")
Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 4
Splenic marginal zone lymphoma: a review of the clinical presentation, pathology, molecular biology, and management
Authors Sena Teixeira Mendes L, Du M, Matutes E, Wotherspoon A
Received 3 April 2014
Accepted for publication 13 May 2014
Published 17 July 2014 Volume 2014:4 Pages 29—38
DOI https://doi.org/10.2147/BLCTT.S49373
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Larissa Sena Teixeira Mendes,1 Ming-Qing Du,2 Estella Matutes,3 Andrew Wotherspoon1
1Histopathology Department, Royal Marsden Hospital, London, UK; 2Molecular Malignancy Laboratory and Department of Histopathology, University Hospital NHS Foundation Trust/Division of Molecular Histopathology, Department of Pathology, University of Cambridge, Cambridge, UK; 3Hematopathology Unit, Hospital Clinic, Barcelona University, Barcelona, Spain
Abstract: Splenic marginal zone lymphoma is a distinct low grade B-cell lymphoma primarily occurring in the spleen and separate from nodal marginal zone lymphoma and extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue. It is characterized by a relative indolent course, splenomegaly, moderate lymphocytosis, and an intrasinusoidal pattern of involvement, especially in the bone marrow. It is postulated that the neoplastic clone originates from persistent antigenic stimulation of marginal zone B-cells. Molecular and cytogenetic studies have failed to show specific alterations. There is no standard criterion to initiate treatment, which may include a watch and wait policy, splenectomy, or chemo/immunotherapy. This review highlights the main features of this entity, reassessing the guidelines for diagnosis, prognostic factors, staging, and management published by the SMZL Working Group (2008).
Keywords: splenectomy, villous lymphocytes, guidelines
Introduction
The term splenic marginal zone lymphoma (SMZL) was first used by Schmid et al1 to describe a pathological entity seen in patients who presented with a leukemic blood picture that had been recognized by hematologists as having circulating villous lymphocytes. Although it is now acknowledged that not all patients with splenomegaly and circulating villous lymphocytes will have SMZL and not all SMZL patients will have circulating villous lymphocytes, there is considerable overlap between what had been termed splenic lymphoma with villous lymphocytes and SMZL. While there are some morphological similarities and genetic overlap, in the current World Health Organization classification of lymphoid malignancies, SMZL is recognized as a distinct clinicopathologic entity separate from nodal marginal zone lymphoma and extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue.1,2 SMZL is a rare entity, accounting for about 2% of all lymphoid malignancies, but it is the most common primary splenic lymphoma. Several reports have accumulated reliable information on the clinical presentation and disease course of patients with this particular entity, but the cell of origin remains unclear.2–4 The pathogenetic mechanisms remain hypothetical, but some features suggest that a chronic persistent antigenic stimulus may have an initial role in the clonal B-cell expansion.5
In this review we outline the main features of SMZL, including clinical presentation, morphology, pathology, and molecular genetics.
Epidemiology and etiology
SMZL is a rare hematological malignancy, often with a chronic clinical course, affecting mainly elderly people with a median age of 65 years at diagnosis and no incidence differences between sexes.2
Chronic antigenic stimulus is thought to be the initiating event in the pathogenesis of this lymphoma. Murakami et al reported a review of primary splenic lymphoma showing a high prevalence of chronic liver disease among these cases and the hepatitis C virus (HCV) in 36%.6 Since then, several reports have suggested that HCV presence is more than a casual association with SMZL. HCV antigen epitope appears to have a role in B-cell selection, stimulating a proliferating pool of naïve B-cells with altered growth and apoptosis mechanisms.5–8 Areas highly endemic for HCV, such as Japan, Italy, and southern parts of the United States, show a more frequent association with marginal zone lymphomas.9,10
At the molecular level, SMZL shows a significant heterogeneity regarding IgVH gene mutations with biased use of selected VH1 genes (VH1.2), which suggests a role of unknown antigens stimulating the neoplastic proliferation.4,11,12
Hyperreactive malarial splenomegaly in West Africa is demographically, clinically, and immunologically very similar to SMZL. The malaria parasite is also thought to have a role as a chronic antigenic stimulus in lymphoproliferation by altering the immune response and promoting tumor cell growth.8,13
Clinical features
