")
Back to Journals » International Journal of Nanomedicine » Volume 16
Specific Cytotoxic Effect of an Auristatin Nanoconjugate Towards CXCR4+ Diffuse Large B-Cell Lymphoma Cells
Authors Falgàs A, Pallarès V, Unzueta U, Núñez Y, Sierra J, Gallardo A, Alba-Castellón L, Mangues MA , Álamo P, Villaverde A, Vázquez E, Mangues R , Casanova I
Received 31 October 2020
Accepted for publication 29 January 2021
Published 5 March 2021 Volume 2021:16 Pages 1869—1888
DOI https://doi.org/10.2147/IJN.S289733
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Aïda Falgàs,1– 3 Victor Pallarès,1– 3 Ugutz Unzueta,1– 4 Yáiza Núñez,1,2 Jorge Sierra,2,5 Alberto Gallardo,1 Lorena Alba-Castellón,1,2 Maria Antonia Mangues,3,6 Patricia Álamo,1– 3 Antonio Villaverde,3,4,7 Esther Vázquez,3,4,7 Ramon Mangues,1– 3 Isolda Casanova1– 3
1Biomedical Research Institute Sant Pau (IIB-Sant Pau), Hospital de la Santa Creu i Sant Pau, Barcelona, 08025, Spain; 2Josep Carreras Leukaemia Research Institute (IJC), Barcelona, 08916, Spain; 3CIBER de Bioingeniería Biomateriales y Nanomedicina (CIBER-BBN), Madrid, 28029, Spain; 4Department of Genetics and Microbiology, Universitat Autònoma de Barcelona, Barcelona, 08193, Spain; 5Department of Hematology, Hospital de la Santa Creu i Sant Pau, Barcelona, 08025, Spain; 6Department of Pharmacy, Hospital de la Santa Creu i Sant Pau, Barcelona, 08025, Spain; 7Institute of Biotechnology and Biomedicine (IBB), Universitat Autònoma de Barcelona, Barcelona, 08193, Spain
Correspondence: Esther Vázquez
Institute of Biotechnology and Biomedicine (IBB) and Department of Genetics and Microbiology, Universitat Autònoma de Barcelona and CIBER-BBN, Barcelona, Spain
Tel +34 935812148
Email [email protected]
Ramon Mangues
Biomedical Research Institute Sant Pau (IIB-Sant Pau), Josep Carreras Leukaemia Research Institute (IJC) and CIBER-BBN, Barcelona, Spain
Tel +34 935537918
Email [email protected]
Background and Purpose: Around 40– 50% of diffuse large-B cell lymphoma (DLBCL) patients suffer from refractory disease or relapse after R-CHOP first-line treatment. Many ongoing clinical trials for DLBCL patients involve microtubule targeting agents (MTAs), however, their anticancer activity is limited by severe side effects. Therefore, we chose to improve the therapeutic window of the MTA monomethyl auristatin E developing a nanoconjugate, T22-AUR, that selectively targets the CXCR4 receptor, which is overexpressed in many DLBCL cells (CXCR4+) and associated with poor prognosis.
Methods: The T22-AUR specificity towards CXCR4 receptor was performed by flow cytometry in different DLBCL cell lines and running biodistribution assays in a subcutaneous mouse model bearing CXCR4+ DLBCL cells. Moreover, we determined T22-AUR cytotoxicity using cell viability assays, cell cycle analysis, DAPI staining and immunohistochemistry. Finally, the T22-AUR antineoplastic effect was evaluated in vivo in an extranodal CXCR4+ DLBCL mouse model whereas the toxicity analysis was assessed by histopathology in non-infiltrated mouse organs and by in vitro cytotoxic assays in human PBMCs.
Results: We demonstrate that the T22-AUR nanoconjugate displays CXCR4-dependent targeting and internalization in CXCR4+ DLBCL cells in vitro as well as in a subcutaneous DLBCL mouse model. Moreover, it shows high cytotoxic effect in CXCR4+ DLBCL cells, including induction of G2/M mitotic arrest, DNA damage, mitotic catastrophe and apoptosis. Furthermore, the nanoconjugate shows a potent reduction in lymphoma mouse dissemination without histopathological alterations in non-DLBCL infiltrated organs. Importantly, T22-AUR also exhibits lack of toxicity in human PBMCs.
