")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Spirolactone provides protection from renal fibrosis by inhibiting the endothelial–mesenchymal transition in isoprenaline-induced heart failure in rats
Authors Zhou H, Xi D, Liu J, Zhao J, Chen S, Guo Z
Received 8 November 2015
Accepted for publication 22 February 2016
Published 5 May 2016 Volume 2016:10 Pages 1581—1588
DOI https://doi.org/10.2147/DDDT.S100095
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Hao Zhou, Dan Xi, Jichen Liu, Jinjin Zhao, Si Chen, Zhigang Guo
Division of Cardiology, Huiqiao Medical Center, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong, People’s Republic of China
Background: Fibrosis results in excessive accumulation of extracellular matrix proteins, collagen component alteration, and abnormalities in structure and is partly derived from a process called the endothelial–mesenchymal transition involving transforming growth factor β (TGF-β). We investigated whether spironolactone, an aldosterone receptor blocker, attenuated isoprenaline (Iso)-induced heart failure in rats and also studied the mechanism for the same.
Methods: Sprague–Dawley rats were subcutaneously injected with Iso to induce heart failure, which promoted renal fibrosis; rats with spironolactone treatment were given a gavage of spironolactone (30 or 60 mg/kg/d, for 21 days). Cardiac function and fibrosis indices were measured. Pathological alterations and expression of Type I and III collagen, α-smooth muscle actin, cluster of differentiation-31, and TGF-β were examined.
Results: In Iso-induced heart failure in rats, spironolactone significantly improved cardiac function and decreased myocardial fibrosis, reduced collagen fibrous proliferation in kidney, reduced expression of Type I and III collagen, increased the expression of cluster of differentiation-31, and decreased the expression of α-smooth muscle actin and TGF-β.
Conclusion: Spironolactone may prevent renal fibrosis by inhibiting the endothelial–mesenchymal transition.
Keywords: spironolactone, heart failure, renal fibrosis, endothelial–mesenchymal transition
Introduction
The direct and indirect effects of heart failure are identified as leading to acute kidney injury and dysfunction.1 Cardiorenal syndrome Type 1 is characterized by acute kidney injury and dysfunction in the patient with acute deterioration of the cardiac function. The degree of renal function impairment relates to increased short- and long-term mortality.2 It has been linked to cardiac and renal fibrosis. Acute and chronic damage induce expression of cell signaling proteins from immune cells and inflammatory cell infiltrates, activation of fibroblasts and myofibroblasts, deposition of Type I, III collagen, and the activation of the final common pathway into the extracellular matrix, leading to irreversible cardiac and renal fibrosis.3
Recently, the endothelial–myofibroblast transition or endothelial–mesenchymal transition (EndMT) has been proved to promote fibrosis.4 Similar to the endothelial–myofibroblast transition, during EndMT, endothelial cells lose their characteristics and obtain the specialty of mesenchymal cells, which possess high migration and invasive properties and also have an elongated spindle shape. The EndMT progress can be demonstrated by specific biochemical changes, including decreased expression of endothelial makers, mainly cluster of differentiation (CD31), vascular endothelial-cadherin, von Willibrand factor, and increased expression of mesenchymal markers such as α-smooth muscle actin (α-SMA), vimentin, and fibroblast-specific protein 1.4 The EndMT program has been suggested to contribute to the development and progression of pulmonary fibrosis, hepatic fibrosis, corneal fibrosis, intestinal fibrosis and wound healing, and cardiac fibrosis, in addition to renal fibrosis.5,6 Transforming growth factor β (TGF-β) is the most important mediator; it increases the synthesis of matrix proteins and decreases the production of matrix metalloproteinase in the renal fibrosis process.7 Other studies have shown that the EndMT is activated mainly by TGF-β.4,8,9
Activation of the renin–angiotensin–aldosterone system has been identified as a risk factor for the development of fibrosis. Studies have demonstrated the importance of aldosterone in oxidative, inflammatory and fibrotic processes in kidney diseases.10 Spironolactone is the most commonly used aldosterone receptor antagonist. As treatment for congestive heart failure, liver cirrhosis, and hypertension, the drug blocks aldosterone by competitively inhibiting the binding of aldosterone to the mineralocorticoid receptor.11 Recent clinical studies show that spironolactone reduced the combined end point of death or hospitalization for heart failure in non-African-Americans but had no effect on mortality, death, or hospitalization in African-Americans.12 Spironolactone was found to be safe in patients with early stages of chronic kidney disease, with a strict monitoring of renal function and electrolytes over the first month of treatment.13
From these observations, we aimed to investigate whether spironolactone affects renal function in isoprenaline (Iso)-induced heart failure in rats, affects EndMT and the expression of TGF-β, as well as study the relation between these changes and renal fibrosis.