SMZL presents most commonly with abdominal discomfort due to moderate/massive splenomegaly or with symptoms associated with cytopenias. Some patients are asymptomatic, with an isolated lymphocytosis discovered on routine tests performed for unrelated reasons. This latter form may be regarded as an early manifestation of SMZL preceding overt disease.14–16 Cytopenias are more commonly related to hypersplenism than to bone marrow replacement. Moderate anemia is present in 65% of the cases, mainly related to hypersplenism and rarely to bone marrow infiltration or hemolysis. Thrombocytopenia, mild neutropenia, and small M-band (immunoglobulin [Ig]M or IgG) can be observed in up to 50% of patients.16,17 Autoimmune phenomena (primary biliary cirrhosis, rheumatoid arthritis, immune thrombocytopenia, autoimmune hemolytic anemia, etc,) are reported in about 20% of patients.18,19 Although bone marrow and splenic hilar lymph nodes are almost always involved, peripheral lymphadenopathy is infrequent. A few patients may have extranodal disease at presentation or during the evolution. Transformation to diffuse large B-cell lymphoma occurs in approximately 10% of the cases, and it should be suspected when B-symptoms and elevated lactate dehydrogenase (LDH) are found at presentation.2,19
Morphologic and immunophenotypic aspects
Macroscopically, the spleen is commonly enlarged, with a median weight of 1,750 g (270–5,500 g). The cut surface shows multiple small, grayish nodules throughout the parenchyma.5
Histologically, there is expansion of the white pulp by a population of neoplastic cells that surround and eventually replace the germinal centers (Figure 1A). These nodules are composed of an inner zone of small- to medium-sized B-cells with round nuclei, clumped chromatin, and scant cytoplasm that in the early stages resemble mantles. External to this area is an outer zone comprising predominantly medium-sized cells with more irregular nuclear contours, dispersed chromatin, and moderate clear cytoplasm (Figure 1B). In this zone, there are scattered large cells with prominent nucleoli resembling immunoblasts.2,20 The overall appearances closely resemble the normal architecture of splenic white pulp. The initial recognition that the outer zone was neoplastic while the inner zone was considered reactive led to the assumption that this was a lymphoma derived from marginal zone B cells. It is now recognized that both inner and outer zones are neoplastic and have variable size (Figure 1C and D). As the disease progresses, the central reactive germinal centers become effaced and more difficult to identify. The red pulp is invariably involved to a variable degree by small aggregates of the larger cells and sheets of the small cells, which often occupy sinuses and cords (Figure 1E–G).2,3 Epithelioid granulomas can be seen within or in the vicinity of the infiltrating nodules. Plasmacytic differentiation can occur, but sheets of plasma cells are rare. This might be a more frequent finding in patients with a monoclonal serum component and autoimmune disorders.21
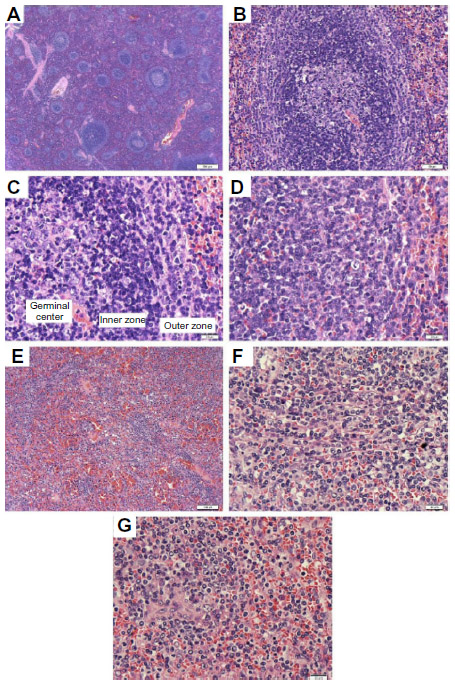 | Figure 1 Spleen H&E. |
The bone marrow is almost always infiltrated. There may be a nodular pattern with cell populations similar to that found in the spleen resembling the arrangement and composition of splenic white pulp nodules. Initially the involvement may be very subtle, corresponding to less than 20% of total marrow cellularity.19 The pattern of interstitial involvement characteristically includes an intrasinusoidal component with a single file arrangement in the smallest sinusoids more easily highlighted with CD20 staining (Figure 2), but more diffuse interstitial and paratrabecular nodular patterns might also be seen. Intrasinusoidal infiltration is more commonly found and evident in the early phases of the disease, while nodular aggregates are generally seen in more advanced disease.22 Intrasinusoidal disease is not specific to SMZL and may be seen in other lymphomas predominantly involving the spleen. Hematopoietic reserve may be expanded in case of hypersplenism.2,5
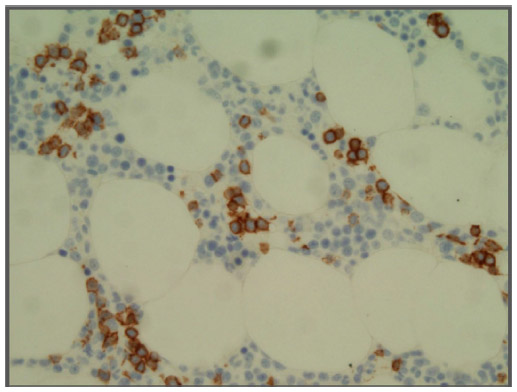 | Figure 2 Bone marrow. |
Splenic hilar lymph nodes are commonly infiltrated by small B-cells in a micronodular pattern, lacking the characteristic sinusoidal involvement, which might be explained by the expression of different adhesion molecules in this particular microenvironment. The sinuses are often spared and dilated. A marginal zone pattern may rarely be seen. Involvement of lymph nodes at other localizations is rarely observed and shows a similar pattern.2,5
The peripheral blood is less frequently affected than the bone marrow, but can be involved by a variable amount of circulating tumor cells, more commonly by small numbers. Cytologically, these lymphocytes present round nuclei with condensed chromatin and basophilic cytoplasm. Some of them show cytoplasmic projections at one or both poles of the cell (villous lymphocytes).9
Liver infiltration is reported in up to 90% of cases showing intrasinusoidal and nodular infiltrate in portal tracts.20,22–24 Other organs are rarely involved. Chacón et al reported that nonhematopoietic sites are affected in 6.6% of cases of SMZL.18 There are reports of dermal, pleural, thoracic, and perirenal soft tissue and meningeal involvement.25–29