Conclusion: T22-AUR exerts in vitro and in vivo anticancer effect on CXCR4+ DLBCL cells without off-target toxicity. Thus, T22-AUR promises to become an effective therapy for CXCR4+ DLBCL patients.
Keywords: nanomedicine, targeted drug delivery, MMAE, DLBCL
Introduction
Diffuse Large B-cell lymphoma (DLBCL) is the most frequent non-Hodgkin lymphoma (NHL).1 Although the addition of rituximab to the standard chemotherapy has improved the outcome of DLBCL patients, about 40–50% of them develop relapsed/refractory (R/R) disease after treatment with rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP).2,3 This is usually managed with salvage chemotherapy and, after a complete or partial response, patients undergo a hematopoietic stem cell transplant (HSCT). However, patients younger than 60 years old with high International Prognostic Index (IPI) and disseminated DLBCL do not have a standard first-line therapy. Moreover, some DLBCL patients do not respond to the high-dose chemotherapy or they are not initially candidate for HSCT (second-line therapy), or even fail to the HSCT (needing third-line therapy).4 Furthermore, current chemotherapy can induce life-threatening side effects in DLBCL patients.5 Thus, new treatments are urgently needed to enhance their antineoplastic effect in lymphoma cells without damaging normal cells. Actively targeted nanoparticles, containing a ligand that allows their specific targeting to a receptor overexpressed in cancer cells, have appeared as a promising option.6,7 Here, we chose the CXCR4 receptor to target lymphoma cells, since around 30–50% of DLBCL biopsies overexpress this receptor (CXCR4+)8,9 and its expression in lymphoma cells is much higher than in normal B cells.10 Moreover, CXCR4 overexpression is a poor prognostic factor in DLBCL patients.9,11
Interestingly, microtubule-targeting agents (MTAs) promise to become potent drug-payloads in anticancer treatments. Microtubules are tubulin polymers and essential components of the cytoskeleton that undergo constant assembling and disassembling within the cell. The main role of microtubules is the segregation of the chromosomes to the spindle poles during mitosis with the consequent formation of daughter cells.12 However, they are involved in many other cellular processes, such as intracellular trafficking, migration and angiogenesis.13,14 Therefore, their dynamic features and their implication in the cancer cell survival renders them good antineoplastic targets.
Our group designed a T22-AUR therapeutic nanoconjugate that includes the T22-GFP-H6 protein nanocarrier, previously demonstrated to selectively target CXCR4 receptor-overexpressing DLBCL cells,15 and is conjugated with the MTA monomethyl auristatin E (MMAE), aiming at selectively killing CXCR4+ DLBCL cells. Furthermore, this nanoconjugate has recently demonstrated efficacy in treating an aggressive acute myeloid leukemia disseminated mouse model.16
MMAE has been tested in different clinical trials always conjugated with monoclonal antibodies in the form of antibody-drug conjugates (ADCs).17 Currently, there are two ADCs-MMAE approved by the FDA, being both indicated for the treatment of hematological malignancies.18 Brentuximab vedotin targets CD30, which is expressed in anaplastic large cell lymphoma and Hodgkin’s lymphoma cells,19 whereas polatuzumab vedotin targets the CD79b marker and was recently approved for the treatment of R/R DLBCL patients.20 Nevertheless, all ADCs, including these two ADCs-MMAE that have reached the clinical setting, share the issue of limited efficacy because less than 1% of the total administered ADC dose reaches the tumor.21,22 Here, we tested the T22-AUR nanoconjugate that displays around twelve CXCR4 ligands per drug-loaded nanoparticle,16 which enhances tumor uptake and selective CXCR4+ DLBCL cell killing and leads to a potent blockade of lymphoma dissemination while reducing systemic toxicity in non-infiltrated organs.