Methods
Animals and treatments
A total of 50 male Sprague–Dawley rats (200–220 g; ~6 weeks old), supplied by Wenzhou Medical University Laboratory Animal Center (Wenzhou City, Zhejiang Province, People’s Republic of China), were housed and maintained under standard conditions. The study was approved by the institutional research ethics committee of Wenzhou Medical University. All animal-handling procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and followed the guidelines of the Animal Welfare Act. Rats were separated into four groups (n=10 per group): 1) control, 2) heart failure model (Iso treatment), 3) low-dose spironolactone (30 mg/kg/d), and 4) high-dose spironolactone (60 mg/kg/d). Subcutaneous injection of Iso (5 mg/kg, Sigma-Aldrich, St Louis, MO, USA, 7 days) induced heart failure, while the control group was injected with normal saline. In the therapeutic groups, Iso administration was the same as in the Iso-model group, and spironolactone at different concentrations was gavaged at the same time as Iso injection and lasted for 7 days, then was continued for another 14 days. The same volume of saline was gavaged for controls.
Cardiac functional measurements
Rats were weighed and an appropriate dose of anesthesia was administered by intraperitoneal injection with 1% sodium pentobarbital (40 mg/kg), and then the left ventricle was catheterized to monitor the changes in left ventricle end diastolic pressure (LVEDP), LV mean systolic pressure (LVSP), and the maximum rate of change in LV pressure (+dp/dtmax, −dp/dtmax) through the right common carotid artery.
Organ weight index
The rats were injected with chilled brine (4°C) into the left ventricle of the heart and kidneys until etiolating. Heart and kidneys were quickly removed, rinsed, and weighed. The heart was then divided into left and right ventricles and weighed. The LV weight indices (LVWI) and RV weight indices (RVWI) were calculated as the LV and RV free wall mass (mg)/by body mass (g), respectively. The renal weight indices (KWI) were calculated as the mean value of mass (mg)/by body mass (g) of two kidneys.
Hematoxylin and eosin
Cardiac apex samples of the ventricle and renal tissue samples were embedded in paraffin and cut into slices. Paraffin sections (4 μm thick) were stained with hematoxylin and eosin. Sections were analyzed by light microscopy and photographed at ×200 magnification.
Masson trichrome staining
The sections were stained with Masson trichrome staining and photographed at ×40 and ×200 magnifications. Fibrous tissue was stained in blue, cytoplasm in red, and cell nucleus in black. The sections were examined using light microscopy, and photographs were taken at ×200 magnification. Five nonrepeating visual fields were randomly selected, myocardial and renal collagen areas were measured using Image-Pro Plus (Media Cybernetics, Rockville, MD, USA), and the collagen content in all areas was averaged.
Enzyme-linked immunosorbent assay
Pieces of the LV myocardium tissue and kidney tissue (100 mg) were cut into pieces and milled on ice with 1 mL phosphate-buffered saline (pH 7.4). The mixture was centrifuged at 3,000 rpm for 20 minutes and the supernatant was separated to measure collagen I and III by using an ELISA kit (Shanghai Boyun Biotech, Shanghai, People’s Republic of China).
Immunofluorescence assay
Dual immunofluorescence staining of CD31 and α-SMA was performed. Sections of kidney tissue were treated with 3% methanol-H2O2 to blocked endogenous peroxidase activity, and the nonspecific sites were treated with 10% fetal bovine serum. Sections were then incubated with primary mixed anti-CD31 (Santa Cruz Biotechnology, Dallas, TX, USA, 1:30) and anti-SMA (Wuhanboshide, People’s Republic of China, 1:100) antibodies. The secondary antibodies of DyLight 594 AffiniPure goat anti-rabbit IgG (H+L) and DyLight 488 AffiniPure goat anti-mouse IgG (H+L) (EarthOX, Millbrae, CA, USA, 1:300) were also added as a mixture. After washing, 4′,6′-diamidino-2-phenylindole hydrochloride (DAPI) staining solution was added and washed. Photographs were taken at 200× magnification. The results were analyzed by fluorescence microscopy (Nikon Corp., Tokyo, Japan) and processed with Image-Pro Plus (Media Cybernetics). Negative control sections were incubated only with phosphate-buffered saline and showed no positive staining.
Western blot analysis
A piece of kidney tissue (100 mg) was homogenized in 1 mL radio immunoprecipitation assay lysis buffer and 10 μL phenylmethanesulfonyl fluoride (100 mmol/L). After centrifugation at 12,000× g for 10 minutes at 4°C, the supernatant was collected. The total protein concentrations were determined by using a bicinchoninic acid protein assay kit. Protein samples (50 μg) were run on sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. After blocking with 5% skim milk for 1 hour, membranes were incubated with primary antibodies (anti-CD31, Santa Cruz Biotechnology, 1:1,000; anti-α-SMA, Wuhanboshide, 1:1,000; anti-TGF-β1, Bioworld Technology, Nanjing, People’s Republic of China, 1:1,000) overnight at 4°C. Immunoreactive bands were detected by the use of chemiluminescent horseradish peroxidase substrate, and scans were obtained using the Bio-Rad gel image analysis system (Bio-Rad Laboratories Inc., Hercules, CA, USA) and processed with Image-Pro Plus (Media Cybernetics, Inc.).