Immunophenotypically, the neoplastic cells typically show positive staining for CD45, CD20 (Figure 3A and B), CD79a, PAX5, IgD (Figure 3C), and bcl-2, while there is variable positivity for DBA-44 (CD72), TRAP, and CD38.30 They may express CD11c and CD25, but are, as a rule, CD103 and CD123 negative. They are negative for CD3, CD5, CD10, bcl-6, CD23, CD43, cyclin-D1, and annexin-A1. Proliferation is usually low but shows a characteristic targetoid pattern, with higher proliferation in the outer zone, low proliferation in the inner small cell zone, and normal high proliferation in the central residual normal germinal center (Figure 3D). Residual germinal centers can be highlighted with CD10 and bcl-6 staining. Staining for CD38 or CD138 and expression of immunoglobulin more easily highlights plasmacytic differentiation. Light chain restriction can be demonstrated and is most evident in the plasma cell component. Transformation is associated with a higher proliferation index and increased expression of bcl-6 and MUM-1.2,22
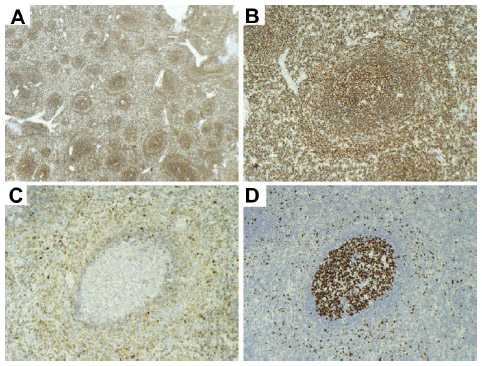 | Figure 3 Spleen immunohistochemistry. |
Diagnostic features
The establishment of SMZL diagnosis has spleen histology as the gold standard. When its assessment is not available (clinically unfit patients, or lack/mild splenomegaly), diagnosis relies on the combination of other laboratorial investigations, including analysis of lymphocyte morphology and immunophenotype and bone marrow histology. Cytogenetic/fluorescence in situ hybridization (FISH) studies may be contributory. Although SMZL does not have a ubiquitous recurrent chromosomal abnormality, cytogenetics/FISH can rule out translocations that are common to other B-cell lymphomas such as t(14;18) in follicular lymphoma and t(11;14) in mantle cell lymphoma.2,19 Diagnostic criteria consist of 1) spleen histology + immunophenotype with a chronic lymphocytic leukemia (CLL) score of 2 or less; or 2) typical blood findings with bone marrow morphology and immunophenotype (intrasinusoidal infiltration by CD20-positive cells) (Table 1).19
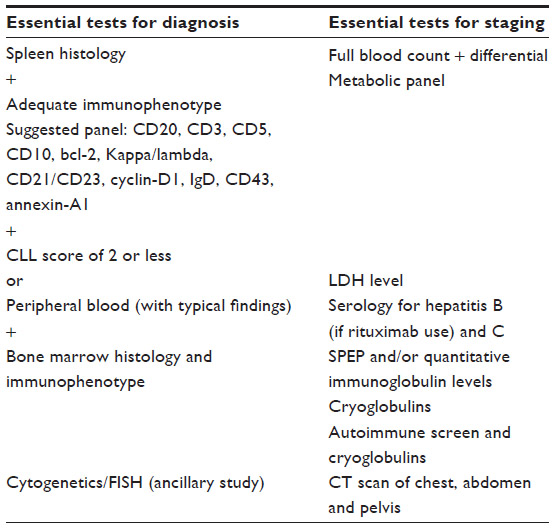 | Table 1 SMZL diagnosis and staging |
Tests that have an ancillary role in the staging of SMZL are full blood count/blood film with differentials and platelets; a comprehensive metabolic panel; LDH levels; liver biochemistry with testing for hepatitis B (if rituximab use is being considered) and C; computerized tomography scan of the chest, abdomen, and pelvis; bone marrow biopsy with or without aspirate; serum protein electrophoresis and/or quantitative immunoglobulin levels; beta2 microglobulin; autoimmune screen (direct antiglobulin test, rheumatoid factor, anti-DNA antibody, AMA (antimitochondrial antibody), and antithyroid antibodies); and cryoglobulins (Table 1).9,19
Differential diagnosis
Other small B-cell lymphomas must be excluded, since the micronodular pattern of splenic involvement, marginal zone differentiation, and the presence of villous lymphocytes in peripheral blood are not features restricted to SMZL. Immunohistochemistry is a valuable tool to correctly classify these entities.
B-cell chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) is composed of small B-cell lymphocytes, often admixed with prolymphocytes and paraimmunoblasts involving peripheral blood, bone marrow, lymph nodes, liver, and spleen. Morphologically, the proliferation rarely shows a marginal zone pattern in the spleen or intrasinusoidal pattern of infiltration in the bone marrow, and there is never true paratrabecular infiltration. Immunohistochemically, CLL/SLL is positive for CD5 and CD23 and shows lower expression of CD20.5,15
Hairy cell leukemia (HCL), although exhibiting some similar lymphocyte morphology to SMZL in the peripheral blood, can be easily discriminated based on the patchy bone marrow infiltration that replaces the normal hematopoietic tissue with increased reticulin fibrosis and the characteristic red pulp infiltration in the spleen with atrophied germinal centers, and expression of CD25, CD103, CD123, and annexin-A1.2,9
Mantle cell lymphoma, although clinically different because of the common peripheral lymphadenopathy, may occasionally present with minimal lymphadenopathy and massive splenomegaly with bone marrow involvement. Cytologically, however, the cells resemble centrocytes with irregular/cleaved nuclei and the characteristic immunophenotype that includes expression of CD5, cyclin-D1, and SOX-11.2,5
Distinction from lymphoplasmacytic lymphoma can be a difficult differential when lymphoplasmacytic differentiation is abundant in SMZL. A nodal biopsy and additional tests, including genetic analysis for MYD88 mutation might be needed.2,3
Distinction from hairy cell leukemia-variant (HCL-v) is usually straightforward when the spleen histology is available for review because they are quite different cytologically, with the cells from HCL-v cases showing prominent nucleoli. The differential from splenic diffuse red pulp small B-cell lymphoma (SDRPSBCL) may be difficult in the absence of splenic histology. The marrow infiltration in both HCL-v and SDRPSBCL is usually more strikingly intrasinusoidal. The cells of SDRPSBCL and SMZL can be very similar, with plasmacytoid nuclear features emphasizing the importance of splenic histology for an accurate diagnosis.2,5
Benign lymphoid hyperplasia associated to HCV and persistent polyclonal B-lymphocytosis are both reactive conditions that may mimic SMZL, but can be distinguished following ancillary laboratory studies and the demonstration of clonality in cases of SMZL.
