")
Back to Journals » Journal of Pain Research » Volume 15
Spoken and Unspoken Matters Regarding the Use of Opioids in Cancer
Authors Baker Rogers J, Higa GM
Received 6 January 2022
Accepted for publication 24 March 2022
Published 5 April 2022 Volume 2022:15 Pages 909—924
DOI https://doi.org/10.2147/JPR.S349107
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Amitabh Gulati
Janna Baker Rogers,1 Gerald M Higa2
1Sections of Geriatrics, Palliative Medicine and Hospice, Department of Medicine, School of Medicine, West Virginia University, Morgantown, WV, USA; 2Departments of Clinical Pharmacy and Medicine, Schools of Pharmacy and Medicine, West Virginia University, Morgantown, WV, USA
Correspondence: Gerald M Higa, Departments of Clinical Pharmacy and Medicine, Schools of Pharmacy and Medicine, West Virginia University, Morgantown, WV, USA, 26506, Email [email protected]
Abstract: Pain is among the most debilitating symptoms in patients with cancer. Except for their relatively frequent use during end-of-life care, opioids are often, though not routinely, prescribed during the course of the disease. Whereas the clinical phenomena of tolerance, dependence, and addiction are invariably recognized, the molecular mechanisms which effect these outcomes are not fully understood, even among health care professionals. Also uncertain is the possible unfavorable effect of these agents on cancer progression and survival, an association that may be related to the expression of opioid receptors in some tumors. An intriguing corollary of the latter finding is that cancer cells may also manifest equivalents of the three maladaptive phenomena. Accordingly, instead of re-addressing the societal and epidemiological impact of opioids, this paper has three alternative foci. The first, and most subordinate, focuses on the mu opioid receptor; the second, centers on the unresolved question regarding the potential adverse effect of opioids on tumor growth; the third, and most compelling, concentrates on the cellular apparatus and influences that modulate tolerance, dependence, and addiction in certain cancers exposed to opioids.
Keywords: addiction, cancer, dependence, MOR, opioids, tolerance
Introduction
Reduction in cancer mortality is usually attributed to early diagnosis, increased understanding of tumor biology, and the development of new drugs. Less apparent is the contribution supportive care may have in improving cancer survivorship;1 and one of the most affecting patient-focused supportive interventions is the management of cancer-related pain.
Approximately two-thirds of all patients diagnosed with cancer will experience pain associated with diagnostic procedures, tumor size and dissemination, and treatment-related side-effects. Pain may even persist for months after treatment is completed. Though severity will differ, symptomatic relief may require the use of opioids, especially in those with advanced or metastatic disease. However, the association between opioids and cancer may extend beyond alleviation of pain.
In spite of their potency, utilization of these analgesics seldom translates to complete analgesia. And equally, or even more, problematic is the development of tolerance, dependence and addiction with chronic opioid therapy. Although well accepted, biological understanding of these three phenomena remains superficial, even among medically-trained personnel. Not surprising also is the lack of appreciation that these consequences could be an intrinsic part of cancer itself. The latter belief was the major premise for exploring the adverse repercussions related to the use of opioids in cancer, a supportive care matter of immense therapeutic concern. To accomplish this goal, numerous topic-related publications were accessed and critically reviewed. Segments of published information were transposed to fortify the validity of the textual content of the manuscript, while areas of ambiguity provided opportunities for cogent author insight.
This invited review was organized to furnish the reader with: 1) a descriptive primer of the opioid receptor family, 2) a systematic discourse related to opioid tolerance, dependence, and addiction, 3) a judicious critique of the oncogenic potential of opioids, and 4) a tenable consideration regarding opioid-like adaptations in cancer cells.
A Descriptive Primer
Revelations regarding the opioid receptor occurred a few years before reports of the nicotinic acetylcholine receptor,2 though molecular cloning of the receptor subtypes did not occur till the early 1990ʹs.3,4 Even though not as profound as the theory of evolution which predated these findings by more than 100 years, the receptor, too, has evolved conceptually from mere chemical structures to integral molecular components capable of transducing external signals and effecting specific cellular, tissue, and systemic responses.
Genealogy
Opioid receptors belong to the superfamily of seven transmembrane Gi/G0-protein coupled receptors (GPCRs).5 Initially, receptor expression was reportedly confined to nociceptive neurons. A few years later, three opioid receptor subtypes were recognized with high levels of mu (MOR), delta (DOR), and kappa (KOR) receptors found in multiple areas of the brain including the cerebral cortex, hippocampus, substantia nigra, putamen, nucleus accumbens, and caudate nucleus. Subsequently, the cDNA encoding an opioid receptor-like 1 (ORL1) protein was isolated; the fourth opioid receptor subtype has been designated Nociceptin Opioid Peptide (NOP);6 and for completeness, the gene for a fifth opioid receptor was cloned in 2000.7 Known as zeta (ζ) or opioid growth factor receptor, the protein’s endogenous ligand is [Met(5)]-enkephalin. Despite these findings, the notion of centrally-restricted receptor expression was later debunked with detection of the receptors in tissues of the cardiovascular, gastrointestinal, reproductive, and immune systems.8
Although encoded by five different genes, all of the subtypes exhibit substantial structural homology in the transmembrane domains and intracellular loops; greater divergence is found in the N- and C-termini as well as the extracellular loops.3 The receptor’s ligand binding region is localized to the multiplex transmembrane domain. The well-conserved lower portion of the binding “pocket” has three unique features: first, an anionic orthosteric (ie active) site which interacts with positively charged amines (common to most ligands); second, a cavity for ligand-containing amino groups; and third, a relatively planar surface that accommodates structural aromatic components. The upper portion of the pocket, which differs among subtypes, modulates ligand selectivity. Collectively, this critical segment plus the C-terminus tail contribute to G-protein coupling.
A common feature of most, if not all, GPCRs relates to their cell surface expression, though the receptors can be localized to pre- and post-synaptic terminals on both central and peripheral neurons. At the level of the plasma membrane, opioid receptors can function as monomers, homodimers, or heterodimers with other opioid receptor subtypes or other GPCRs.9 Heterodimeric coupling can alter ligand properties, modify ligand selectivity to Gi/Go, Gz (a guanine nucleotide-binding protein), or β-arrestin-2 signaling pathways, and affect intracellular trafficking of the receptor.10
The three major receptor subtypes (MOR, DOR, and KOR) are activated by endogenous peptides including endorphins, enkephalins, and dynorphins.11 Although these ligands interact with all three receptors, the peptides exhibit variable receptor affinities and potencies.12 In particular, β-endorphin is the most potent endogenous ligand for the MOR, while enkephalins and dynorphins exhibit greater affinity for DORs and KORs, respectively. NOP is activated by native nociception/orphanin FQ and does not bind other endogenous opioid ligands at low concentrations or naloxone.13 Even though surface activation is predominant, receptor stimulation may not occur exclusively at the plasma membrane as ligand-induced activation of MORs has also been observed in certain cytoplasmic compartments such as the endosome.9
Functional Complexity of MOR
The extensive use of morphine as well as its higher affinity for the mu opioid receptor contributed to the MOR being the most well-studied subtype. The MOR is encoded by the OPRM1 gene which contains at least 20 exons. Several single nucleotide polymorphisms (SNPs) in the coding regions have been identified which may explain some of the variability in clinical responses to exogenous MOR agonists.14 For example, the most common SNP, N40D (asparagine replaced by aspartic acid), which affects the N-terminus, has been associated with alterations of pain threshold levels, sensitivity to exogenous opioids, and increased risk of opioid-induced respiratory depression. In addition, two promoters which regulate nearly all of the gene’s transcriptional activity are highly sensitive to epigenetic modification due to the presence of numerous Cytosine phosphate followed by Guanine (CpG) islands.15 Methylation of various CpG sites can suppress OPRM1 gene expression and subsequently decrease MOR expression.
The OPRM1 gene can also undergo alternative pre-mRNA splicing to a degree thought to be unusual for GPCRs. That this level of atypia may parallel the evolutionary importance of OPRM1 gene-splicing is strengthened by the preservation of alternative splicing events observed in rodents and humans.16 Distinguished by the number of transmembrane domains, at least 30 splice variants have been identified; the variants are aggregated into three groups based on their OPRM1 isoform. The first group, generated by 3’ splicing, expresses the traditional seven transmembrane domains with variability in the C-terminus region.17 The potential relevance of this group may relate to observed differences in mu agonist-induced G-protein coupling, B-arrestin-2 recruitment, receptor phosphorylation, and degree of internalization.16,18 The second group contains six transmembrane domains generated through 5’ splicing. In preclinical models of thermal and neuropathic pain, members of this group appear to have a role in opioid-induced analgesia without producing respiratory depression, dependence, or reward.9,19 Interestingly, even if endogenous opioids may not bind or activate members in this group, these variants are able to heterodimerize with, and facilitate expression of, seven transmembrane-containing MORs.16 The third group has only one transmembrane domain yet appears to be functionally important as molecular chaperones by reducing the turnover and increasing the potency of wild-type MORs.16
A Systematic Discourse
The preceding information represents only a small portion of accumulated data addressing the inherent complexity of the MOR. What is apparent is that exogenous opioids can affect neurotransmitter release, collateral signaling pathways, and even expression of the mature form of the OPRM1 transcript. As a result of the latter, post-translational modifications of the MOR can alter receptor binding and signaling properties. Conceivably, mutations or alterations of the MOR gene or function may even have a role in the development of opioid-associated tolerance, dependence, and addiction.