Statistical analysis
Data are expressed as mean ± standard error of mean. All statistical analyses involved use of SPSS v16.0 (SPSS Inc., Chicago, IL, USA) with one-way analysis of variance and Student’s t-test. P-values <0.05 were considered statistically significant.
Results
Effect of spironolactone on cardiac function in rats
Rats were subcutaneously injected with Iso to induce heart failure. Iso significantly decreased LVSP, heart rate, +dp/dtmax, and −dp/dtmax and increased LVEDP when compared with controls (P<0.01). Spironolactone attenuated ventricular function, increased LVSP, heart rate, +dp/dtmax, and −dp/dtmax, and decreased LVEDP compared with Iso treatment (P<0.05; Table 1).
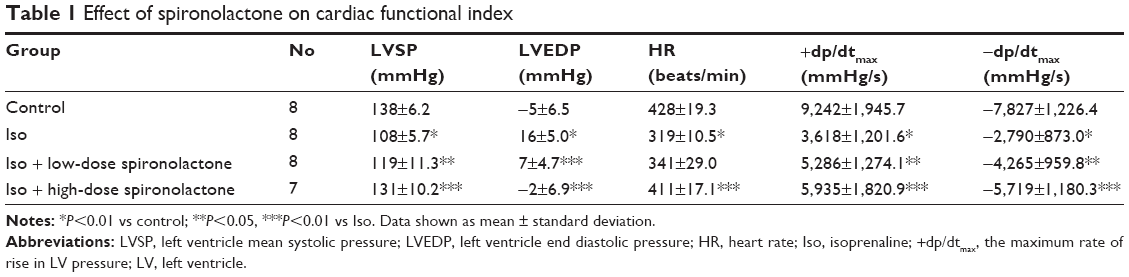 | Table 1 Effect of spironolactone on cardiac functional index |
Effect of spironolactone on organ weight index (LVWI, RVWI, and KWI) in rats
LVWI, RVWI, and KWI were significantly higher with Iso than control treatment (P<0.01); treatment with spironolactone decreased LVWI, RVWI, and KWI as compared with Iso treatment (P<0.05; Table 2).
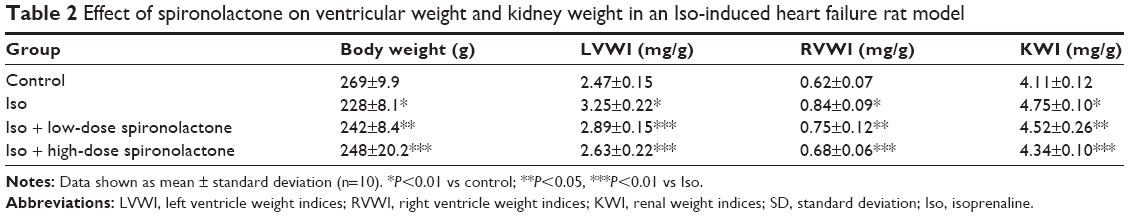 | Table 2 Effect of spironolactone on ventricular weight and kidney weight in an Iso-induced heart failure rat model |
Histopathological observations of heart and kidney in rats
Heart tissues from Iso-treated rats showed collagen fiber hyperplasia, disordered myocardial structure, and leukocyte infiltration when compared with controls (Figure 1). Treatment with spironolactone ameliorated the Iso-induced myocardial fibrosis. Iso also induced fibrous tissue proliferation and leukocyte infiltration in renal interstitial tissue. Treatment with spironolactone alleviated these changes (Figure 1).