Materials and Methods
T22-AUR Nanoconjugate Production and Characterization
T22-GFP-H6 nanocarrier was produced in E. coli Origami B strain as previously described in Int J Nanomedicine. 2012;7:4533–44. Maleimide functionalized Monomethyl Auristain E (MC-MMAE) was acquired as custom synthesis from Levena Biopharma (Levena Biopharma, San Diego, CA, USA). T22-GFP-H6-MMAE (T22-AUR) nanoconjugate was synthetized by the covalent binding of MC-MMAE to T22-GFP-H6 through protein lysine amines (generation of Alkylamine bonds) in a one-pot reaction. For that, T22-GFP-H6 was incubated in presence of a 1:50 molar excess of MC-MMAE for 4 h at R.T in sodium carbonate buffer (166 mM NaCO3H, 333 mM NaCl pH=8). T22-AUR nanoconjugate was then re-purified by IMAC affinity chromatography in order to remove non-reacted free MC-MMAE molecules. Finally, re-purified nanoconjugates were dialyzed against sodium carbonate buffer and filtered through 0.22 µm pore filter. Conjugation efficiency was checked by MALDI-TOF mass spectrometry and nanoconjugate final concentration determined by Bradford assay.
Volume size distribution and zeta potential of parental T22-GFP-H6 nanocarrier and T22-AUR nanoconjugate was determined by Dynamic Light Scattering (DLS) and Electrophoretic Light Scattering (ELS), respectively, in a Zetasizer Nano ZS (Malvern instruments) at 633 nm.
Average molar mass of T22-GFP-H6 nanoparticle and T22-AUR nanoconjugate was determined by size exclusion chromatography coupled to a multi angle light scattering (SEC-MALS). For that, 200 µg of each sample was injected in a Superdex 200 increase 10/300 GL column (GE Healthcare, Chicago, Illinois, USA) and run in a sodium carbonate buffer supplemented with zinc (166 mM NaCO3H, 333 mM NaCl, 0.1 mM ZnCl2 pH=8). Eluent was monitored by an in-line UV-Vis detector, a Dawn Heleos MALS detector and an Optilab rEX RI detector (Wyatt Technology Corporation, Santa Barbara, California, USA). All data were then analyzed by Astra 6.0.2.9 software (Wyatt Technology Corporation) using dn/dc value of 0.185 (mL/g) and protein UV molar extinction coefficient value of 1.099 (mL/mg.cm).
DLBCL Cell Lines and Human PBMCs
The human Toledo and U-2932 DLBCL cell lines were cultured with RPMI 1640 medium whereas the human SUDHL-2 DLBCL cell line was cultured in IMDM medium. All cell lines were supplemented with 10% fetal bovine serum (FBS), 1% glutamine and 100 U/mL penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA, USA) and were incubated at 37°C and 5% CO2 in humidified atmosphere. Toledo cell line was purchased from the American Type Culture Collection (ATCC, Manassas, Virginia, USA) and U-2932 cell line from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). Finally, SUDHL-2 was kindly provided by Dr L. Pasqualucci (Columbia University, NY, USA) and its use approved by IIB-Sant Pau research ethics committee.
U-2932 cell line was transfected with the Luciferase gene (pPK-CMV- F3, Promokine, TE Huissen, The Netherlands) by electroporation (Nucleofector TM 2b Device, Lonza, Basel, Switzerland). Then, transfected cells were selected with 0.4 mg/mL of geneticin (Thermo Fisher Scientific) in order to achieve stable clones.
Fresh peripheral blood was obtained from healthy donors after acquiring informed consent and approval of the Hospital de la Santa Creu i Sant Pau Ethical Committee for Clinical Research. Human PBMCs were isolated by Lymphoprep gradient centrifugation (Stem Cell Technologies Vancouver, BC, Canada) followed by red blood cell lysis (Thermo Fisher Scientific) according to the manufacturer’s instructions. Human PBMCs were maintained in culture for 48 h in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 3% heat inactivated FBS, 2 mM L-glutamine (Thermo Fisher Scientific), 20% BIT 9500 Serum Substitute (StemCell Technologies), 5 ng/mL IL-3 (Peprotech, Rocky Hill, NJ, USA), 5 × 10−5 M β-mercaptoethanol (Sigma-Aldrich, St Louis, MO, USA), 1 mM sodium pyruvate and 0.1 mM non-essential amino acids (Thermo Fisher Scientific).
