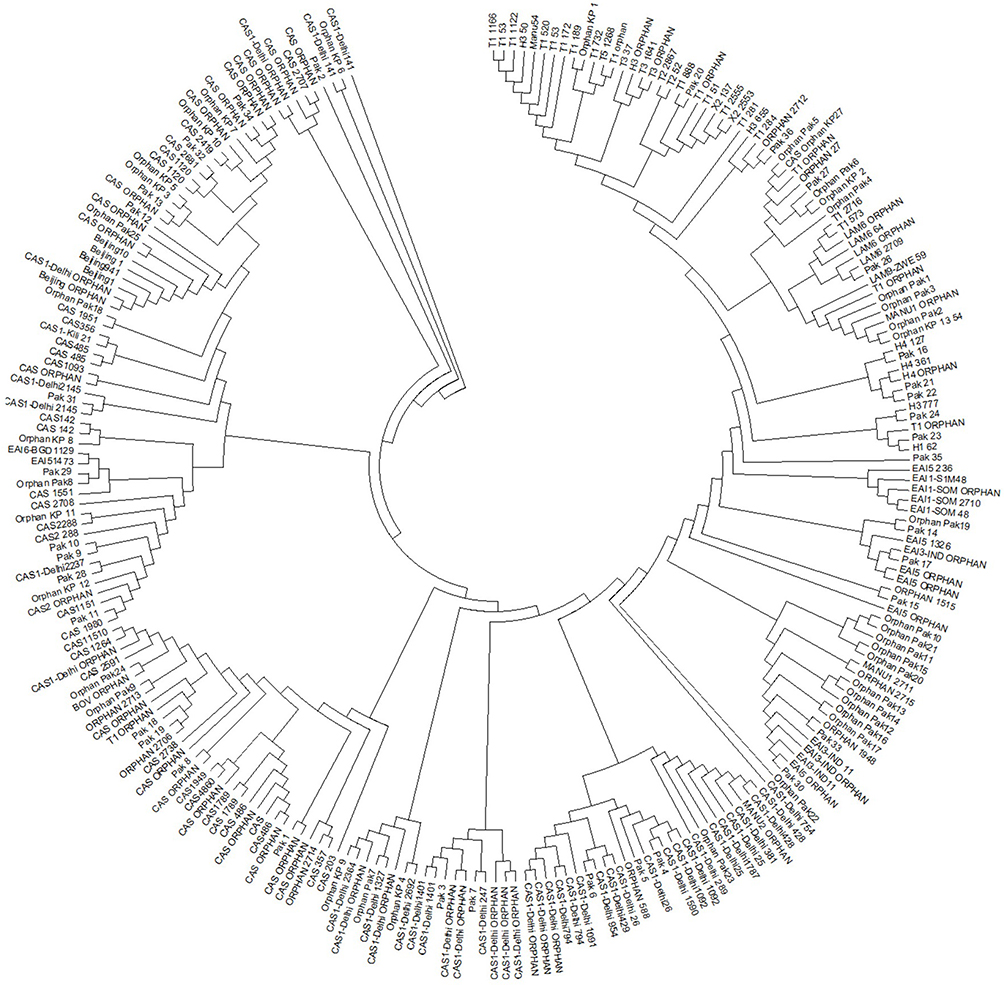 | Figure 2 Maximum parsimony analysis of new unclustered spoligotypes. |
The tree shows the relationship of new unclustered spoligotypes in this study to the previously published spoligotype patterns from Pakistan.15–19 The new spoligotype patterns in this study were numbered Orphan KP1 to Orphan KP13.