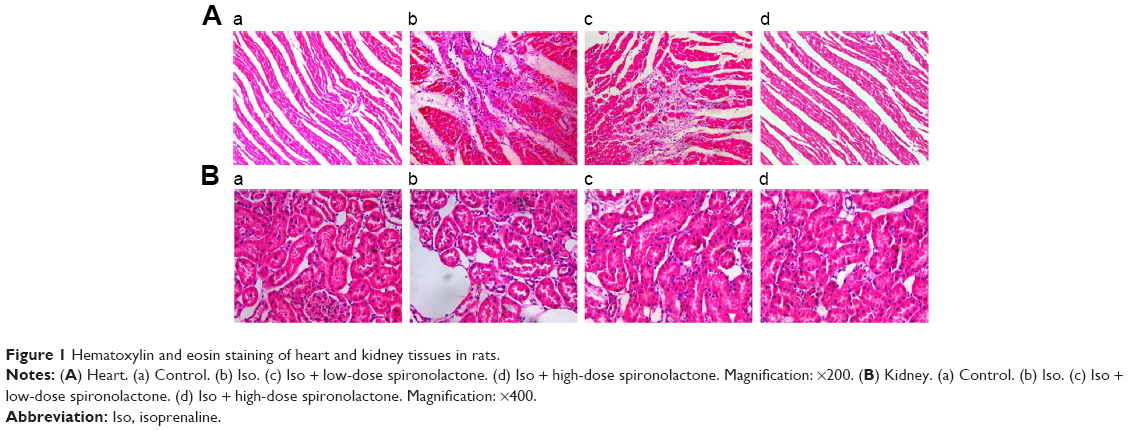 | Figure 1 Hematoxylin and eosin staining of heart and kidney tissues in rats. |
Effect of spironolactone on collagen area in heart and kidney
Stained sections of myocardia and kidney in Iso-treated rats showed more blue collagen fibers as compared with controls (P<0.01; Figure 2). Treatment with spironolactone decreased fibrous tissue proliferation as compared with Iso treatment (P<0.01).
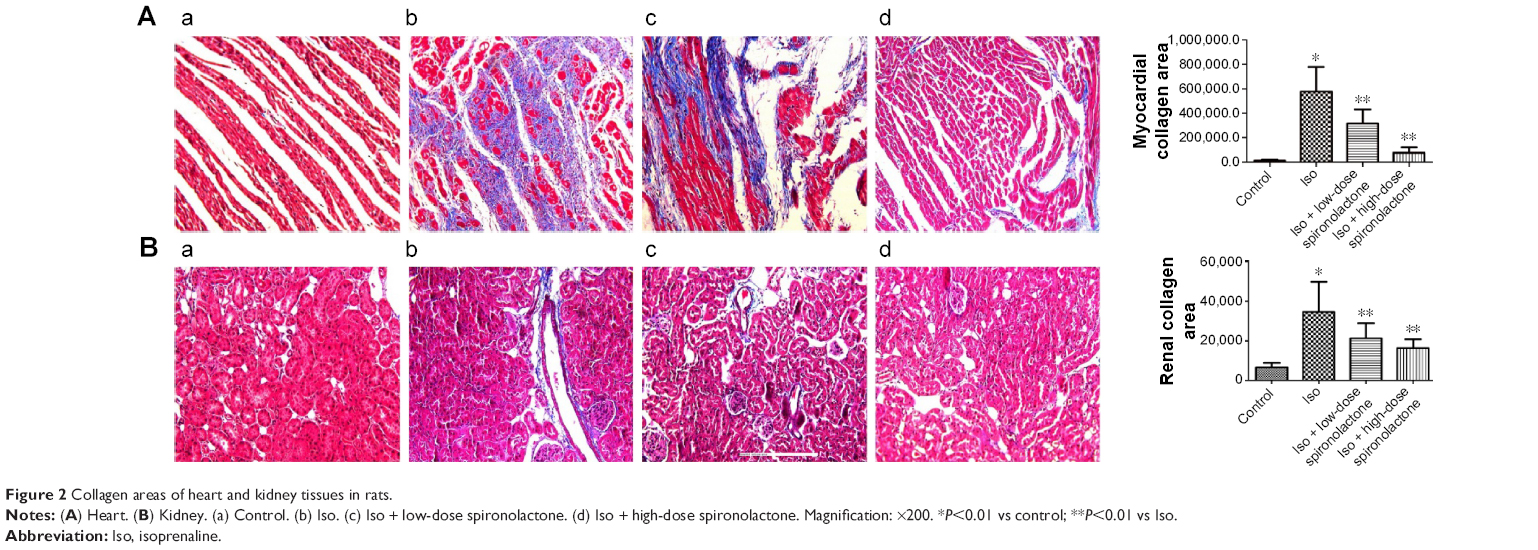 | Figure 2 Collagen areas of heart and kidney tissues in rats. |
Effect of spironolactone on Type I and III collagen content in heart and kidney
Content of Type I and III collagen in cardiac and kidney tissue homogenate was higher with Iso treatment than control treatment (P<0.01; Table 3). Treatment with spironolactone significantly decreased the collagen content as compared with Iso treatment (P<0.01).