Tolerance
Tolerance is defined by the National Institute on Drug Abuse (NIDA) as
… what happens when a person no longer responds to a drug in the way they did at first, so it takes a higher dose of the same drug to achieve the same effect as when the person first used it.
The unique aspect about this definition is its reference to essentially only one drug-induced effect, analgesia. While tolerance develops to nearly all opioid-related effects, the largely qualitative description is also devoid of relative differences in time frame for some of the sequelae to occur. In addition, a “steady state” of tolerance has been observed in patients on chronic stable doses of opioid therapy.20
Agonist-bound MORs engage a number of processes that result not only in receptor-mediated analgesia and reward but also receptor desensitization and re-localization (Figure 1). Briefly, signal termination following ligand binding requires receptor phosphorylation. However, unlike other GPCRs which are phosphorylated by G-protein–coupled receptor kinases (GRKs), mitogen-activated protein kinase phosphorylates early endosome antigen-1 (EEA-1) and Rabenosyn-5 of the MOR followed by recruitment of a key regulatory protein known as β-arrestin-2.21,22 Even though phosphorylated and arrestin-bound receptors are believed to be functionally desensitized, other processes that regulate ion channels or enzyme production can also subvert an operational receptor. Receptor desensitization can also occur rapidly, in some instances less than a minute. Furthermore, the degree or extent of receptor desensitization is inconsistent as different patterns have been observed in the central nervous system after chronic morphine administration.10 Findings supportive of the latter consideration are brain areas that mediate analgesia and respiratory depression underwent nearly complete desensitization compared to regions mediating rewards. Desensitization in the former is believed to be mediated by inhibitory β-arrestin-2.
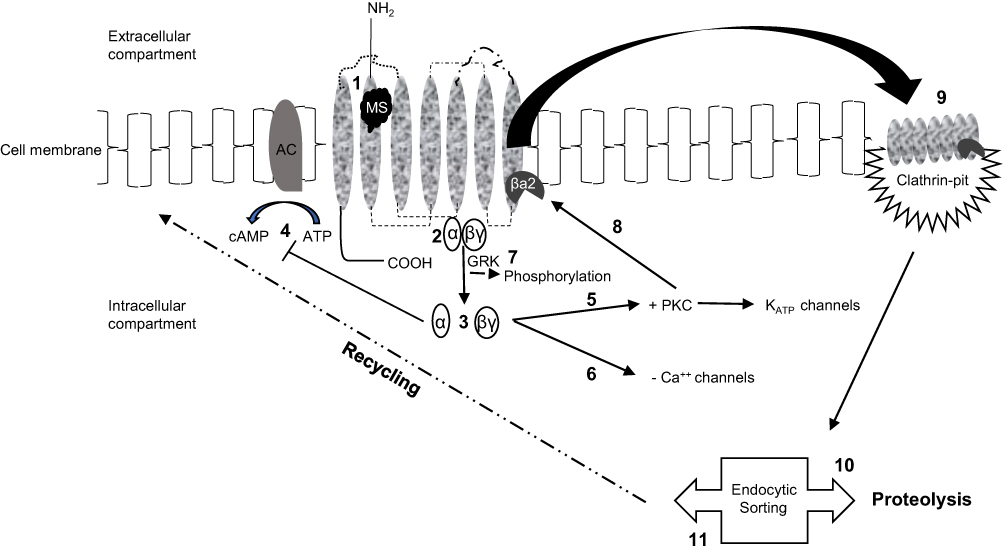 |
Figure 1 Schematic rendition of the opioid receptor-signaling pathways. Morphine (MS)-binding (1) to a monomeric receptor initiates intracellular signaling by activating the trimeric G-protein (2) and causing dissociation of α- and βγ-subunits (3). The G-protein subunits can promote different cellular responses such as α-inhibition of adenylyl cyclase and the generation of cAMP (4) or βγ- induction of protein kinase C (PKC)-mediated stimulation of K+ channels (5) or inhibition of Ca++ channels (6). Notably, ligand-bound receptors lacking the G-protein subunits are functionally desensitized by GPCR kinase (GRK)-induced phosphorylation (7). PKC prompts recruitment of β-arrestin-2 (βa2) (8) driving receptors to clathrin-coated pits which enable the endocytic process (9). Further sorting of the internalized receptors affects cellular signaling in additional ways. Receptors marshalled to lysosomes (10) are degraded while receptors sorted to endosomes may undergo rapid recycling to the cell surface, effectively re-sensitized (11) and able to be activated again. |
Arrestin binding also causes structural changes which not only inhibit further receptor activation but also promote co-internalization of the bound receptor via clathrin-mediated endocytosis (Figure 1).23 Importantly, divergent sorting of internalized receptors produces marked differences on cell signaling. Accordingly, receptors sorted to lysosomes are degraded thus preventing, or prolonging attenuation of, further receptor activation; conversely, endosomal uptake can lead to rapid recycling of receptors back to the plasma membrane, effectively re-sensitizing the cell-signaling pathway.10 Receptor fate likely depends on multiple kinases though their relative importance in regulating opioid receptor activity is only partially resolved. Consequently, partnering desensitization with internalization may be inaccurate because the two processes may not always accompany one another, especially in cells activated by morphine.24 Even so, the importance of post-stimulation trafficking of the receptor and consequential development (and degree) of tolerance appears to be associated with decreased cell surface recycling and re-sensitization. This belief is reinforced by observations indicating that less desensitization (and tolerance) occurred with ligands that induced receptor endocytosis. Further evidence that this may indeed be valid is the finding that compared to native MOR ligands, morphine-bound receptor complexes can elude GRK-induced phosphorylation and subsequent endocytosis.10 As such, two agonists may demonstrate similar efficacy, yet have dissimilar signaling effects due to variable abilities to promote receptor endocytic desensitization. Variations in efficacy and endocytosis have spawned the terms “relative activity versus endocytosis” or RAVE.25
The lack of clarity regarding whether desensitization depends on the added component of internalization led to two credible, though separable, hypotheses. First, membrane-bound MORs are desensitized immediately after phosphorylation and arrestin binding; and second, internalization of the deactivated receptor plays a vital role in receptor re-sensitization. Nonetheless, receptor desensitization appears to be an obligatory precursor for the development of tolerance and decreased analgesic activity.
Dependence
Although dependence is described as the continued administration of opioids in order to prevent withdrawal symptoms, some mechanisms may be unique to the development of this vulnerable state.21 One refers to the onset of allostasis or the maintenance of stability, outside of a normal homeostatic range, necessitated by chronic demands on physiologic systems.26 Chronic opioid use alters the neuronal circuitry and cultivates a compensatory “new norm” predicated on drug presence to maintain “normal” function; removal of opioid stimulation results in disruption of the adaptive state and the onset of withdrawal symptoms.27 Despite proof that consequent dependence (and tolerance) are determined primarily by engagement of MOR-associated regulatory events, these observable aftereffects appear to be molecularly well-defined phenomena. An explanation why this is so is proffered. Often linked to receptor desensitization and endocytosis, decreased expression of membrane-bound MORs has been the fundamental premise underlying the development of tolerance. Notwithstanding this well-grounded argument, findings related to this assumption have not been consistently observed. For example, some investigators have found that GRK-induced phosphorylation and β-arrestin-2-recruitment appear to be important components in signal termination and cellular uptake by facilitating dissociation of G-protein kinase from the receptor. Because the fate of uncoupled and internalized receptors is either endosomal reclamation or lysosomal degradation, development of tolerance could indeed be, though only partially, attributable to fewer numbers of recycled receptors. However quantitative expression alone does not appear to provide a plausible mechanism intrinsic to the development of dependence. Rather, cellular dependence is more likely an outcome related to alterations of crucial regulatory components. This notion may certainly be well-founded as other investigators have observed that prolonged stimulation of morphine-bound MORs by “evading” or “avoiding” recruitment of GRK and arrestin promotes the development of withdrawal potential (and tolerance).28 What is intriguing about this finding is the inference that insensate MORs do not undergo canonical desensitization. If so, mechanisms that curtail prolonged signaling such as phosphorylation and internalization would reduce the likelihood of developing (or at least decrease the severity of) both adverse phenomena. As antithetical as this seems, there is some support for this concept as studies demonstrated that methadone- but not morphine-bound receptors underwent internalization, yet morphine produces a higher degree of cellular tolerance (and withdrawal).29
Arguably complex, these seemingly distinct, yet connected, mechanisms need to be reconciled. As indicated previously, if restoration of functional MORs is largely dependent on endocytic processing of the morphine-receptor complex, then greater dependence and cellular tolerance should emerge if desensitized, plasma-bound receptors fail to be internalized. On the other hand, continued signaling in receptors that do not undergo internalization may actuate other cellular adaptations which promote the development of both conditions. One cellular alteration of note involves cAMP. Biochemical regulation of cyclic adenosine monophosphate is stringently controlled by two G protein α subunits. Normally, morphine-bound MORs coupled to Gαi subunits inhibit adenylyl cyclase (AC) resulting in decreased formation of cAMP. However, chronic morphine exposure paradoxically upregulates the cAMP-dependent cascade, a phenomenon not related to uncoupling of inhibitory G-proteins but rather associated with increased expression and/or super-sensitization of some AC isoforms.10,27,30 As a consequence of the rebound effect, regulation of cAMP cannot be accomplished as proficiently as in the naïve state.10 And incidentally, hyperactivated cAMP is a hallmark manifestation of opioid withdrawal.31 Exemplary studies by Finn and Whistler further affirm this revelation.32 Compared to two mutant MORs, super-activation of cAMP was observed only in wild-type MOR (which did not undergo appreciable internalization) chronically exposed to morphine. These, and other, data strongly implicate not only the contribution of chronic morphine exposure and endocytosis but also the role of cellular adaptation in effecting dependence.