Genetics and molecular biology
Antigenic stimulation in pathogenesis of SMZL
There are several strands of evidence suggesting a role of antigenic stimulation in the pathogenesis of SMZL. Approximately 20% of patients with SMZL present with autoimmune phenomena. A small proportion of cases, particularly those from Italy, are associated with HCV infection and could be effectively treated by antiviral therapy.10 Sequencing analysis of the rearranged immunoglobulin heavy chain variable gene (IGHV) demonstrated a biased usage of IGHV genes, with IGHV1-2, IGHV4-34, and IGHV3-23 accounting for 45% of cases.31,32 Most of the rearranged IGHV genes show somatic mutations, although at considerably variable frequencies. The IGHV1-2 rearrangements in SMZL are characterized by minimal somatic mutations and longer complementarities determining region 3 sequence with common motifs, suggesting possible selection by superantigens.31 Taken together, the above findings suggest a role of antigenic stimulation in the pathogenesis of SMZL.
Genetic basis of SMZL
Unlike other low grade B-cell lymphomas, SMZL lacks any specific and recurrent chromosome translocation, although CCND1-, CCND3-, BCL6-, MUM1-, or PAX5-involved translocations were reported in isolated cases of SMZL.33,34 Comparative genomic hybridization (CGH) analyses identify several recurrent chromosomal gains and losses in SMZL, with 7q deletion being preferentially associated with this lymphoma entity.
7q deletion in SMZL
A number of CGH studies including both early metaphase and, more recently, array-based analyses consistently showed recurrent gains of 3q25-ter (22%–32%), 6p (10%), 8q (8%–20%), 12q13–21 (~10%), and 18 (12%), and losses of 6q (8%–16%), 7q (18%–44%), 8p (~15%) and 17p (8%–14%).35–40 Among these recurrent genomic gains and losses, 7q deletion is the most frequent alteration. The minimal common region deleted in 7q has been mapped to a ~3Mb region at 7q32.1–32.2 by array CGH analyses.39,40 Extensive screening of the 7q32 deletion by interphase FISH analysis identified the deletion in 34% of SMZL and in ~33% of splenic B-cell lymphoma/leukemia unclassifiable, but not or rarely in other lymphomas involving the spleen, including CLL and HCL.39,40
Although an earlier study suggested an association between 7q32 deletion and unmutated IGHV status in SMZL,11 recent studies showed no correlation between 7q32 deletion and a range of genetic and clinicopathological parameters, including the IGHV mutation status.39,40 In addition, gene expression profiling analyses did not reveal any evidence, suggesting that SMZL with 7q deletion may constitute a distinct subgroup.41 These findings suggest that a common molecular mechanism may underscore the pathogenesis of SMZL with and without 7q32 deletion.
The genetic target(s) of 7q32 deletion in SMZL is unknown, despite the extensive research for potential tumor suppresser gene(s) at this genomic locus. There are no recurrent breakpoints or cryptic deletion, nor homozygous deletion in the minimal deleted region at 7q32 in SMZL.39,40 There are a number of genes in this region, including both coding genes and noncoding microRNAs, which could be the potential candidate genes targeted by the deletion. Sequencing of these candidate genes so far, such as IRF5, FAM71F1, FLNC, TNP03, AHCYL2, and CPA4, did not identify any recurrent pathogenic mutations.39,40 Nonetheless, the expression of many of these candidate genes was reduced in SMZL with 7q32 deletion in comparison with those without the deletion. It remains to be investigated whether a haplodeficiency of one or more of these candidate genes may have a role in the pathogenesis of SMZL. The target of 7q deletion in SMZL remains obscure.
Somatic mutation profiles in SMZL
To date, there are four studies that have investigated somatic mutation profile in a total of 36 cases of SMZL by whole exome or genome sequencing, and these genome mutation screenings have uncovered a wide range of potential pathogenic mutations.42–45 These mutation screenings, together with candidate gene mutation analyses, have shown that several signaling pathways and molecular processes are frequently targeted by pathogenic mutations in SMZL.
Mutation in the NOTCH pathway
The NOTCH family members are highly conserved cell surface receptor proteins that are fundamental for diverse organogenesis and cellular differentiation. Activation of NOTCH receptor by ligand binding initiates a cascade of proteolytic cleavages of NOTCH and liberates its intracellular domain, which translocates to the nucleus and activates transcription of downstream target genes.46 NOTCH2 is a master regulator of marginal zone B-cell development.47,48
NOTCH2 mutations are the most frequent somatic mutations found in SMZL, although at variable frequencies among different studies (6.5%–25%).42–45 Extensive screening of other low-grade B-cell lymphomas including CLL and HCL showed no or very rare NOTCH2 mutation, suggesting that NOTCH2 mutation appeared to be specific to SMZL.42,43 The vast majority of NOTCH2 mutations are frameshift indels or nonsense mutations, and clustered in a hotspot region upstream or within the C-terminal PEST domain that is important for regulating degradation of the NOTCH2 intracellular domain.42–45,49 These mutations would eliminate or truncate the C-terminal PEST domain and thus impair the degradation of the active NOTCH2 intracellular domain, potentially enhancing its function.42–45,49
In addition to NOTCH2, several other NOTCH signaling regulators including NOTCH1, SPEN, and DTX1 are also targeted by somatic mutations, but at much lower frequencies. Interestingly, mutations in these genes are largely mutually exclusive.42,43 There is also a tendency of a mutually exclusive pattern between the NOTCH and NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) pathway mutations.42
Mutation in the NF-κB pathway
Activation of NF-κB is normally transient and plays a critical role in lymphocyte development, activation, and differentiation, including the generation and/or maintenance of marginal zone B-cells. Mutations in both the canonical and noncanonical NF-κB pathways have been extensively investigated in other B-cell lymphoma characterized by constitutive NF-κB activities,50–52 and these findings have provided a framework for mutation analyses in SMZL.
Through largely candidate gene mutation analyses, several NF-κB regulators have been shown to be targeted by somatic mutations in SMZL. These included activating mutations in positive regulators such as CARD11 (~7%), MYD88 (~5%–13%), IKBKB (7%), and MAP3K14, and inactivating mutations in negative regulators such as TNFAIP3 (7%–13%), BIRC3 (6.3%) and TRAF3 (5%).42,53,54 These mutations are largely mutually exclusive,42,54 and most likely enhance NF-κB activity.