Discussion
Many techniques have been used for MTB genotyping, including restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP), mycobacterial interspersed repetitive unit–variable number tandem repeat (MIRU-VNTR) and spoligotyping. The spoligotyping technique has been used worldwide for genotyping and as a first-line method in epidemiological studies of MTB, in conjunction with VNTR analysis. It was originally developed by Kamerbeek et al, based on identical DRs interspersed by short spacers of variable length.4 A public database, SITVIT2, is available for the analysis of spoligotyping patterns, which consists of 111,635 clinical isolates from 169 countries of patient origin.20 The distribution of these spoligotypes was analyzed among the 20 studied subregions. It is evident from the literature that these genotypes are specific to certain geographical regions; for example, L4.3/LAM is geographically widely distributed in South America, followed by L4.1/T and L4.1.2/Haarlem lineage, at 49.3%, 26.7% and 15.7%, respectively. Similarly, L1/EAI is geographically distributed in South-East Asia and L3/CAS in Middle-East Asia.11,21
Our study represents the first molecular characterization of TB bacilli in the study area and may be useful for better management of TB and drug resistance.22 Considering the spoligotyping patterns of the majority of the strains, the current study confirmed a diverse population structure with a total of 55 different types, whether clustered or not. However, by far the most dominant lineage was L3/CAS-1 Delhi, which belongs to the L3/CAS lineage. Overall, the distribution of genotypes in Khyber Pakhtunkhwa was in line with other spoligotyping analysis in Pakistan.16,19,21 However, we do not report any genotype of L4.5 “NEW-1”, an emerging genotype in Pakistan, since no SIT127 types were observed.23 Spoligotyping, in some cases, is not able to identify L4.5, which is better characterized by RD122, specific VNTR signatures and specific single-nucleotide polymorphisms.13,23 A rather important population of 12% of strains could not be matched to the internationally defined genotypes in the database and was declared “new”. In previous investigations, the spoligotyping patterns of new genotypes were underestimated and left unstudied.16,19 In-depth analysis revealed clear identification of L3/CAS by the presence of the first three spacers followed by a set of four absent spacers (4–7) and two more spacers (33–34). We confirmed that the majority of the new spoligotypes in this study were likely to have evolved from L3/CAS, characterized by the absence of spacers 4–7 and 23–34, as changes in the DR locus are most likely to occur via consecutive irreversible deletions of either single or contiguous blocks.3,24
The current study provides basic information on the geographical distribution of MTB strains in Khyber Pakhtunkhwa, Pakistan. It has some limitations for the time being, since spoligotyping-based clusters were not further studied using VNTR. However, such comparisons of spoligotypes may not be representative of the true phylogenetic relationships owing to genetic convergence. By contrast, DNA sequencing can be used to determine the true evolution.
Conclusion
We found that L3/CAS is the predominant lineage, which is likely to be epidemiologically linked in many cases, and the majority of the unclustered L3 strains are likely to have evolved from pre-existing L3/CAS strains based on the MP analysis tree construct. Other tools, such as MIRU-VNTR in combination with spoligotyping, would help us to further explore the molecular epidemiology of MTB in our study area.
Acknowledgments
This paper was published in Journal of Biological Regulators and Homeostatic Agents, 2018;32(2):195–198 (PMID 29685006;
Disclosure
The authors report no conflicts of interest in this work.
References
1. Khan MT, Malik SI, Ali S, et al. Pyrazinamide resistance and mutations in pncA among isolates of Mycobacterium tuberculosis from Khyber Pakhtunkhwa, Pakistan. BMC Infect Dis. 2019;19(1):116. doi:10.1186/s12879-019-3764-2
2. Khan MT, Malik SI, Bhatti AI, et al. Pyrazinamide-resistant mycobacterium tuberculosis isolates from Khyber Pakhtunkhwa and rpsA mutations. J Biol Regul Homeost Agents. 2018;32(3):705–709.
3. van Embden JD, van Gorkom T, Kremer K, Jansen R, van Der Zeijst BA, Schouls LM. Genetic variation and evolutionary origin of the direct repeat locus of Mycobacterium tuberculosis complex bacteria. J Bacteriol. 2000;182(9):2393–2401.