Even though compelling, the above cannot be the sole mechanism underlying the development of dependence as incessant receptor activation may also be partly related to pharmacokinetic differences between endogenous and exogenous receptor ligands. Compared to the phasic or pulsatile release patterns of the former, continuous dosing of the latter compounds sustains receptor activation due to the extended presence of extracellular opioid concentrations.10 In addition, other molecular adaptations may also contribute to this rebound effect including increased expression of protein kinase A and cAMP response element binding-protein. Generation of these adaptive molecules may be related to coupling of Gi/G0 and Src signaling pathways. Ultimately, super-activated cAMP is capable of side-stepping native regulatory effects of morphine.
Aside from cAMP the emergence of dependence may also be a consequence of the interconnectivity between opioid, dopamine, and GABA pathways. In this scenario, the rewarding effects of morphine result from activated MOR-induced disinhibition of GABAergic neurons and subsequent activation of dopamine neurons in the ventral tegmental area (VTA) of the brain. However, chronic morphine exposure promotes alterations in both pre-synaptic and post-synaptic neurons of the VTA.27 Presynaptic adaptations involve cAMP-dependent and -independent effects on GABA and glutamate release while post-synaptic changes may involve activated KORs or modifications in afferent transmission. The end result of these various changes is substantial and durable decrease in dopaminergic activity.
Physiologically, morphine withdrawal has a paradoxical effect on several signaling molecules. On one hand, increased expression of two stress-associated endogenous signaling molecules, dynorphin and corticotropin-releasing factor (CRF) has been observed in the striatum;33 and dynorphin’s high affinity for the KOR reversibly contributes to the negative emotional states of withdrawal by promoting aversion and depression. KOR-mediated signaling also inhibits dopamine release in the nucleus accumbens, which further induces stress-associated dysphoria and increases vulnerability to drug relapse. Activation of CRF and other noradrenergic stress-modulating pathways also accentuates the negative affective state.33 On the other hand, decreased serotonin transmission occurring in the nucleus accumbens may further contribute to depression and mood disturbances.33
Addiction
Central to opioid addiction are changes within the neuronal signaling mainframe that mediate the evolution from initial controlled use for analgesia to unconstrained consumption caused by several heteromorphic behavioral disturbances. A few of the most maladaptive behaviors are 1) amplified incentive reinforcement and habit development, 2) reward deficits and stress accumulation, and 3) impaired decision-making capacity. Not only do the three functional domains establish a neurochemical basis of addiction but also a physio-psychological explanation that underlie the continued use of a substance despite negative consequences. That the opioid receptor system could instigate such deviant behavior is strengthened by the proximity of these receptors to neurocircuitry at risk for pernicious alterations occurring over time. Transpiring sequentially and additively, these neuroplastic conversions are believed to be stimulated by ongoing use of increasing doses of opioids. And not inconceivably, transitioning through these changes appear to parallel the emergence of escalating compulsivity and ensuing addiction.33
At the molecular level, opioid activity in the basal ganglia, particularly the mesolimbic system, mediates pleasure and reward. This system also plays a key role by strengthening self-motivation to meet basic survival needs.27 In addition, endogenous opioid peptides promote a steep and rapid increase of dopamine (D) in the ventral striatum which stimulate low affinity D1 receptors so as to effect reward, commandeer attention, direct motivation, and promote arousal and conditioned learning. However, a notable paradox has also been observed regarding opioids and dopamine. While the neurotransmitter is required for the opioid-induced reward effect in the opioid-dependent state, decreased dopamine activity occurs in chronic opioid users.33 The outcome of changes in the dopaminergic system in the mesolimbic and ventral striatum/nucleus accumbens distorts incentive reinforcement towards habit-forming use of opioids. Combined with dysregulation of brain stress systems, these changes result in decreased reward experience and provide negative reinforcement for drug use.33
In addition, maladaptive changes in the amygdala and prefrontal cortex impact multiple aspects of executive function.33 Deregulation of cognitive, emotional, and memory processes brings about distorted valuation of drug-taking and natural reinforcers. As a result, inhibitory control of drug responses declines. These changes may also be compounded by alterations in MOR and DOR functions in decision-making processes. Triggers for drug reinstatement can be cue- and context- induced, drug-induced, or stress-induced. Cue- and context-dependent craving for previously neutral stimuli stem from neurocircuitry changes in the ventral and dorsal striatums contribute to habit formation and incentive “wanting” directed toward drug-seeking. Cues associated with yearning also increase dopamine release in the prefrontal cortex and opioid peptide release in the frontal cortex. MORs in the nucleus accumbens core are necessary for the former while DORs in the nucleus accumbens shell are essential for the latter.
While opioids and their receptors have a foundational role in affecting neuronal transfiguration, genetic, developmental, and environmental factors may transform the neuronal circuitry as well. The contributory role of the latter components in addiction is expanded on later. Still, the apparent ubiquity of opioid receptor expression suggests these unique integrants have functions beyond modulating sensory perception of pain. In reality, these receptors have also been reported to affect feeding habit, brainwave activity, cognitive behavior, cardiovascular performance, neuroprotection and immune function. Still, the significance of these effects in cancer patients may not supersede the promiscuous role opioids (and their receptors) may have on the tumorigenic process.34,35 The relevance of the latter may offset some of the real-life benefits associated with the management of cancer pain. So salient is this matter that it raises the question whether this aspect of duality extends to opioid receptors, and if so, how strong is the evidence.
A Judicious Critique
Early, though inconsistent, data in animal models and humans suggested an unfavorable effect of opioids on cancers. Accordingly, numerous investigators reported that concurrent use of anesthesia and opioids during surgical resection of primary tumors involving the breast, prostate, colon and skin (melanoma) was negatively associated with disease control and survival.36–39 Even though material proof of the biological and molecular mechanisms to support study conclusions was not presented, corollary evidence providing some insight regarding the observed phenomena was forthcoming. Using murine- and human-derived non-small cell lung cancer (NSCLC) cell lines exposed to morphine, Singleton et al observed up to 10-fold greater intensity of MOR expression in tumor compared to control (ie, non-tumorous human lung) cells.40 Comparative in-vitro assays of the effect of morphine on MOR-bearing and MOR-poor Lewis Lung Carcinoma (LLC) cells indicated 50% to 70% decreases in proliferative and invasive capacities in the receptor-deficient subset. In-vivo, silencing of MOR expression (siRNA) or function (methylnaltrexone) in the LLC mouse model significantly reduced tumor size and lung metastases even in the absence of exogenous administration of opioids. That some of these findings may be clinically relevant are further supported by data from a follow up study of the same group. Analyses of archived samples of tissue obtained from 34 subjects with NSCLC demonstrated significantly increased expression of MORs in tumor compared to adjacent normal cells. In addition, opioid receptor expression was two-fold greater among patients with metastases than those without distant disease. Another group of investigators reported an inverse association between severity of pain/opioid dosage requirements and duration of survival in patients with NSCLC.41 Despite these complementary laboratory findings, a number of other retrospective human studies have conflicting results.
Interestingly, the apparent link between opioids and several cancer-related processes in NSCLC may not be connected exclusively with the MOR. Using the human-derived H2009 NSCLC cell line which overexpressed EGFR (epidermal growth factor receptor), as well as MORs and DORs, investigators from the University of Minnesota suggested that overexpression of these proteins introduced the possibility of receptor cross-talk.42 In point of fact, morphine and epidermal growth factor independently promoted phosphorylation of EGFR followed by time-dependent activation of downstream kinases including protein kinase B (Ak strain-transforming, Akt) and mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK). These findings not only coincided with ligand-induced phosphorylation of the kinases but also enhanced proliferation and invasiveness of tumor cells. Furthermore, the obtrusive manifestations of the cells could be significantly abrogated by naloxone, a non-selective opioid receptor antagonist, or erlotinib, an EGFR receptor tyrosine kinase inhibitor.