Mutation in epigenetic regulation
Apart from mutations seen in the NOTCH and NF-κB pathways, whole exome or genome sequencing has also identified mutations in several epigenetic regulators.42–45 They include MLL2 (a histone methyltransferase), ARID1A (a member of the SWI/SNF adenosine triphosphate-dependent chromatin-remodeling complexes), EP300 (histone and nonhistone acetyltransferases), SIN3A (a core component of the SIN3-HDAC1/2 histone deacetylase complex), and TBL1XR1 (an intrinsic component of the SMRT-N-CoR transcription corepressor machinery). The incidence of these epigenetic regulator mutations and their role in the pathogenesis of SMZL remain to be investigated. Nonetheless, these genes are also mutated in a range of hematological and solid tumors.
Although whole exome or genome sequencing identified a wide spectrum of mutations in SMZL, there are few common mutations identified among the four published studies.42–45 A high proportion of cases showed no clearly pathogenic mutations. It is highly likely that the genetic bases of SMZL are not yet fully unraveled.
Cell of origin
Non-neoplastic marginal zone cells are germinal center experienced B-cells with mutated IGHV chain genes.55 Intriguingly, a great number of SMZL cells are IgD positive, with a significant molecular heterogeneity related to IGHV mutations (almost 1/3 of cases lack somatic hypermutation).9 SMZL cells show an overuse of VH1-2 gene segment in mutated and unmutated cases, and some share almost indistinguishable stereotyped B-cell receptors. These features support the possibility of a highly selected subpopulation of B-cells by a common antigen.32
Clinical course and predictive factors
Most SMZL show a sustained uncomplicated evolution with a median survival around 10 years.15,16,18 Time to progression generally exceeds 5 years developing in about 10% of the cases.18 Deaths are most commonly unrelated to the lymphoma, occurring in the event of secondary neoplasm or vascular conditions.2,56
Factors associated with a poorer survival have been listed and can be divided in clinical and biological parameters. Several studies in multivariate analysis have proposed that older age, high tumor burden (massive splenomegaly) anemia, lymphocytosis, high LDH, low albumin level, failure to respond to treatment, and involvement of nonhematopoietic sites are predictive factors of a shorter survival.9,57 Arcaini et al16 proposed a prognostic model with three categories (low risk, no adverse factors; intermediate risk, one adverse factor; high risk, two to three adverse factors) based on the presence of the following adverse factors: hemoglobin <12 g/dL, elevated LDH, and albumin <35 g/dL. The 5-year survival reported was 88%, 73%, and 50%, respectively (Table 2).
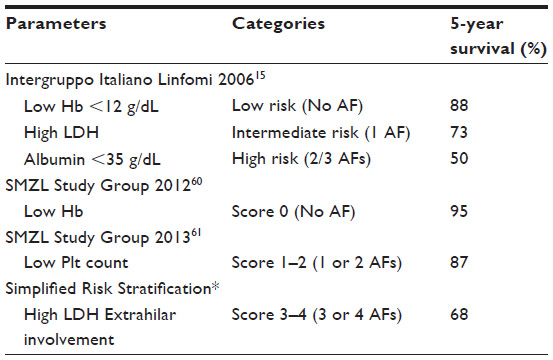 | Table 2 SMZL risk stratification for a shorter cause-specific survival |
The SMZL Working group58 also suggested a score based on parameters to discriminate lower survival (low hemoglobin, low platelet count, high LDH, and extrahilar lymphadenopathy). This stratification divided the population analyzed into three groups – score zero (no adverse factors); score 1–2 (one or two adverse factors) and score 3–4 (three or four adverse factors), with a 5-year survival of 95%, 87%, and 68%, respectively. This group listed factors that seemed to relate to need for treatment initiation: low hemoglobin, extranodal disease, and positive serology for HCV. Montalban et al, in an attempt to simplify the risk stratification and make it more pragmatic for clinical use, proposed cut-offs for hemoglobin (9.5 g/dL) and platelet counts (80×109/L) (Table 2).59
Biological parameters such as mutation and/or overexpression of TP53 seemed to be associated with a more aggressive clinical course and poorer survival.11,18,30,60
Management
Currently, there is no standard treatment for SMZL or criteria to initiate it, as most patients are asymptomatic at diagnosis and some may never require conventional therapy.5 Due to the lack of prospective therapeutic trials and definite prognostic scores, management is based mainly on disease manifestations/stage and patient characteristics.9
Management strategies include a watch-and-wait policy, splenectomy, splenic radiation, and chemotherapy/immunotherapy. The watch-and-wait policy is reserved for patients with early/asymptomatic disease defined by lack of B-symptoms, nonbulky splenomegaly, good hematopoietic reserve, absent/mild cytopenias, and no significant lymphadenopathy. This should include follow-up every 3–6 months of a physical examination, full blood counts, and biochemistry.16,61,62
Patients with symptomatic/advanced disease defined by splenomegaly, presence of cytopenias, progressive lymphocytosis, and B-symptoms should receive treatment according to their clinical status/staging.2,9
Splenectomy is becoming rarer as a primary treatment, mostly related to the success of immunotherapy, but may be considered for patients fit for surgery with a bulky spleen or hypersplenism, minimal bone marrow involvement, and negative serology for HCV. This procedure will debunk tumor burden and correct/improve cytopenias. A clinical response to splenectomy is reported in 75%–100% of patients, with an overall survival of 72%–88%.56,63,64 The bone marrow will remain infiltrated; thus, all responses will be partial.16,56
Splenic irradiation (6–8 grays in one or two fractions) can be used as a palliative modality in the minority of patients unsuitable for surgery or standard therapy.