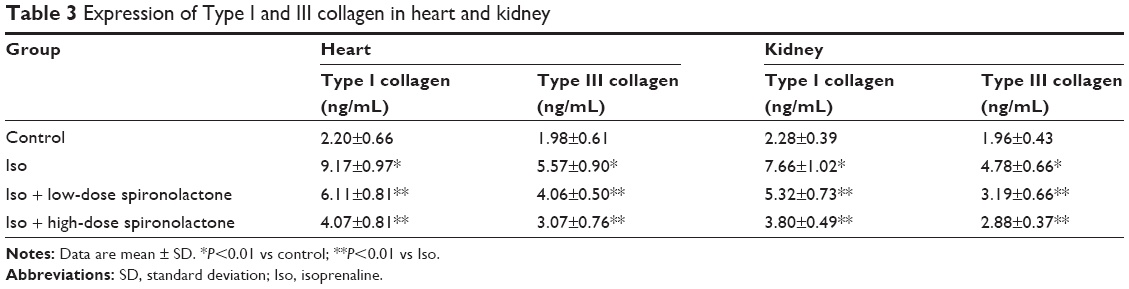 | Table 3 Expression of Type I and III collagen in heart and kidney |
Effect of spironolactone on expression of CD31 and α-SMA
Immunofluorescence staining and Western blot analyses revealed that Iso decreased CD31 protein level and increased α-SMA level as compared with the control (P<0.01) but spironolactone reversed these changes as compared with Iso treatment (P<0.05; Figure 3A–D).
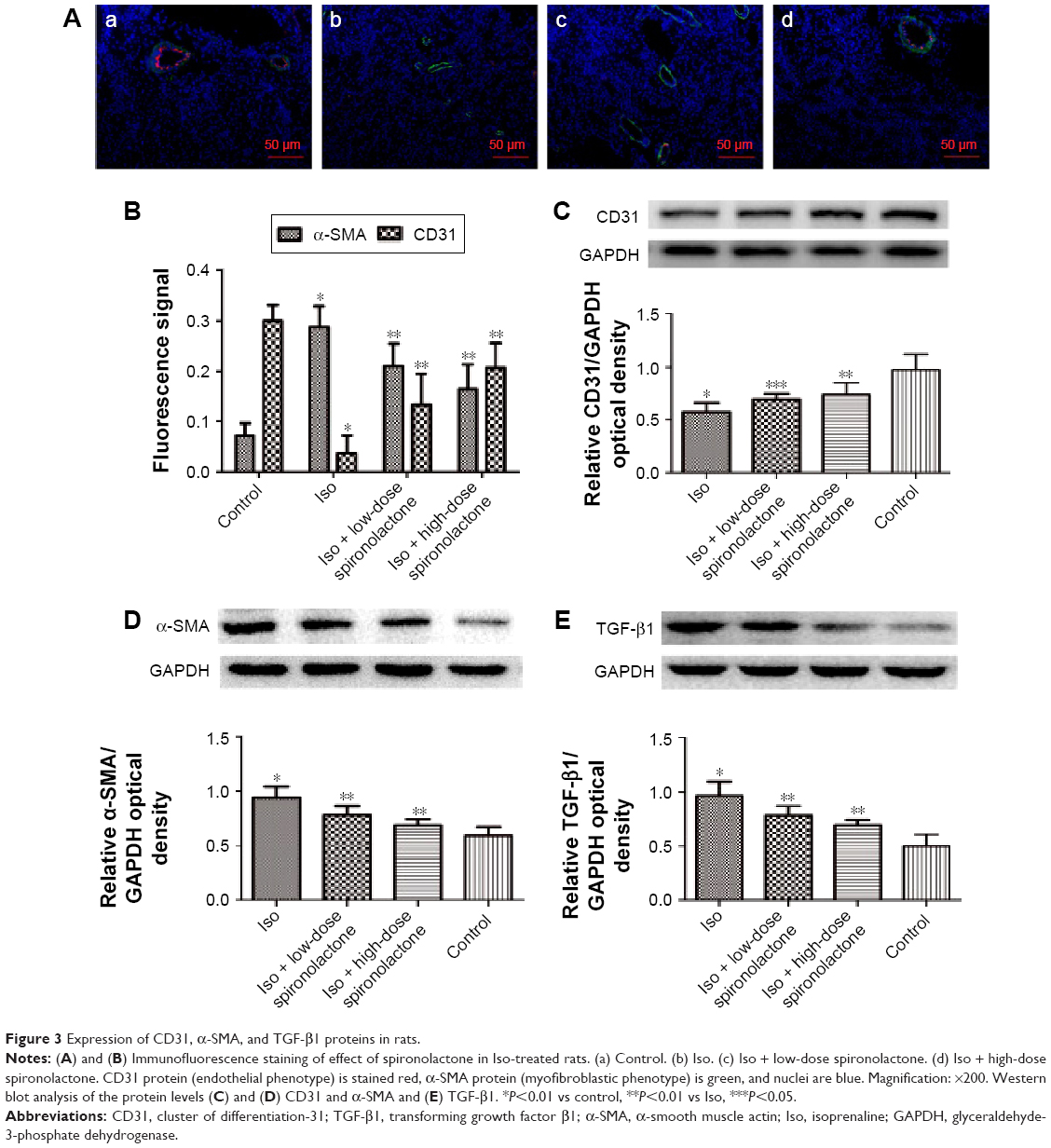 | Figure 3 Expression of CD31, α-SMA, and TGF-β1 proteins in rats. |
Effect of spironolactone on expression of TGF-β1 protein
The expression of endogenous TGF-β1 is greater with Iso than with control treatment (P<0.01). Treatment with spironolactone reduced the level of TGF-β1 as compared with Iso treatment (P<0.01; Figure 3E).