In-vitro and in-vivo laboratory studies also indicated reciprocal interactions between opioids and vascular endothelial growth factor (VEGF). While the latter is a mediator involved in embryogenesis and wound healing, VEGF also has a prominent role in pathological processes such as macular degeneration and tumorigenesis. However, the interplay between the two signaling pathways in tumor angiogenesis is not only extraordinarily complex but, at times, also confounding. Even though a causal relationship between opioids and angiogenesis has been demonstrated, the precise mechanism remains unclear. The apparent connectivity stems from the paracrine-like manner whereby VEGF promotes MOR expression in endothelial cells and morphine activates the VEGF-mediated angiogenic pathway.43 The question whether morphine’s vasculogenic effect is mediated through the MOR is not frivolous as logic does not always prevail. In their elegantly-designed laboratory studies using human dermal microvascular endothelial cells (HDMEC), Gupta and colleagues demonstrated that morphine, at normal serum concentrations found in humans, induced a number of physiological effects including MAPK phosphorylation, nitric oxide production, Akt activation, and cell-cycle progression.44 In addition to angiogenesis, tumor cell proliferation was also observed in morphine-treated mice transfected with an estrogen receptor-positive breast cancer cell line (MCF-7). Predictably, increased tumor volume closely paralleled tumor vascularization. The fact that MCF-7 cells do not express opioid receptors strongly suggests that the angiogenic process facilitated tumor growth and disease progression. One other key revelation related to naloxone which, unexpectedly, had no inhibitory effect on angiogenesis in the same MCF-7 xenograft mouse model. While these findings argue against the belief that angiogenesis (and tumor progression) are mediated via the MOR, antibodies directed against VEGF receptors 1 and 2 completely blocked these morphine-associated tumor-promoting effects.45
Even though much of the focus has been on the MOR, other classical opioid receptors may also be important. Based on previous reports of the presence of the DOR in lung and colon cancers, samples of surgically-resected human breast tumors and adjacent tissue were analyzed for tissue expression and clinical correlation of the DOR.46 Of the 62 subjects evaluated, 54 demonstrated high tumor levels of the DOR. Clinically, tumor DOR was correlated with node involvement at diagnosis, higher stage disease, and poorer overall survival. While data regarding opioid use were absent, it should be emphasized that none of the exogenous opioids have high affinity for the DOR though the latter characteristic does not exclude any negative impact of the opioids. In an attempt to explain the potential molecular mechanism(s) underlying the poor prognostic tumor features and cancer outcomes, the investigators selected two breast cancer cell lines (MCF-7 and SKBR-3) which exhibited high-level expression of the DOR. Incubation with DADLE ([D-Ala2, D-Leu5]-Enkephalin), a derivative of an endogenous DOR-specific agonist, resulted in phosphorylation of protein kinase C (PKC) and ERK, and proliferation of both cell lines. While the role of these signaling molecules is not conclusive evidence of their link to cancer prognosis and disease progression, activated PKC had been previously shown to stimulate the growth of breast cancer cells in-vitro.47 Furthermore, activated PKC induces ERK 1/2,48 which are reportedly involved in multiple tumorigenic processes.49
The previous findings were partially corroborated by another group of investigators.50 Analyses of DOR gene expression performed on clinically available tumor data sets indicated a strong association between increased receptor levels and tumor recurrence/cancer survival. For example, among breast cancer patients, extent of DOR mRNA was directly correlated with disease stage and progression; an accompanying, and consistent, finding was the receptor’s direct relationship with tumor grade. Further inspection of the data indicated DOR expression was also associated with poorer disease outcomes in patients with adenocarcinoma of the lung, head and neck carcinomas, ovarian and kidney cancers, and melanoma. More importantly, these investigators also provided compelling insight regarding the logistics and mechanics of the DOR in cancer metastases. Using a model that behaves clinically like human breast cancer, MDA-MB-23 cells were embedded in one of the mammary fat pads of fully immunocompromised or wild-type mice. The animals were then treated with either DADLE or saline (control) for 10 days. At necropsy, substantially more lung metastases were observed in DADLE-treated mice compared to control regardless of immune status. Concomitant ex-vivo studies demonstrated that tumor cells exposed to the modified native enkephalin exhibited significantly greater transwell cell movement without affecting the cell cycle or cell proliferation. While a selective DOR antagonist suppressed the locomotive activity suggests opioid-induced tumor cell migration is mediated through the receptor, this observation does not specify the intracellular mechanism. However, based on ample evidence supporting an association between epithelial-to-mesenchymal transition (EMT) and enhanced migratory and invasive characteristics, these same investigators focused their efforts on several critical oncogenic pathways that have been shown to promote EMT. In contrast to minimal upregulation of the β-Catenin/WNT-signaling pathways, JAK1-2/STAT3 were rapidly activated in DADLE-exposed tumor cells. Partially confirming the association between JAK/STAT and EMT was the observation that tumor cells cultured in an opioid-conditioned medium induced STAT-mediated transcription of SNAIL and SLUG. Protein products of the two genes are known repressors of E-cadherin, an important cell-adhesion molecule which impedes EMT. Additional proof that activation of JAK/STAT signaling contributes to cell migration was substantiated by co-administration of specific JAK1/2 or DOR inhibitors, which blocked the pro-migratory effects of the opioid. These findings were consistent with earlier reports demonstrating that activated STAT3 promotes the E to M transition process.51
While these data provide compelling arguments of opioid involvement in the oncogenic process and cancer outcomes, research findings have not always been uniform. The lack of consistency, especially in humans, may be partially attributable to data being collected retrospectively, uncertainty created when quantification of use (consumption) rely on opioid prescription records, and the confusing effects of tumor burden on pain, opioid use, and disease outcomes. Furthermore, opioids such as methadone and fentanyl may have anti-tumor effects in certain cancers. For example, methadone’s anti-cancer effect appears to be mediated by down-regulating anti-apoptotic proteins52 or enhancing the efficacy of chemotherapy.53 Even fentanyl has been reported to suppress tumor growth, an effect that may be mediated, in part, by inhibition of angiogenesis.54
In addition, pain may independently affect tumor growth by stimulating the release of native endorphins which can bind peripheral MORs as well as interact with other oncogenic pathways such as EGFR and VEGFR. Hence, in the presence, or even absence, of exogenous opioids uncontrolled pain could impact certain cancers. Of relevance also is the finding that pain may mediate surgery-associated immune suppression by altering the balance of helper T cells subsets,55 decreasing natural killer cell activity,56 and suppressing cell-mediated immunity.57 Parenthetically, pain-effected immunosuppression can be intensified by opioids. Numerous publications provide evidence that activation of opioid receptors disrupts elements of innate immunity (ie, reduced number and phagocytic activity of macrophages) and adaptive immunity (ie, decreased ratios of Th1/Th2 and CD4+/CD8+ cells; decreased antibody production).58,59 Pain also affects cell migration through cyclooxygenase 2 (COX2), a critical mediator of inflammation. Prostaglandins E2 and F2α, end-products of the inducible COX2 pathway, upregulate matrix metalloproteinase 1 (MMP1).60 And MMP1-mediated degradation of adhesion molecules has been shown to create gaps in tight junctions thus impinging on the integrity of blood barriers and facilitating metastasis to the brain.61 These confounding manifestations do not negate the plausible assertion that a correlation between opioids and poorer cancer outcomes exists though a causal relationship has not yet been proven.
A Tenable Consideration
Still, even in the absence of definitive conclusions research in humans unequivocally indicates that opioid receptors, particularly the MOR, are associated with the development of pharmacologic, physiologic, and neuropsychiatric consequences. While the context of tolerance, dependence, and addiction has been studied in animal models, these effects are particularly important in humans. Although the basal mechanisms appear to be known, one other notable aspect is whether these negative adaptive phenomena could also be manifested, at a cellular level, by malignant tumors. Despite what would be considered as inconceivable, it could be within the realm of possibility.
Tumor Tolerance
The emergence of tolerance following chronic opioid exposure has been associated with decreased numbers of receptors, aberrant receptor coupling, and/or alterations of effector-mediated signaling through the receptor. Because opioid receptor expression has been established in various types of tumors and demonstrated to enhance the malignant phenotype, the question whether tolerance can be manifested, at least mechanistically, by tumors cells is not a rhetorical one.