The efficacy of IFN-α with or without ribavirin in the setting of HCV infection has been demonstrated in several reports. Complete responses have been associated with loss of detectable HCV RNA load.65–67
Chemotherapy is suggested for patients unfit for surgery and those without bulky splenomegaly, with generalized lymphadenopathy, progressive lymphocytosis, or extranodal involvement. There is no current standard regimen. Most studies suggest that the use of purine analogs (fludarabine, 2′ chlorodeoxyadenosine, and 2′ deoxycoformycin) is more promising than alkylating agents regarding response.9,17,19,61 Tsimberidou et al in a retrospective study have shown that the use of fludarabine alone is effective but inferior to rituximab alone or in combination regimen.63 The use of rituximab, a monoclonal antibody that targets CD20 antigen expressed in normal and neoplastic B-cells either as a single agent or in combination with chemotherapy, has been shown to have a remarkable efficacy, superior to the purine analogs in terms of response rates, complete responses, and duration of responses. Therefore, rituximab is becoming the treatment of choice for these patients. There have not been randomized clinical studies to compare rituximab as single agent with schedules that combine it with chemotherapy. At present, treatment with one or the other modality may be tailored to an individual patient, considering the extent of disease and his/her fitness.63,68–70 Patients with autoimmune disorders should receive conventional treatment for these disorders.
Complementary treatment is based on vaccinations for Pneumococcus, Haemophilus, and Meningococcus and antibiotic prophylaxis in patients undergoing splenectomy and splenic irradiation. Patients who receive purine analogs should undergo prophylaxis for herpes zoster and pneumocystosis.19,70
New potential treatments that are being used in other B-cell lymphomas include potent anti-CD20 monoclonal antibodies other than rituximab, drugs targeting the NF-κB pathway such as the proteasome inhibitor bortezomib, and Bruton´s tyrosine kinase.
Inhibitors or NOTCH inhibitors may well be considered in the future, particularly in refractory patients.9
Conclusion
SMZL is a distinct entity with heterogeneous biological and clinical features. Pathogenesis remains hypothetical, with the neoplastic clone possibly arising from persistent antigenic stimulation of marginal zone B-cells. Prognosis is favorable, with a median overall survival of approximately 12 years. Due to its biological heterogeneity, SMZL lacks any specific/recurrent chromosome translocation. Treatment is dictated by clinical parameters and may include a watch-and-wait policy for asymptomatic patients, splenectomy, splenic radiotherapy, or immunotherapy with or without chemotherapy selected according to clinical status/staging. In the future, molecular and cytogenetic data may dictate personalized treatments based on abnormalities in the NOTCH or NF-κB pathways.
Acknowledgement
We acknowledge support from the National Institute for Health Research (NIHR) Royal Marsden Hospital (RMH)/Institute of Cancer Research (ICR) Biomedical Research Center.
Disclosure
The authors declare no conflicts of interest in this work.
References
Schmid C, Kirkham N, Diss T, Isaacson PG. Splenic marginal zone lymphoma. Am J Surg Pathol. 1992;16(5):455–466. | |
Isaacson PG, Piris MA, Berger F, et al. In: Swerdlow SH, Campos E, Harris NL, et al. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues, 4th ed. Lyon, France: IARC Press, 2008. 289–291. | |
Papadaki T, Stamatopoulos K, Belessi C, et al. Splenic marginal-zone lymphoma: one or more entities? A histologic, immunohistochemical, and molecular study of 42 cases. Am J Surg Pathol. 2007;31(3):438–446. | |
Arcaini L, Zibellini S, Passamonti F, et al. Splenic marginal zone lymphoma: Clinical clustering of immunoglobulin heavy chain repertoires. Blood Cells Mol Dis. 2009;42(3):286–291. | |
Franco V, Florena AM, Iannitto E. Splenic marginal zone lymphoma. Blood. 2003;101(7):2464–2472. | |
Murakami H, Irisawa H, Saitoh T, et al. Immunological abnormalities in splenic marginal zone cell lymphoma. Am J Hematol. 1997;56(3):173–178. | |
Satoh T, Yamada T, Nakano S, et al. The relationship between primary splenic malignant lymphoma and chronic liver disease associated with hepatitis C virus infection. Cancer. 1997;80(10):1981–1988. | |
Bates I, Bedu-Addo G, Rutherford TR, Bevan DH. Circulating villous lymphocytes – a link between hyperreactive malarial splenomegaly and splenic lymphoma. Trans R Soc Trop Med Hyg. 1997;91(2):171–174. | |
Matutes E. Splenic marginal zone lymphoma: disease features and management. Expert Rev Hematol. 2013;6(6):735–745. | |
Arcaini L, Bruno R. Hepatitis C virus infection and antiviral treatment in marginal zone lymphomas. Curr Clin Pharmacol. 2010;5(2):74–81. | |
Kalpadakis C, Pangalis GA, Dimitriadou E, et al. Mutation analysis of IgVH genes in splenic marginal zone lymphomas: correlation with clinical characteristics and outcome. Anticancer Res. 2009;29(5):1811–1816. | |
Algara P, Mateo MS, Sanchez-Beato M, et al. Analysis of the IgVH somatic mutations in splenic marginal zone lymphoma defines a group of unmutated cases with frequent 7q deletion and adverse clinical course. Blood. 2002;99(4):1299–1304. | |