Discussion
In this study, Iso-treated rats showed worse heart function, impaired renal structure, and higher levels of renal collagen deposition (ie, fibrosis) and TGF-β, indicating EndMT, than control rats. Spironolactone treatment significantly improved cardiac function and reversed renal fibrosis and TGF-β level, thereby suppressing the EndMT.
Myocardial infarction (MI) is often associated with renal dysfunction,14 which indicates the “cardiorenal syndrome Type 1”. Renal dysfunction acts as an independent risk factor for adverse cardiovascular events and death in patients after MI. During MI admission, even mild renal dysfunction may be transient and can independently affect long-term (10 years) survival.15 Venous congestion, anemia, sympathetic nervous system dysfunction, disruption of the hypothalamic–pituitary axis, activation of the renin–angiotensin–aldosterone system, and a marked alteration of immune and somatic cell signaling have an influence on the pathogenesis of renal injury beyond the classic hemodynamic mechanisms. Therefore, the complexity of this syndrome poses huge challenges on diagnostic and treatment approaches.
In our study, rats in the Iso-treated group showed worse heart function, higher weight index (LVWI and RVWI), and increased collagen fiber hyperplasia and Type I/III collagen as compared with controls, for damaged cardiac function. Rats also showed higher KWI and increased renal fibrosis and Type I/III collagen as compared with controls, which suggested renal injury.
Neurohormonal activation may play a key role in renal fibrosis. After acute MI, neurohormonal activation occurs rapidly, which is likely or at least in part related to hemodynamic derangements. This activation is commonly sustained in patients with overt heart failure.16 In terms of subsequent fibrosis, neurohormonal activation is closely related to the TGF-β system. TGF-β can be activated by angiotensin II both in heart17 and kidney.18 TGF-β-mediated fibrosis has been reported to be associated with sympathetic and endothelin systems.19,20 Finally, blocking these systems not only retards the progression of chronic kidney disease but also inhibits renal inflammation, TGF-β expression, and fibrosis.21,22 These researches show that aldosterone induces renal injury through inflammation, oxidative stress, fibrosis, and hemodynamic alterations.9 Spironolactone is a high-affinity, but nonspecific, mineralocorticoid receptor antagonist that, with structural similarity to progesterone, binds to progesterone, androgen, and glucocorticoid receptors, but with reduced affinity.23 The kidney and renal vasculature are potential targets of aldosterone-mediated pathology. Activation of the mineralocorticoid receptor has also been implicated in cardiac hypertrophy observed in uremic rats, and spironolactone has been shown to attenuate cardiac hypertrophy and prevent oxidative stress in this model.24
In this study, the administration of spironolactone alleviated Iso-induced changes. The effect is even greater with high-dose spironolactone (60 mg/kg/d over 30 mg/kg/d), and so the antifibrosis effect of spironolactone may be dose related.