While the development of tolerance to analgesia as well as some common side effects including nausea, vomiting, and sedation is believed to be a centrally-mediated phenomenon, not all opioid-associated tolerance appear to be linked solely to central neuronal adaptation. An example of the latter relates to peripheral tissue such as the gastrointestinal tract, which at first glance appears aimless because of the claim that material tolerance does not occur in the bowels. Regardless, this assertion may, surprisingly, not be entirely accurate. Despite engagement of processes that actuate desensitization, studies in murine models showed that tolerance to the effects of morphine does indeed occur in the ileum but not the colon.62 Here, the mechanisms contributing to these disparate phenomena appear to be localized to opioid receptor-expressing neurons of the enteric nervous system.63 Recall that cellular tolerance is partially contingent on β-arrestin-2-dependent MOR desensitization and internalization. These processes, however, differ between small and large intestines.64 In contrast to β-arrestin-2 degradation in ileal epithelial cells chronically exposed to morphine (tolerance develops), arrestin in the large bowel is rapidly recycled to the plasma membrane (no tolerance). That tolerance could be induced in colonic tissue of β-arrestin-2 knockout mice suggests a critical role for this particular protein, which is expressed extensively in human tissue.65
Discouragingly, if exogenous opioids do encroach on the malignant process, the absence of tolerance (at least in the large bowel) raises the possibility that these compounds could promote tumor growth without the need for increased dosages. Yet an even more unfavorable consequence relates to the mechanism underlying the preservation of gastrointestinal immotility. In this framework, conserved β-arrestin-2 binds to, and facilitates trafficking of, the phosphorylated (desensitized) receptor to the endosome. Intra-endosomic-signaling causes receptor dephosphorylation and arrestin dissociation which prompts recycling of the protein to the cell membrane and, unfortunately, renewed opioid stimulation.66
Tumor Dependence
If opioid tolerance can be manifested or, perhaps even worse, attenuated, it is plausible to ask whether signature signs consistent with dependence could also be demonstrated in tumor cells. Traditionally, (opioid) dependence refers to the association whereby drug discontinuation effects the onset of physical (and psychological) withdrawal symptoms. Even though both types of symptoms will not be perceptible, tumors may, nevertheless, exhibit cellular mechanics of the withdrawal backwash.
Perhaps even more striking, index signs of withdrawal are systematically characterized by compensatory upregulation or rebound activation of key regulatory components. Notably, cAMP and GABA may not be the only “super-activated” messengers. In fact, abstinence-induced reversal of opioid-induced inhibition of enteric Ca++ channels result in acetylcholine, vasoactive intestinal polypeptide, and nitric oxide release and consequent withdrawal-associated diarrhea. In addition, precipitation of opioid withdrawal jerking and jumping in morphine-dependent mice has been shown to be related to excitation of central neurons. Despite the prominent role of the MOR, the involvement of DOR, therefore, cannot be totally excluded.67–69
These data highlight at least three possibilities. First, different aspects of the withdrawal syndrome such as agitated jumping and unconstrained diarrhea may not be mediated solely through the central nervous system but may involve the autonomic nervous system as well; second, dependence and withdrawal may not be mediated exclusively through the MOR; and third, adaptive cellular processes may be a crucial cell survival mechanism that, nonetheless, contributes to the development of tolerance and dependence. The latter has, perhaps, the most relevance to cancer and is highlighted as follows. Even though coupled to two cation channels, morphine-bound MORs activate one and inhibits the other. Despite the opposing reactions, the effect on gut motility (ie, constipation) are additive rather than antagonistic. Whereas, activation of K+ channels hyperpolarizes the membrane and neutralizes the action potential, inhibition of Ca++ channel function decreases neurotransmitter release.70,71 Consider another well described action of morphine regarding the effect on adenylate cyclase and consequent diminution of cAMP followed by restorative upregulation of the second messenger. What these examples highlight is that inhibitory and compensatory effects of chronic morphine exposure can generate both “loss-of-function” and “gain-of-function” phenotypes, arguably two of the most well accepted features of cancer cells. These concepts lead to addiction, the final and most devastating adverse effect of opioids and its possible connection to malignant tumors.
Tumor Addiction
Defining (substance) addiction is not as simple as it appears. Different from tolerance and dependence, addiction is either psychometrically vilified as a “voluntary, self-indulgent act in pursuit of pleasure” or pathologically classified as an “acquired disease of the brain”.72 These disparate perspectives, notwithstanding, current research increasingly supports the view that addiction is a manifestation of a brain disease. If so, presentation of reasoned evidence to support the notion that “addiction” occurring in tumors would be improbable. Still, a number of biological parallels or correlates inherent in the development of addiction and cancer are worthy of discussion.
Unquestionably, both addiction and cancer susceptibility have genetic connections; and known for many years, the most prevalent genetic variations found in the human genome are SNPs.73 A number of single nucleotide polymorphisms have been reported to be associated with addiction, while many others have been linked to various cancers. Because dopaminergic and opioid receptor-signaling either produce or mediate satisfaction or gratification, as well as pleasurable, stress-coping, and anxiety-calming effects, certain dopamine and opioid genes harboring specific SNPs may have significant roles in the development of addiction. Candidate genes include DRD2 (dopamine receptor D2) and OPRM1 (opioid receptor µ1). The relevance of the DRD2 receptor pathway is possibly related to induction of pleasure.74 In particular, the reference SNP (rs)1,800,497 polymorphism located downstream of DRD2 has been associated with increased heroin use and enhanced susceptibility to substance addiction among the most populous ethnic group in mainland China as well as Caucasians residing in Australia and the United States.75,76 Genetic alterations in the opioid signaling pathway may be equally, if not more, important than that of dopamine. One of the most frequently identified SNPs in OPRM1 is rs1799971, though ethnic variability regarding opioid dependence appears to exist. For example, a higher frequency of the reference SNP was found among opioid-dependent compared to control subjects from the Indian subcontinent.77 In contrast, other investigators found no significant differences in the frequency of the rs1799971 allele among non-Hispanic white, African-American, and Hispanic ethnic groups. However, analyses based on ethnicity alone indicated a higher frequency of the reference SNP in non-opioid dependent Hispanic subjects, suggesting the allele may impart a preventive effect.78
Although corollary findings regarding SNPs and cancer are too numerous to list, a review of the topic has been published.79 Still, it may be beneficial to highlight a few noteworthy single nucleotide aberrations. One of the most well accepted genetic variations is the breast and ovarian cancer type 1 (BRCA1) protein.80 Although characterized most frequently as a “tumor suppressor”, BRCA1 has a critical role in DNA synthesis and damage repair, as well as maintaining genomic integrity.81 Because of these “gatekeeper-like” responsibilities, it is conceivable that mutated BRCA1ʹs applicability may extend beyond susceptibility to breast and ovarian cancers. In particular, the rs799917 T>C associated with breast cancer risk82 has also been connected to gastric, esophageal, and possibly lung cancers.83–85
While epidemiological studies suggest that genetic factors contribute up to 50% of the risk of addiction, regulation of gene expression can be influenced in the absence of alterations in DNA sequence86 via inborn abnormalities of the epigenome. As it happens, growing emphasis has focused on how the epigenetic machinery alters gene expression.87 Included among the most important epigenetic mechanisms are DNA methylation, post-translational histone modifications, and chromatin re-composition.88,89 Furthermore, epigenetic changes can accumulate over time and translate to germline mutations. Notwithstanding the contribution of multiple interacting factors which converge to influence vulnerability, the MOR may have a pivotal role in the development of opioid addiction. The latter assumption is supported in both laboratory and clinical settings. In-vitro studies have shown that increased methylation of the OPRM1 promoter and reduction of histone deacetylase activity resulted in decreased gene transcription.90 Corollary findings have been observed in humans as well. From a total of 329 Caucasians, Nielsen et al found significantly higher levels of hypermethylated CpG sites in the MOR gene promoter and reduced gene expression among former heroin addicts compared to control subjects.91 These details reinforce the idea that epigenetic modifications in DNA can be induced by societal experiences and substances of abuse.
Data regarding the impact of epigenetics in cancer are even more profound. Not only have epigenetic changes been ascribed to each hallmark of cancer,92 but mutations in numerous epigenetic modifiers have been associated with specific cancers. For example, epigenetic alterations including methylation of the E cadherin promoter and the RASSFIA gene have been linked to two hallmarks of cancer, activating invasion and metastasis and sustaining proliferative signaling, respectively.93,94 Of note, because the DNA methyltransferase 1 (DNMT1) gene encodes an enzyme with a critical role in preserving the stability of the epigenome, hypermethylation of the gene has been associated with colorectal cancer.95
Though genetic and epigenetic factors increase susceptibility, heritable genomic variations (alone) do not ultimately result in addiction or cancer.86 Just as environmental factors such as early exposure, or easy access, to drugs, socially deviant surroundings, and poor family and social support systems can increase individual vulnerability to substance addiction, external factors may also contribute to the development of cancer. Recall the involvement of BRCA1 in DNA damage repair and the effect of one SNP (rs799917 CC) on reducing levels of the encoded protein. Conceivably, the negative effect of the SNP can be accentuated by cigarette smoking which can trigger DNA single and double strand breaks.96 Interestingly, termination of smoking for 30 days reportedly decreased strand breaks to levels observed in non-smokers.97 Helicobacter pylori is another external factor that has been shown to not only induce DNA strand breaks and compromise DNA repair mechanisms but also have a causal role in gastric cancer.98,99 That extrinsic factors are potentially modifiable suggest that altering components of the external milieu could reduce addiction and cancer risks.