4. Kamerbeek J, Schouls L, Kolk A, et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35(4):907–914.
5. Jansen R, van Embden JDA, Gaastra W, Schouls LM. Identification of a novel family of sequence repeats among prokaryotes. Omics J Integr Biol. 2002;6(1):23–33. doi:10.1089/15362310252780816
6. Ruettger A, Nieter J, Skrypnyk A, et al. Rapid spoligotyping of Mycobacterium tuberculosis complex bacteria by use of a microarray system with automatic data processing and assignment. J Clin Microbiol. 2012;50(7):2492–2495. doi:10.1128/JCM.00442-12
7. Song EJ, Jeong HJ, Lee SM, et al. A DNA chip-based spoligotyping method for the strain identification of Mycobacterium tuberculosis isolates. J Microbiol Methods. 2007;68(2):430–433. doi:10.1016/j.mimet.2006.09.005
8. Gomgnimbou MK, Hernández-Neuta I, Panaiotov S, et al. Tuberculosis-spoligo-rifampin-isoniazid typing: an all-in-one assay technique for surveillance and control of multidrug-resistant tuberculosis on Luminex devices. J Clin Microbiol. 2013;51(11):3527–3534. doi:10.1128/JCM.01523-13
9. Bespyatykh JA, Zimenkov DV, Shitikov EA, et al. Spoligotyping of Mycobacterium tuberculosis complex isolates using hydrogel oligonucleotide microarrays. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2014;26:41–46. doi:10.1016/j.meegid.2014.04.024
10. Brudey K, Driscoll JR, Rigouts L, et al. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006;6:23. doi:10.1186/1471-2180-6-23
11. Demay C, Liens B, Burguière T, et al. SITVITWEB – a publicly available international multimarker database for studying Mycobacterium tuberculosis genetic diversity and molecular epidemiology. Infect Genet Evol. 2012;12(4):755–766. doi:10.1016/j.meegid.2012.02.004
12. Coll F, Preston M, Guerra-Assunção JA, et al. PolyTB: a genomic variation map for Mycobacterium tuberculosis. Tuberc Edinb Scotl. 2014;94(3):346–354. doi:10.1016/j.tube.2014.02.005
13. Stucki D, Brites D, Jeljeli L, et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat Genet. 2016;48(12):1535–1543. doi:10.1038/ng.3704
14. Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;msw054. doi:10.1093/molbev/msw054
15. Gascoyne-Binzi DM, Barlow REL, Essex A, et al. Predominant VNTR family of strains of Mycobacterium tuberculosis isolated from South Asian patients. Int J Tuberc Lung Dis Off J Int Union Tuberc Lung Dis. 2002;6(6):492–496. doi:10.5588/09640569512995
16. Hasan Z, Tanveer M, Kanji A, Hasan Q, Ghebremichael S, Hasan R. Spoligotyping of Mycobacterium tuberculosis isolates from Pakistan reveals predominance of Central Asian Strain 1 and Beijing isolates. J Clin Microbiol. 2006;44(5):1763–1768. doi:10.1128/JCM.44.5.1763-1768.2006
17. Ali A, Hasan Z, Tanveer M, et al. Characterization of Mycobacterium tuberculosis Central Asian Strain1 using mycobacterial interspersed repetitive unit genotyping. BMC Microbiol. 2007;7:76. doi:10.1186/1471-2180-7-76
18. Ayaz A, Hasan Z, Jafri S, et al. Characterizing Mycobacterium tuberculosis isolates from Karachi, Pakistan: drug resistance and genotypes. Int J Infect Dis. 2012;16(4):e303–e309. doi:10.1016/j.ijid.2011.12.015
19. Yasmin M, Gomgnimbou MK, Siddiqui RT, Refrégier G, Sola C. Multi-drug resistant Mycobacterium tuberculosis complex genetic diversity and clues on recent transmission in Punjab, Pakistan. Infect Genet Evol. 2014;27:6–14. doi:10.1016/j.meegid.2014.06.017
20. Couvin D, David A, Zozio T, Rastogi N. Macro-geographical specificities of the prevailing tuberculosis epidemic as seen through SITVIT2, an updated version of the Mycobacterium tuberculosis genotyping database. Infect Genet Evol. 2018. doi:10.1016/j.meegid.2018.12.030
21. Tanveer M, Hasan Z, Siddiqui AR, et al. Genotyping and drug resistance patterns of M. tuberculosis strains in Pakistan. BMC Infect Dis. 2008;8(1):171. doi:10.1186/1471-2334-8-171
22. Khan MT, Malik SI, Ali S, et al. Prevalence of pyrazinamide resistance in Khyber Pakhtunkhwa, Pakistan. Microb Drug Resist Larchmt N. 2018;24:1417–1421. doi:10.1089/mdr.2017.0234
23. Mokrousov I, Shitikov E, Skiba Y, Kolchenko S, Chernyaeva E, Vyazovaya A. Emerging peak on the phylogeographic landscape of Mycobacterium tuberculosis in West Asia: definitely smoke, likely fire. Mol Phylogenet Evol. 2017;116:202–212. doi:10.1016/j.ympev.2017.09.002
24. Sola C, Filliol I, Gutierrez MC, Mokrousov I, Vincent V, Rastogi N. Spoligotype database of Mycobacterium tuberculosis: biogeographic distribution of shared types and epidemiologic and phylogenetic perspectives. Emerg Infect Dis. 2001;7(3):390. doi:10.3201/10.3201/eid0703.0107304
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.