The mechanisms of how renal dysfunction occur following heart failure are poorly understood. Kidney diseases, regardless of the nature of the initial injury, ultimately cause renal interstitial fibrosis and lead to disturbed renal function.25
Several growth factors and cytokines participate in the inflammatory process; in particular, TGF-β appears to play a chief role in renal fibrosis.26 The expression of several proinflammatory molecules that may contribute to the pathogenesis of cardiac remodeling is modulated by aldosterone, in association with NaCl: aldosterone increases levels of monocyte chemoattractant protein 1, TGF-β1, collagen, and metalloproteases through mineralocorticoid-receptor-dependent mechanisms.27
The hallmark of renal fibrosis is the activation and accumulation of fibroblasts or myofibroblasts and the deposition of abundant extracellular matrix. Renal myofibroblasts appear de novo in renal fibrosis, and they most likely represent a stressed and dedifferentiated phenotype of fibroblasts.28 There is a significant heterogeneity among fibroblasts.29 These activated fibroblasts or myofibroblasts are derived from residential quiescent tissue fibroblasts, vascular pericytes, epithelial–mesenchymal transition, and bone marrow (circulating fibrocytes).30 Recently, increasing evidence has suggested that endothelial cells may undergo EndMT or endothelial–myofibroblast transition, which has also been suggested to promote fibrosis and is now recognized as a novel mechanism for the generation of myofibroblasts.4 EndMT was first discovered in heart development31 and was confirmed to be crucially important in forming the valves and septa of the heart during embryogenesis.32,33 In the adult organism, pathological conditions such as injury, inflammation, or aging can reactivate the EndMT and induce fibrosis of involved organs.4,5 Zeisberg et al7 first confirmed the contribution of EndMT to renal fibrosis. The authors found that a considerable proportion of myofibroblasts coexpress the endothelium marker CD31, also known as platelet endothelial cell adhesion molecule-1, and the (myo) fibroblast markers α-SMA and fibroblast-specific protein-1 in all three models. Li et al8 also confirmed that EndMT occurs and contributes to the generation of myofibroblasts in early diabetic renal fibrosis. TGF-β is the key factor in regulating the EndMT, which increases the production of extracellular matrices and promotes the fibrosis of cardiac and renal tissues.35,36
In our study, the expression of CD31 was decreased, and that of Type I/III collagen and α-SMA were increased in Iso-treated rats as compared with controls, which suggests subdued endothelial property and enhanced fibroblasts characteristic, thereby suggesting EndMT in Iso-induced renal fibrosis. Meanwhile, the level of TGF-β, the key factor regulating EndMT, was increased in Iso-treated rats. Treatment with spironolactone attenuated these effects, and so spironolactone may improve renal fibrosis by inhibiting EndMT. TGF-β may play a role as the key factor. There is also a study which showed that spironolactone could reduce fibrosis, and this improvement of renal function is possibly due to a dual inhibitory effect on the renin-angiotensin system, and suppression of TGF-β.34 Previous research showed that the protective effects of spironolactone are mediated to a large extent by its ability to reverse differentiation and inhibit the subsequent ability of mesenchymal cells to synthesize collagen, regardless of their origin.28 Spironolactone is considered to play a crucial role in inhibiting the pathogenesis of tissue fibrosis, and it also efficiently stimulates the expression of many genes through the EndMT and helps in avoiding the generation into myofibroblasts.35
Conclusion
Our results showed that spironolactone prevents Iso-induced renal fibrosis by inhibiting the renal EndMT.
Study limitations
First, our in vitro study examined only the expression of CD31 and α-SMA, which do not adequately represent the EndMT. The increased CD31 and decreased α-SMA can only indicate that spironolactone can inhibit the fibrosis while improving the endothelial expression. Unfortunately, we cannot determine the EndMT definitely from this data. However, these problems could be solved if we consider more specific markers in the fibrotic renal system in future. Second, further research should be done to confirm the role of spironolactone on the intervention of renal interstitial fibrosis and investigate the inhibition of renal EndMT, so that it can be used for the treatment of renal fibrosis, thereby providing a new class of drugs with a novel mechanism.
Acknowledgments
The authors thank Professor Chaoshu Tang for the valuable discussion. This study was supported by National Natural Science Foundation of People’s Republic of China (81570364).
Author contributions
Hao Zhou and Zhigang Guo contributed to the conception and study design. Hao Zhou, Dan Xi, Jichen Liu, Jinzhen Zhao, and Si Chen contributed to data acquisition. Dan Xi, Jichen Liu, Jinzhen Zhao, and Si Chen contributed to data analysis. Hao Zhou, Dan Xi, and Zhigang Guo drafted the manuscript. Hao Zhou and Zhigang Guo critically revised the manuscript. Hao Zhou, Dan Xi, Jichen Liu, Jinzhen Zhao, Si Chen, and Zhigang Guo gave final approval. All authors contributed toward data analysis, drafting, and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Ronco C, Cicoira M, McCullough PA. Cardiorenal syndrome type 1 pathophysiological crosstalk leading to combined heart and kidney dysfunction in the setting of acutely decompensated heart failure. J Am Coll Cardiol. 2012;60:1031–1042. | ||