Fittingly, a complementary view of dependence, tolerance, and addiction germane to cancer is put forward. In addition to the mechanistic workings previously discussed, genomic clues have led to the concept that the same three phenomena observed with opioids are also distinctly apparent in cancer. If one subscribes to Darwin’s theory of evolution, the process associated with the coined terms “survival of the fitter” is remarkably similar to the transformative development of cancer. Rather than leaving these evolutionary processes to providence, both are dependent on variability and heritability, two elements which are encoded in the human and tumor genomes. Well accepted also is the paradigm that most human cancers evolve over a relatively protracted period of time; time during which multiple genetic mutations or abnormalities not only accumulate but also endow tumor cells with growth and survival advantages. Yet in the face of considerable derangements inherent in the tumor genome, clinical findings demonstrate that targeted inhibition of a single aberrant gene can induce significant tumor cell death and improve cancer survival. Two (of many) examples supportive of the latter two outcomes include mutated EGFR in non-small cell lung cancer and overexpressed HER2 (ErbB2) in breast cancer.100–102 The presence of these molecular anomalies underscores the apparent “dependence” some cancers have on one gene to preserve the malfeasant phenotype. Moreover, subsequent alterations such as the T790M exon mutation in EGFR and truncated p95ErbB2 result in diminished activity of the original therapies. While the emergence of “tolerance” could be attributed to different oncogenes, inhibition of the same receptors with a different therapeutic agent can restore tumor response and achieve clinically meaningful improvement in overall survival.103,104 What these data indicate is the reliance some tumors have on one gene, a concept Weinstein et al have introspectively labeled “oncogene addiction”.105
One terminal musing relates to the potential lethality of addiction. As discussed in a recent publication, treatment of mutation-driven anaplastic lymphoma kinase (ALK)-large-cell lymphoma with a specific ALK inhibitor produced a paradoxical form of resistance characterized by overexpression of the targeted kinase.106 While chronic therapy promoted tumor cell resistance, drug withdrawal unexpectedly caused tumor cell death. However, instead of a mechanism confined to the ALK-signaling pathway, inhibitor addiction fomented a lethal cellular adaptation induced by super-activation of a tumor suppressive gene program. This exciting finding may have added relevance. If chronic opioid exposure can also elicit comparable adaptive changes in tumor cells, the development of a similar deadly phenotype breathes a modicum of life into explaining the lack of consistency regarding the effects of opioids in cancer.
A Lasting Relationship
Although the molecular mechanisms of opioid-induced analgesia have been largely elucidated, the same cannot be said concerning the development of tolerance, dependence, and addiction in humans; and even more so, the overall impact of these drugs on cancers themselves. Nonetheless, laboratory and clinical correlates suggest opioids may affect tumor “behavior” in two ways. Juxtaposed to the unresolved, though possibly detrimental, effect of opioids on disease outcomes is the perception that cancer cells can manifest signs of the drug-associated dysfunctional aftermath. The clinical implication of the former is the inability to provide patients with complete details of this adverse effect, information of such import that would contribute to making a fully informed decision regarding the use of opioids in the management of pain; and what the latter lacks in terms of clinical application may be supplanted by its instructive revelation.
Finally, the use of opioids in the management of cancer pain represents a covenant between supportive care and oncology. While the seemingly incompatible pairing of opioid-associated beneficial effects and drug-related repercussions is an enduring relationship, the coupling also evokes the nuptial sentiment “for better and for worse”.
Author Contributions
Both authors contributed equally to the intellectual pursuit, in conception, context design, and data acquisition, analysis and interpretation; engaged in drafting, revising and critically reviewing the manuscript; gave final approval of the version to be submitted; agreed on the journal to which the article has been submitted; and assented to accountability for all aspects of the work herein.
Disclosure
The authors do not have any relationship, financial or otherwise (ie, support in the form of employment, consultancies, honoraria, stock ownership and options, expert testimony, grants or patents received or pending, or royalties) with manufacturers of all agents described that influenced the writing of this manuscript.
References
1. Sullivan DR, Chan B, Lapidus JA, et al. Association of early palliative care use with survival and place of death among patients with advanced lung cancer receiving care in the Veterans Health Administration. JAMA Oncol. 2019;5(12):1702–1709. doi:10.1001/jamaoncol.2019.3105
2. Weiland G, Frisman D, Taylor P. Affinity labeling of the subunits of the membrane associated cholinergic receptor. Mol Pharmacol. 1979;15:213–226.
3. Chen Y, Mestek A, Liu J, Yu L. Molecular cloning of a rat kappa opioid receptor reveals sequence similarities to the mu and delta opioid receptors. Biochem. 1993;295(Pt3):625–628. doi:10.1042/bj2950625
4. Yasuda K, Raynor K, Kong H, et al. Cloning and functional comparison of kappa and delta opioid receptors from mouse brain. Proc Natl Acad Sci USA. 1993;90(14):6736–6740. doi:10.1073/pnas.90.14.6736
5. Vass M, Kooistra AJ, Yang D, Stevens RC, Wang MW, de Graaf C. Chemical diversity in the G protein-coupled receptor superfamily. Trends Pharmacol Sci. 2018;39(5):494–512. doi:10.1016/j.tips.2018.02.004
6. Mollereau C, Parmentier M, Mailleux P, et al. ORL1, a novel member of the opioid receptor family: cloning, functional expression and localization. FEBS Lett. 1994;341(1):33–38. doi:10.1016/0014-5793(94)80235-1
7. Zagon IS, Verderame MF, Allen SS, McLaughlin PJ. Cloning, sequencing, chromosomal location, and function of cDNAs encoding an opioid growth factor receptor (OGFr) in humans. Brain Res. 2000;856(1–2):75–83. doi:10.1016/S0006-8993(99)02330-6
8. Peng J, Sarkar S, Chang SL. Opioid receptor expression in human brain and peripheral tissues using absolute quantitative real-time RT-PCR. Drug Alcohol Depend. 2012;124(3):223–228. doi:10.1016/j.drugalcdep.2012.01.013
9. Valentino RJ, Volkow ND. Untangling the complexity of opioid receptor function. Neuropsychopharmacology. 2018;43(13):2514–2520. doi:10.1038/s41386-018-0225-3
10. Waldhoer M, Bartlett SE, Whistler JL. Opioid receptors. Annu Rev Biochem. 2004;73(1):953–990. doi:10.1146/annurev.biochem.73.011303.073940
11. Shang Y, Filizola M. Opioid receptors: structural and mechanistic insights into pharmacology and signaling. Eur J Pharmacol. 2015;763(Pt B):206–213. doi:10.1016/j.ejphar.2015.05.012
12. Gomes I, Sierra S, Lueptow L, et al. Biased signaling by endogenous opioid peptides. Proc Natl Acad Sci USA. 2020;117(21):11820–11828. doi:10.1073/pnas.2000712117
13. Toll L, Bruchas MR, Calo’ G, Cox BM, Zaveri NT. Nociceptin/orphanin FQ receptor structure, signaling, ligands, functions, and interactions with opioid systems. Pharmacol Rev. 2016;68(2):419–457. doi:10.1124/pr.114.009209
14. Manglik A. Molecular basis of opioid action: from structures to new leads. Biol Psych. 2020;87(1):6–14. doi:10.1016/j.biopsych.2019.08.028
15. Jang HS, Shin WJ, Lee JE, Do JT. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes. 2017;8(6):2–20. doi:10.3390/genes8060148
16. Abrimian A, Kraft T, Pan Y-X. Endogenous opioid peptides and alternatively spliced mu opioid receptor seven transmembrane carboxyl-terminal variants. Int J Mol Sci. 2021;22(7):343. doi:10.3390/ijms22073779
17. Zhang T, Xu J, Pan Y-X. A truncated six transmembrane splice variant mor-1g enhances expression of the full-length seven transmembrane μ-opioid receptor through heterodimerization. Mol Pharmacol. 2020;8(4):518–527. doi:10.1124/mol.120.119453
18. Jullié D, Gondin AB, von Zastrow M, Canals M. Opioid pharmacology under the microscope. Mol Pharmacol. 2020;98(4):425–432. doi:10.1124/mol.119.119321