In vitro Nanoconjugate Internalization
The evaluation of the T22-AUR internalization in different DLBCL cell lines was performed measuring the GFP levels of the nanoconjugate and using the FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA). Cells were incubated with 10 nM T22-AUR for 1 h. Then, cells were washed with PBS and trypsinized for 15 min (1 mg/mL trypsin, Thermo Fisher Scientific) in order to remove nonspecific binding of nanoconjugates to the cell membrane. The competition assays were done by preincubating the cells for 1 h with 100 nM of the CXCR4 antagonist AMD3100 (Sigma-Aldrich, St-Louis, MO, USA). Results were analyzed with Cell Quest Pro software (BD Biosciences) and expressed as mean fluorescence intensity (MFI).
Cytotoxicity Assays
The in vitro cytotoxic effect of T22-AUR was evaluated using the colorimetric cell proliferation kit (XTT) after the exposure of the three DLBCL cell lines (3.5·105 cells/mL) or human PBMCs (10·105 cells/mL) to buffer (166 mM NaCO3H 333 mM NaCl, pH=8) or different concentrations of T22-AUR for 48 h in 96 well plates. Then, XTT reagent (Roche Diagnostics, Basel, Switzerland) was added and, after 4 h incubation, cell viability was quantified by measuring the absorbance at 492 nm wavelength using a FLUOstar OPTIMA spectrophotometer (BMG Labtech, Ortenberg, Germany). Moreover, the cytotoxic effect of free MMAE (Levena Biopharma) or vehicle DMSO (Sigma-Aldrich) in the DLBCL cell lines was also evaluated following the same procedure. IC50 of free MMAE for all DLBCL cell lines was calculated using the GraphPad Prism 6 program. Competition viability assays were done by 1 h pretreatment of U-2932 cells with AMD3100 followed by the addition of the T22-AUR nanoparticle (ratio 10 AMD300 – 1 T22-AUR). Data were shown as percentage of cell death in relation to its control (buffer, DMSO or AMD3100).
CXCR4 Flow Cytometry
CXCR4 membrane determination was done by flow cytometry in human PBMCs and DLBCL cells, after washing with PBS 0.5% BSA and incubating with either PE-Cy5 mouse anti-human CXCR4 monoclonal antibody (BD Biosciences) or PE-Cy5 Mouse IgG2a isotype (BD Biosciences) as negative control. Results were analyzed using the FACS Calibur flow cytometer with the Cell Quest Pro software and expressed as ratio of CXCR4 MFI divided by isotype MFI.
Cell Cycle Analysis
Cells were treated with buffer or 125 nM of T22-AUR for 24 h and 48 h. Then, 1.2·106 cells were centrifuged and washed with PBS. Afterwards, cells were fixed with cold 66% ethanol for 2 h at 4°C, centrifuged and washed with PBS. Finally, propidium iodide (Merck Millipore, Burlington, Massachusetts, USA) and RNAse (Thermo Fisher Scientific) were added at a final concentration of 40 µg/mL and 20 µg/mL, respectively, and cells were incubated at 37°C for 20 min. Cells were analyzed using the FACS Calibur flow cytometer and data were obtained using the Cell Quest Pro software (BD Biosciences).
Paraffin-Embedded Cell Blocks
Toledo, U-2932 and SUDHL-2 cells were seeded at 3.5·105 cells/mL in 75 cm2 cell culture flasks. Moreover, U-2932 cells were either treated with buffer or 125 nM of T22-AUR for 24 h and 48 h. Then, paraffin-embedded cell blocks were prepared from the pellet of centrifuged cell suspension by adding five drops of plasma and thrombin to enmesh the cellular material in a clot. Then, cell clots were placed in a cassette, fixed in 4% paraformaldehyde and paraffin-embedded in a tissue processor (Sakura, Tokyo, Japan) for future histology or immunohistochemistry (IHC) evaluations.