Gibson CM, Pinto DS, Murphy SA, et al. Association of creatinine and creatinine clearance on presentation in acute myocardial infarction with subsequent mortality. J Am Coll Cardiol. 2003;42:1535–1543. | ||
Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res. 2011;89:265–272. | ||
He J, Xu Y, Koya D, Kanasaki K. Role of the endothelial-to-mesenchymal transition in renal fibrosis of chronic kidney disease. Clin Exp Nephrol. 2013;17:488–497. | ||
Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. 2011;179:1074–1080. | ||
Rieder F, Kessler SP, West GA, et al. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol. 2011;179:2660–2673. | ||
Zeisberg EM, Potenta SE, Sugimoto H, et al. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymaltransition. J Am Soc Nephrol. 2008;19:2282–2287. | ||
Li J, Qu X, Bertram JF. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am J Pathol. 2009;175:1380–1388. | ||
Medici D, Shore EM, Lounev VY, et al. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. 2010;16:1400–1406. | ||
Namsolleck P, Unger T. Aldosterone synthase inhibitors in cardiovascular and renal diseases. Nephrol Dial Transplant. 2014;29:i62–i68. | ||
Luo JH, Gao X, Peng L, Sun H, Dai G. Effects of hydrochlorothiazide on cardiac remodeling in a rat model of myocardial infarction-induced congestive heart failure. Eur J Pharmacol. 2011;667:314–321. | ||
Vardeny O, Cavallari LH, Claggett B, et al. Race influences the safety and efficacy of spironolactone in severe heart failure. Circ Heart Fail. 2013;6:970–976. | ||
Edwards NC, Steeds RP, Chue CD, et al. The safety and tolerability of spironolactone in patients with mild to moderate chronic kidney disease. Br J Clin Pharmacol. 2012;73:447–454. | ||
Hillege HL, van Gilst WH, van Veldhuisen DJ, et al. Accelerated decline and prognostic impact of renal function after myocardial infarction and the benefits of ACE inhibition: the CATS randomized trial. Eur Heart J. 2003;24:412–420. | ||
Parikh CR, Coca SG, Wang Y, et al. Long term prognosis of acute kidney injury after acute myocardial infarction. Arch Intern Med. 2008;168:987–995. | ||
Sigurdsson A, Held P, Swedberg K. Short- and long-term neurohormonal activation following acute myocardial infarction. Am Heart J. 1993;126:1068–1076. | ||
Leask A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74:207–212. | ||
Wolf G. Renal injury due to renin-angiotensin-aldosterone system activation of the transforming growth factor-beta pathway. Kidney Int. 2006;70:1914–1919. | ||
Akiyama-Uchida Y, Ashizawa N, Ohtsuru A, et al. Norepinephrine enhances fibrosis mediated by TGF-beta in cardiac fibroblasts. Hypertension. 2002;40:148–154. | ||
Berger R, Pacher R. The role of the endothelin system in myocardial infarction – new therapeutic targets? Eur Heart J. 2003;24:294–296. | ||
Takeda Y, Nishikimi T, Akimoto K, Matsuoka H, Ishimitsu T. Beneficial effects of a combination of Rho-kinase inhibitor and ACE inhibitor on tubulointerstitial fibrosis induced by unilateral ureteral obstruction. Hypertens Res. 2010;33:965–973. | ||
Zoja C, Corna D, Gagliardini E, et al. Adding a statin to a combination of ACE inhibitor and ARB normalizes proteinuria in experimental diabetes, which translates into full renoprotection. Am J Physiol Renal Physiol. 2010;299:F1203–F1211. | ||
Struthers A, Krum H, Williams GH. A comparison of the aldosterone-blocking agents eplerenone and spironolactone. Clin Cardiol. 2008;31:153–158. | ||
Michea L, Villagrán A, Urzúa A, et al. Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and prevents oxidative stress in uremic rats. Hypertension. 2008;52:295–300. | ||
Eddy AA. Experimental insights into the tubulointerstitial disease accompanying primary glomerular lesions. J Am Soc Nephrol. 1994;5:1273–1287. | ||
Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000;15:290–301. | ||
Marney AM, Brown NJ. Aldosterone and end-organ damage. Clin Sci (Lond). 2007;113:267–278. | ||
Boor P, Floege J. The renal (myo-)fibroblast: a heterogeneous group of cells. Nephrol Dial Transplant. 2012;27:3027–3036. | ||
Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631–637. | ||
Barnes JL, Glass WF 2nd. Renal interstitial fibrosis: a critical evaluation of the origin of myofibroblasts. Contrib Nephrol. 2011;169:73–93. | ||
Markwald RR, Fizharris TP, Smith WN. Structural analysis of endocardial cytodifferentiation. Dev Biol. 1975;142:160–180. | ||
Mercado-Pimentel ME, Runyan RB. Multiple transforming growth factor-beta isoforms and receptors function during epithelial-mesenchymal cell transformation in the embryonic heart. Cells Tissues Organs. 2007;185:146–156. | ||
Kisanuki YY, Hammer RE, Miyazaki J, et al. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. | ||
Guney I, Selcuk NY, Altintepe L, et al. Antifibrotic effects of aldosterone receptor blocker (spironolactone) in patients with chronic kidney disease. Ren Fail. 2009;31:779–384. | ||
Verrecchia F, Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 2002;118:211–215. | ||
Zhou X, Chen X, Cai JJ, et al. Relaxin inhibits cardiac fibrosis and endothelial-mesenchymal transition via the Notch pathway. Drug Des Devel Ther. 2015;11:4599–4611. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.