19. Lu Z, Xu J, Rossi GC, Majumdar S, Pasternak GW, Pan Y-X. Mediation of opioid analgesia by a truncated 6-transmembrane GPCR. J Clin Invest. 2015;125(7):2626–2630. doi:10.1172/JCI81070
20. Pasternak GW, Pan Y-X. Mu opioids and their receptors: evolution of a concept. Pharmacol Rev. 2013;65(4):1257–1317. doi:10.1124/pr.112.007138
21. Mace G, Miaczynska M, Zerial M, Nebreda AR. Phosphorylation of EEA1 by p38 MAP kinase regulates μ opioid receptor endocytosis. EMBO J. 2005;24(18):3235–3246. doi:10.1038/sj.emboj.7600799
22. Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308(5721):512–517. doi:10.1126/science.1109237
23. Ferguson SS, Downey WEIII, Colapietro AM, Barak LS, Menard L, Caron MG. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science. 1996;271(5247):363–366. doi:10.1126/science.271.5247.363
24. Celver J, Xu M, Jin W, Lowe J, Chavkin C. Distinct domains on the μ-opioid receptor control uncoupling and internalization. Mol Pharmacol. 2004;65(3):528–537. doi:10.1124/mol.65.3.528
25. Whistler JL. Functional dissociation of mu opioid receptor- signaling and endocytosis: implications for the biology of tolerance and addiction. Neuron. 1999;23(4):737–746. doi:10.1016/s0896-6273(01)80032-5
26. Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24(2):97–129. doi:10.1016/S0893-133X(00)00195-0
27. Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81(1):299–343. doi:10.1152/physrev.2001.81.1.299
28. Whistler JL, von Zastrow M. Morphine-activated opioid receptors elude desensitization by beta-arrestin. Proc Natl Acad Sci USA. 1998;95(17):9914–9919. doi:10.1073/pnas.95.17.9914
29. Duttaroy A, Yoburn BC. The effect of intrinsic efficacy on opioid tolerance. Anesthesiology. 1995;82(5):1226–1236. doi:10.1097/00000542-199505000-00018
30. Yuan L, Luo L, Ma X, et al. Chronic morphine induces cyclic adenosine monophosphate formation and hyperpolarization-activated cyclic nucleotide-gated channel expression in the spinal cord of mice. Neuropharmacology. 2020;176:108222. doi:10.1016/j.neuropharm.2020.108222
31. Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17(2):796–803. doi:10.1523/JNEUROSCI.17-02-00796.1997
32. Finn AK, Whistler JL. Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron. 2001;32(5):829–839. doi:10.1016/s0896-6273(01)00517-7
33. Koob GF, Simon EF. The neurobiology of addiction: where we have been and where we are going. J Drug Issues. 2009;39(1):115–132. doi:10.1177/002204260903900110
34. Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14(5):381–395. doi:10.1016/S0898-6568(01)00271-6
35. Sun Y, Liu W-Z, Liu T, Feng X, Yang N, Zhou H-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct. 2015;35(6):600–604. doi:10.3109/10799893.2015.1030412
36. Biki B, Mascha E, Moriarty DC, Fitzpatrick JM, Sessler DI, Buggy DJ. Anesthetic technique for radical prostatectomy surgery affects cancer recurrence: a retrospective analysis. Anesthesiology. 2008;109(2):180–187. doi:10.1097/ALN.0b013e31817f5b73
37. Exadaktylos AK, Buggy DJ, Moriarty DC, Mascha E, Sessler DI. Can anesthetic technique for primary breast cancer surgery affect recurrence or metastasis? Anesthesiology. 2006;105(4):660–664. doi:10.1037/a0030561
38. Christopherson R, James KE, Tableman M, Marshall P, Johnson FE. Long-term survival after colon cancer surgery: a variation associated with choice of anesthesia. Anesth Analg. 2008;107(1):325–332. doi:10.1213/ane.0b013e3181770f55
39. Schlagenhauff B, Ellwanger U, Breuninger H, Stroebel W, Rassner G, Garbe C. Prognostic impact of the type of anaesthesia used during the excision of primary cutaneous melanoma. Melanoma Res. 2000;10(2):165–169. doi:10.1097/00008390-200004000-00009
40. Mathew B, Lennon FE, Siegler JH, et al. Novel role of the mu opioid receptor in lung cancer progression: a laboratory study. Anesth Analg. 2011;112(3):558–567. doi:10.1213/ANE.0b013e31820568af
41. Zylla D, Kuskowski MA, Gupta K, Gupta P. Association of opioid requirement and cancer pain with survival in advanced non-small cell lung cancer. Br J Anaesth. 2014;113(suppl 1):i109–116. doi:10.1093/bja/aeu351
42. Fujioka N, Nguyen J, Chen C, et al. Morphine-induced epidermal growth factor pathway activation in non-small cell lung cancer. Anesth Analg. 2011;113(6):1353–1364. doi:10.1213/ANE.0b013e318232b35a
43. Chen C, Farooqui M, Gupta K. Morphine stimulates vascular endothelial growth factor-like signaling in mouse retinal endothelial cells. Curr Neurovasc Res. 2006;3(3):171–180. doi:10.2174/156720206778018767
44. Gupta K, Kshirsagar S, Chang L, et al. Morphine stimulates angiogenesis by activating proangiogenic and survival-promoting signaling and promotes breast tumor growth. Cancer Res. 2002;62(15):4491–4498.
45. Singleton PA, Lingen M, Fekete M, Garcia J, Moss J. Methylnaltrexone inhibits opiate and VEGF-induced angiogenesis: role of receptor transactivation. Microvasc Res. 2006;72(1–2):3–11. doi:10.1016/j.mvr.2006.04.004
46. Wei Y, Zhang B, Li X, et al. Upregulation and activation of δ–opioid receptors promote the progression of human breast cancer. Oncol Rep. 2016;36(5):2579–2586. doi:10.3892/or.2016.5109
47. Oskoueian E, Abdullah N, Ahmad S. Phorbol esters from Jatropha meal triggered apoptosis, activated PKC‑δ, caspase‑3 proteins and down‑regulated the proto‑oncogenes in MCF-7 and HeLa cancer cell lines. Molecules. 2012;17(9):10816–10830. doi:10.3390/molecules170910816
48. Zhu M, Li M, Yang F, et al. Mitochondrial ERK plays a key role in δ‑opioid receptor neuroprotection against acute mitochondrial dysfunction. Neurochem Int. 2011;59(6):
49. Sancho P, Galeano E, Estañ MC, Ganan‑Gomez I, Boyano‑Adanez MC, Garcia‑Perez AI. Raf/MEK/ERK signaling inhibition enhances the ability of dequalinium to induce apoptosis in the human leukemic cell line K562. Exp Biol Med. 2012;237(8):933–942. doi:10.1258/ebm.2012.011423
50. Tripolt S, Neubauer HA, Knab VM, et al. Opioids drive breast cancer metastasis through the δ-opioid receptor and oncogenic STAT3. Neoplasia. 2021;23(2):270–279. doi:10.1016/j.neo.2020.12.011
51. Wendt MK, Balanis N, Carlin CR, Schiemann WP. STAT3 and epithelial-mesenchymal transitions in carcinomas. JAK-STAT. 2014;3(1):e28975. doi:10.4161/jkst.28975
52. Friesen C, Roscher M, Alt A, Miltner E. Methadone, commonly used as maintenance medication for outpatient treatment of opioid dependence, kills leukemia cells and overcomes chemoresistance. Cancer Res. 2008;68(15):6059–6064. doi:10.1158/0008-5472.CAN-08-1227
53. Friesen C, Hormann I, Roscher M, et al. Opioid receptor activation triggering downregulation of cAMP improves effectiveness of anti-cancer drugs in treatment of glioblastoma. Cell Cycle. 2014;13(10):1560–1570. doi:10.4161/cc.28493
54. He G, Li LI, Guan E, Chen J, Qin YI, Xie Y. Fentanyl inhibits the progression of human gastric carcinoma MGC-803 cells by modulating NF-kappaB-dependent gene expression in vivo. Onc Lett. 2016;12(1):563–571. doi:10.3892/ol.2016.4619
55. Decker D, Schöndorf M, Bidlingmaier F, et al. Surgical stress induces a shift in the type-1/type-2 T-helper cell balance, suggesting down-regulation of cell-mediated and up-regulation of antibody-mediated immunity commensurate to trauma. Surgery. 1996;119(3):316–325. doi:10.1016/s0039-6060(96)80118-8
56. Kutza J, Gratz I, Afshar M, et al. The effects of general anesthesia and surgery on basal and interferon stimulated natural killer cell activity of humans. Anesth Analg. 1997;85(4):918–923. doi:10.1097/00000539-199710000-00037
57. Riboli EB, Terrizzi A, Arnulfo G, et al. Immunosuppressive effect of surgery evaluated by the multitest cell-mediated immunity system. Can J Surg. 1984;27(1):60–63.
58. Filipczak-Bryniarska I, Nowak B, Sikora E, et al. The influence of opioids on the humoral and cell-mediated immune responses in mice. The role of macrophages. Pharmacol Rep. 2012;64(5):1200–1215. doi:10.1016/S1734-1140(12)70916-7
59. Garcia JB, Cardoso MG, Dos-Santos MC. Opioids and the immune system: clinical relevance. Rev Bras Anesthesiol. 2012;62(5):709–718. doi:10.1016/S0034-7094(12)70169-1
60. Nørregaard R, Kwon TH, Frøkiær J. Physiology and pathophysiology of cyclooxygenase-2 and prostaglandin E2 in the kidney. Kidney Res Clin Pract. 2015;34(4):194–200. doi:10.1016/j.krcp.2015.10.004
61. Lee KY, Kim YJ, Yoo H, Lee SH, Park JB, Kim HJ. Human brain endothelial cell-derived COX-2 facilitates extravasation of breast cancer cells across the blood-brain barrier. Anticancer Res. 2011;31(12):4307–4313.