Wallace S, Bedu-Addo G, Rutherford TR, Bates I. Serological similarities between hyperreactive malarial splenomegaly and splenic lymphoma in west Africa. Trans R Soc Trop Med Hyg. 1998;92(4):463–467. | |
Xochelli A, Kalpadakis C, Gardiner A, et al. Clonal B-cell lymphocytosis exhibiting immunophenotypic features consistent with a marginal-zone origin: is this a distinct entity? Blood. 2014;123(8):1199–1206. | |
Thieblemont C, Felman P, Callet-Bauchu E, et al. Splenic marginal-zone lymphoma: a distinct clinical and pathological entity. Lancet Oncol. 2003;4(2):95–103. | |
Arcaini L, Lazzarino M, Colombo N, et al; Integruppo Italiano Linfomi. Splenic marginal zone lymphoma: a prognostic model for clinical use. Blood. 2006;107(12):4643–4649. | |
Troussard X, Valensi F, Duchayne E, et al. Splenic lymphoma with villous lymphocytes: clinical presentation, biology and prognostic factors in a series of 100 patients. Groupe Francais d’Hématologie Cellulaire (GFHC). Br J Haematol. 1996;93(3):731–736. | |
Chacón JI, Mollejo M, Muñoz E, et al. Splenic marginal zone lymphoma: clinical characteristics and prognostic factors in a series of 60 patients. Blood. 2002;100(5):1648–1654. | |
Matutes E, Oscier D, Montalban C, et al. Splenic marginal zone lymphoma proposals for a revision of diagnostic, staging and therapeutic criteria. Leukemia. 2008;22(3):487–495. | |
Hammer RD, Glick AD, Greer JP, Collins RD, Cousar JB. Splenic marginal zone lymphoma. A distinct B-cell neoplasm. Am J Surg Pathol. 1996;20(5):613–626. | |
Duong Van Huyen JP, Molina T, Delmer A, et al. Splenic marginal zone lymphoma with or without plasmacytic differentiation. Am J Surg Pathol. 2000;24(12):1581–1592. | |
Franco V, Florena AM, Stella M, et al. Splenectomy influences bone marrow infiltration in patients with splenic marginal zone cell lymphoma with or without villous lymphocytes. Cancer. 2001;91(2):294–301. | |
Wu CD, Jackson CL, Medeiros LJ. Splenic marginal zone lymphoma. An immunophenotypic and molecular study of five cases. Am J Clin Pathol. 1996;105(3):277–285. | |
Rosso R, Castello A, Colosini G, Astori C, Lazzarino M, Magrini U. Splenic marginal zone cell lymphoma involving liver and bone marrow. Report of a case with protracted follow-up, showing progressive disappearance of the lymphoma after splenectomy. Haematologica. 1996;81(1):44–46. | |
Mollejo M, Algara P, Mateo MS, et al. Splenic small B-cell lymphoma with predominant red pulp involvement: a diffuse variant of splenic marginal zone lymphoma? Histopathology. 2002;40(1):22–30. | |
Berger F, Felman P, Thieblemont C, et al. Non-MALT marginal zone B-cell lymphomas: a description of clinical presentation and outcome in 124 patients. Blood. 2000;95(6):1950–1956. | |
Orero MT, Miguel-Sosa A, Matutes E, et al. A soft tissue extranodal mass as an initial manifestation of splenic lymphoma with circulating villous lymphocytes. Br J Haematol. 1998;103(4):1213–1214. | |
Yamazaki K, Shimizu S, Negami T, et al. Leukemic meningitis in a patient with splenic lymphoma with villous lymphocytes (SLVL). Meningitis as a possible initial manifestation of SLVL. Cancer. 1994;74(1):61–65. | |
Gotlib V, Singareddy S, Gergis U, et al. Leptomeningeal involvement in a patient with splenic lymphoma with villous lymphocytes. Leuk Lymphoma. 2002;43(6):1337–1340. | |
Gruszka-Westwood AM, Hamoudi RA, Matutes E, Tuset E, Catovsky D. p53 abnormalities in splenic lymphoma with villous lymphocytes. Blood. 2001;97(11):3552–3558. | |
Zibellini S, Capello D, Forconi F, et al. Stereotyped patterns of B-cell receptor in splenic marginal zone lymphoma. Haematologica. 2010;95(10):1792–1796. | |
Bikos V, Darzentas N, Hadzidimitriou A, et al. Over 30% of patients with splenic marginal zone lymphoma express the same immunoglobulin heavy variable gene: ontogenetic implications. Leukemia. 2012;26(7):1638–1646. | |
Oscier D, Owen R, Johnson S. Splenic marginal zone lymphoma. Blood Rev. 2005;19(1):39–51. | |
Remstein ED, Law M, Mollejo M, Piris MA, Kurtin P, Dogan A. IGH translocation and 7 deletions are frequent genetic abnormalities in splenic marginal zone lymphoma and are present in mutually exclusive subjects [abstract]. XIII meeting of the European Association for Haematopathology, Vienna, Austria, 2006, LO8. | |
Dierlamm J, Rosenberg C, Stul M, et al. Characteristic pattern of chromosomal gains and losses in marginal zone B cell lymphoma detected by comparative genomic hybridization. Leukemia. 1997;11(5):747–758. | |
Hernández JM, García JL, Gutiérrez NC, et al. Novel genomic imbalances in B-cell splenic marginal zone lymphomas revealed by comparative genomic hybridization and cytogenetics. Am J Pathol. 2001;158(5):1843–1850. | |
Andersen CL, Gruszka-Westwood A, Atkinson S, et al. Recurrent genomic imbalances in B-cell splenic marginal-zone lymphoma revealed by comparative genomic hybridization. Cancer Genet Cytogenet. 2005;156(2):122–128. | |
Boonstra R, Bosga-Bouwer A, van Imhoff GW, et al. Splenic marginal zone lymphomas presenting with splenomegaly and typical immunophenotype are characterized by allelic loss in 7q31-32. Mod Pathol. 2003;16(12):1210–1217. | |
Watkins AJ, Huang Y, Ye H, et al. Splenic marginal zone lymphoma: characterization of 7q deletion and its value in diagnosis. J Pathol. 2010;220(4):461–474. | |
Fresquet V, Robles EF, Parker A, et al. High-throughput sequencing analysis of the chromosome 7q32 deletion reveals IRF5 as a potential tumour suppressor in splenic marginal-zone lymphoma. Br J Haematol. 2012;158(6):712–726. | |