62. Ross G, Gabra B, Dewey W, Akbarali H. Morphine tolerance in the mouse ileum and colon. J Pharmacol Exp Ther. 2008;327(2):561–572. doi:10.1124/jpet.108.143438
63. Nelson AD, Camilleri M. Chronic opioid induced constipation in patients with nonmalignant pain: challenges and opportunities. Therap Adv Gastroenterol. 2015;8(4):206–220. doi:10.1177/1756283X15578608
64. Galligan J, Akbarali H. Molecular physiology of enteric opioid receptors. Am J Gastroenterol Suppl. 2014;2(1):17–21.
65. Kang M, Maguma HT, Smith TH, Ross GR, Dewey WL, Akbarali HI. The role of β-arrestin2 in the mechanism of morphine tolerance in the mouse and Guinea pig gastrointestinal tract. J Pharmacol Exp Ther. 2012;340(3):567–576. doi:10.1124/jpet.111.186320
66. Claing A, Laporte SA, Caron MG, et al. Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and beta-arrestin proteins. Prog Neurobiol. 2002;66(2):61–79. doi:10.1016/s0301-0082(01)00023-5
67. Sofuoglu M, Portoghese PS, Takamori AE. Differential antagonism of delta opioid agonists by naltrindole and its benzofuran analog (NTB) in mice: evidence for delta opioid receptor subtypes. J Pharmacol Exp Ther. 1991;257(2):676–680.
68. Miyamoto Y, Portoghese PS, Takemori AE. Involvement of delta 2 opioid receptors in the development of morphine dependence in mice. J Pharmacol Exp Ther. 1993;264(3):1141–1145.
69. Ananthan S. Opioid ligands with mixed mu/delta opioid receptor interactions: an emerging approach to novel analgesics. AAPS J. 2006;8(1):E118–125. doi:10.1208/aapsj080114
70. North RA, Williams JT, Surprenant A, et al. Mu and delta receptors belong to a family of receptors that are coupled to potassium channels. Proc Natl Acad Sci USA. 1987;84(15):5487–5491. doi:10.1073/pnas.84.15.5487
71. Surprenant A, Shen KZ, North RA, et al. Inhibition of calcium currents by noradrenaline, somatostatin and opioids in Guinea-pig submucosal neurones. J Physiol. 1990;431:585–608. doi:10.1113/jphysiol.1990.sp018349
72. Volkow ND, Koob GF, McLellan AT. Neurobiologic advances from the brain disease model of addiction. N Engl J Med. 2016;374(4):363–371. doi:10.1056/NEJMra1511480
73. Collins FS, Brooks LD, Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998;8(12):1229–1231. doi:10.1101/gr.8.12.1229
74. Volkow ND, Wang GJ, Fowler JS, et al. Brain DA D2 receptors predict reinforcing effects of stimulants in humans: replication study. Synapse. 2002;46(2):79–82. doi:10.1002/syn.10137
75. Hou QF, Li SB. Potential association of DRD2 and DAT1 genetic variation with heroin dependence. Neurosci Lett. 2009;464(2):127–130. doi:10.1016/j.neulet.2009.08.004
76. Lawford BR, Young RM, Noble EP, et al. The D(2) dopamine receptor A(1) allele and opioid dependence: association with heroin use and response to methadone treatment. Am J Med Genet. 2000;96(5):592–598. doi:10.1002/1096-8628(20001009)96:5<592:aid-ajmg3>3.0.co;2-y
77. Kapur S, Sharad S, Singh RA, Gupta AK. A118g polymorphism in mu opioid receptor gene (oprm1): association with opiate addiction in subjects of Indian origin. J Integr Neurosci. 2007;6(4):511–522. doi:10.1142/s0219635207001635
78. Bond C, LaForge KS, Tian M, et al. Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: possible implications for opiate addiction. Proc Natl Acad Sci USA. 1998;95(16):9608–9613. doi:10.1073/pnas.95.16.9608
79. Deng N, Zhou H, Fan H, Yuan Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget. 2017;8(66):110635–110649. doi:10.18632/oncotarget.22372
80. Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71. doi:10.1126/science.7545954
81. Huen MSY, Sy SMH, Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol. 2010;11(2):138–148. doi:10.1038/nrm2831
82. Nicoloso MS, Sun H, Spizzo R, et al. Single-nucleotide polymorphisms inside microRNA target sites influence tumor susceptibility. Cancer Res. 2010;70(7):2789–2798. doi:10.1158/0008-5472.CAN-09-3541
83. Wang K, Xu L, Pan L, Xu K, Li G. The functional BRCA1 rs799917 genetic polymorphism is associated with gastric cancer risk in a Chinese Han population. Tumour Biol. 2015;36(1):393–397. doi:10.1007/s13277-014-2655-9
84. Zhang X, Wei J, Zhou L, et al. Functional BRCA1 coding sequence genetic variant contributes to risk of esophageal squamous cell carcinoma. Carcinogenesis. 2013;34(10):2309–2313. doi:10.1093/carcin/bgt213
85. Liu D, Gao Y, Li L, et al. Single nucleotide polymorphisms in breast cancer susceptibility gene 1 are associated with susceptibility to lung cancer. Oncol Lett. 2021;21(5):424. doi:10.3892/ol.2021.12685
86. Kendler KS, Prescott CA, Myers J, Neale MC. The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Arch Gen Psychiatry. 2003;60(9):929–937. doi:10.1001/archpsyc.60.9.929
87. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11(10):726–734. doi:10.1038/nrc3130
88. Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153(1):38–55. doi:10.1016/j.cell.2013.03.008
89. Maze I, Nestler EJ. The epigenetic landscape of addiction. Ann NY Acad Sci. 2011;1216:99–113. doi:10.1111/j.1749-6632.2010.05893.x
90. Hwang CK, Song KY, Kim CS, et al. Evidence of endogenous mu opioid receptor regulation by epigenetic control of the promoters. Mol Cell Biol. 2007;27(13):4720–4736. doi:10.1128/MCB.00073-07
91. Nielsen DA, Yuferov V, Hamon S, et al. Increased OPRM1 DNA methylation in lymphocytes of methadone-maintained former heroin addicts. Neuropsychopharmacology. 2009;34(4):867–873. doi:10.1038/npp.2008
92. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
93. Lombaerts M, van Wezel T, Philippo K, et al. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br J Cancer. 2006;94(5):661–671. doi:10.1038/sj.bjc.6602996
94. Pfeifer GP, Yoon J-H, Liu L, Tommasi S, Wilczynski SP, Dammann R. Methylation of the RASSF1A gene in human cancers. Biol Chem. 2002;383(6):907–914. doi:10.1515/BC.2002.097
95. Kanai Y, Ushijima S, Nakanishi Y, Sakamoto M, Hirohashi S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett. 2003;192(1):75–82. doi:10.1016/s0304-3835(02)00689-4
96. Nakayama T, Kaneko M, Kodama M, Nagata C. Cigarette smoke induces DNA single-strand breaks in human cells. Nature. 1985;314(6010):462–464. doi:10.1038/314462a0
97. Ishida M, Ishida T, Tashiro S, et al. Smoking cessation reverses DNA double-strand breaks in human mononuclear cells. PLoS One. 2014;9(8):e103993. doi:10.1371/journal.pone.0103993
98. Toller IM, Neelsen KJ, Steger M, et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. PNAS. 2011;108(36):14944–14949. doi:10.1073/pnas.1100959108
99. Kim JJ, Tao H, Carloni E, Leung WK, Graham DY, Sepulvedaal AR. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology. 2002;123(2):542–553. doi:10.1053/gast.2002.34751
100. Soria J-C, Mok TS, Cappuzzo F, Jänne PA. EGFR-mutated oncogene-addicted non-small cell lung cancer: current trends and future prospects. Cancer Treat Rev. 2012;38(5):416–430. doi:10.1016/j.ctrv.2011.10.003
101. O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004. doi:10.1056/NEJMoa022457
102. Carey LA. HER2-a good addiction. Nat Rev Clin Oncol. 2012;9(4):196–197. doi:10.1038/nrclinonc.2012.36
103. Ahn M-J, Tsai C-M, Shepherd FA, et al. Osimertinib in patients with T790M mutation-positive, advanced non–small cell lung cancer: long-term follow-up from a pooled analysis of 2 Phase 2 studies. Cancer. 2019;125(6):892–901. doi:10.1002/cncr.31891
104. Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–2743. doi:10.1056/NEJMoa064320
105. Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68(9):3077–3080. doi:10.1158/0008-5472.CAN-07-3293
106. Rajan SS, Amin AD, Li L, et al. The mechanism of cancer drug addiction in ALK-positive T-Cell lymphoma. Oncogene. 2020;39(10):2103–2117. doi:10.1038/s41388-019-1136-4
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.