Watkins AJ, Hamoudi RA, Zeng N, et al. An integrated genomic and expression analysis of 7q deletion in splenic marginal zone lymphoma. PLoS One. 2012;7(9):e44997. | |
Rossi D, Trifonov V, Fangazio M, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012;209(9):1537–1551. | |
Kiel MJ, Velusamy T, Betz BL, et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med. 2012;209(9):1553–1565. | |
Martínez N, Almaraz C, Vaqué JP, et al. Whole-exome sequencing in splenic marginal zone lymphoma reveals mutations in genes involved in marginal zone differentiation. Leukemia. 2014;28(6):1334–1340. | |
Parry M, Rose-Zerilli MJ, Gibson J, et al. Whole exome sequencing identifies novel recurrently mutated genes in patients with splenic marginal zone lymphoma. PLoS One. 2013;8(12):e83244. | |
Radtke F, MacDonald HR, Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nat Rev Immunol. 2013;13(6):427–437. | |
Kuroda K, Han H, Tani S, et al. Regulation of marginal zone B cell development by MINT, a suppressor of Notch/RBP-J signaling pathway. Immunity. 2003;18(2):301–312. | |
Saito T, Chiba S, Ichikawa M, et al. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity. 2003;18(5):675–685. | |
Trøen G, Wlodarska I, Warsame A, Hernández Llodrà S, De Wolf-Peeters C, Delabie J. NOTCH2 mutations in marginal zone lymphoma. Haematologica. 2008;93(7):1107–1109. | |
Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2(6):a000109. | |
Du MQ. MALT lymphoma: many roads lead to nuclear factor-κb activation. Histopathology. 2011;58(1):26–38. | |
Shaffer AL, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol. 2012;30:565–610. | |
Rossi D, Deaglio S, Dominguez-Sola D, et al. Alteration of BIRC3 and multiple other NF-κB pathway genes in splenic marginal zone lymphoma. Blood. 2011;118(18):4930–4934. | |
Yan Q, Huang Y, Watkins AJ, et al. BCR and TLR signaling pathways are recurrently targeted by genetic changes in splenic marginal zone lymphomas. Haematologica. 2012;97(4):595–598. | |
Tierens A, Delabie J, Michiels L, Vandenberghe P, De Wolf-Peeters C. Marginal-zone B cells in the human lymph node and spleen show somatic hypermutations and display clonal expansion. Blood. 1999;93(1):226–234. | |
Mulligan SP, Matutes E, Dearden C, Catovsky D. Splenic lymphoma with villous lymphocytes: natural history and response to therapy in 50 cases. Br J Haematol. 1991;78(2):206–209. | |
Hockley SL, Else M, Morilla A, et al. The prognostic impact of clinical and molecular features in hairy cell leukaemia variant and splenic marginal zone lymphoma. Br J Haematol. 2012;158(3):347–354. | |
Montalbán C, Abraira V, Arcaini L, et al; Splenic Marginal Zone Lymphoma Study Group. Risk stratification for Splenic Marginal Zone Lymphoma based on haemoglobin concentration, platelet count, high lactate dehydrogenase level and extrahilar lymphadenopathy: development and validation on 593 cases. Br J Haematol. 2012;159(2):164–171. | |
Montalban C, Abraira V, Arcaini L, et al; Splenic Marginal Zone Lymphoma Study Group (SMZLSG). Simplification of risk stratification for splenic marginal zone lymphoma: a point-based score for practical use. Leuk Lymphoma. 2014;55(4):929–931. | |
Salido M, Baró C, Oscier D, et al. Cytogenetic aberrations and their prognostic value in a series of 330 splenic marginal zone B-cell lymphomas: a multicenter study of the Splenic B-Cell Lymphoma Group. Blood. 2010;116(9):1479–1488. | |
Parry-Jones N, Matutes E, Gruszka-Westwood AM, Swansbury GJ, Wotherspoon AC, Catovsky D. Prognostic features of splenic lymphoma with villous lymphocytes: a report on 129 patients. Br J Haematol. 2003;120(5):759–764. | |
Iannito E, Ambrosetti A, Ammatuna E, et al. Splenic marginal zone lymphoma with or without villous lymphocytes. Hematologic findings and outcomes in a series of 57 patients. Cancer. 2004;101(9):2050–2057. | |
Tsimberidou AM, Catovsky D, Schlette E, et al. Outcomes in patients with splenic marginal zone lymphoma and marginal zone lymphoma treated with rituximab with or without chemotherapy or chemotherapy alone. Cancer. 2006;107(1):125–135. | |
Bennett M, Schechter GP. Treatment of splenic marginal zone lymphoma: splenectomy versus rituximab. Semin Hematol. 2010;47(2):143–147. | |
Arcaini L, Paulli M, Boveri E, et al. Splenic and nodal marginal zone lymphomas are indolent disorders at high hepatitis C virus seroprevalence with distinct presenting features but similar morphologic and phenotypic profiles.Cancer. 2004;100(1):107–115. | |
Hermine O, Lefrère F, Bronowicki JP, et al. Regression of splenic lymphoma with villous lymphocytes after treatment of hepatitis C virus infection. N Engl J Med. 2002;347(2):89–94. | |
Kelaidi C, Rollot F, Park S, et al. Response to antiviral treatment in hepatitis C virus-associated marginal zone lymphomas. Leukemia. 2004;18(10):1711–1716. | |
Bennett M, Yegena S, Dave HP, Schechter GP. Re: Rituximab monotherapy is highly effective in splenic marginal zone lymphoma. Hematol Oncol. 2008;26(2):114. | |
Kalpadakis C, Pangalis GA, Dimopoulou MN, et al. Rituximab monotherapy is highly effective in splenic marginal zone lymphoma. Hematol Oncol. 2007;25(3):127–131. | |
Kalpadakis C, Pangalis GA, Angelopoulou MK, et al. Treatment of splenic marginal zone lymphoma with rituximab monotherapy: progress report and comparison with splenectomy. Oncologist. 2013;18(2):190–197. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.