DAPI Staining
U-2932 cells (3.5·105 cells/mL) were incubated with buffer or 125 nM T22-AUR for 24 h and 48 h. After, cells were washed with PBS, resuspended with 3.7% of paraformaldehyde (Thermo Fisher Scientific) and fixed for 10 min at −20°C. Then, cells were resuspended with 10 μL of PBS and were let air dry on a slide. Finally, cells were stained with DAPI mounting medium (Thermo Fisher Scientific) and visualized using a fluorescence microscope (Olympus BX53, Olympus, Tokyo, Japan). Representative pictures were taken using an Olympus DP73 digital camera and processed with the cellSens Dimension 1.9 software (Olympus) at 400x. Apoptotic cells were visualized as chromatin condensation and/or formation of apoptotic bodies, while mitotic catastrophe (MC) cells as aberrant mitosis formation.
Annexin/PI Staining
DLBCL cells were seeded at 3.5·105 cells/mL and treated with buffer or 125 nM T22-AUR for 24 h or 48 h. The percentage of apoptosis (early/late) for each condition was evaluated using the Annexin V-CF Blue/PI apoptosis detection kit (Abcam, Cambridge, UK) and following the manufacturer’s instructions. Data were analyzed by MACSQuant analyzer flow cytometry using the MACS Quantify version 2.3 software (Miltenyi Biotec).
Western Blotting
DLBCL cells were seeded at 3.5·105 cells/mL and treated with buffer or 125 nM T22-AUR for 24 h and 48 h. Then, cells were washed with PBS and resuspended in lysis buffer (1 M Tris/acetate, 1 M sucrose, 100 mM EDTA, 100 mM EGTA, 10% Triton-X-100, 100 mM Naorto, 100 mM Naβglycerol, 0.5 M NaF, 100 mM Napyro, β-mercapto, 100 mM Benzamidine, 1.74 mg/mL PMSF and 2 mg/mL leupeptin). The cell suspension was sonicated and rested for 20 min on ice and centrifuged for 10 min at 18000g at 4°C. Protein concentration in supernatant was determined using the Bradford protein assay (BioRad, Hercules, California, USA), according to the manufacturer’s instructions. Cell lysates (50 μg) were separated using 12–15% SDS-PAGE and transferred to a nitrocellulose blotting membrane. Membranes were blocked with 5% skim milk in TBST for 2 h at room temperature, and then incubated with primary antibodies, 1:2000 PARP (556494, BD) O/N, 1:1000 caspase-3 (610322, BD) O/N, 1/1000 cleaved caspase-3 (9661, Cell Signaling Technology, Danvers, Massachusetts, USA) O/N, 1:10000 GAPDH (MAB374, Merck Millipore) for 1 h or 1:1000 α/β tubulin (2148, Cell Signaling Technology) for 1 h. Membranes were washed with TBST and then incubated with the appropriate secondary antibody (1:10000, Jackson Immune Research, West Grove, Pennsylvania, USA). Western blot visualization was performed using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) and the Universal Hood II Gel Doc Imaging System (BioRad).
Animal Maintenance
Four-week-old female NOD/SCID mice were obtained from Charles River Laboratories. Mice were maintained in specific pathogen-free (SPF) conditions with sterile food and water ad libitum. All animal experiments were approved by the Hospital de la Santa Creu i Sant Pau Animal Ethics Committee (project number 10108 by the Government of Catalonia) and performed following the European Union Directive 2010–63-EU for welfare of the laboratory animals.
In vivo Nanoconjugate Biodistribution
The nanoconjugate biodistribution was assessed in a subcutaneous (SC) DLBCL mouse model. Firstly, 10 million DLBCL Toledo cells were injected in both dorsal flanks of NOD/SCID mice. Tumor growth was monitored twice a week with bilateral caliper measurements. The mean tumor volumes were calculated using the equation (length·width2)/2. When tumors reached a volume of 600–800 mm3, mice received a single intravenous (IV) dose of 325 μg T22-AUR (n=3/time point) or 166 mM NaCO3H 333 mM NaCl pH=8 buffer (n=3). Fluorescence intensity (FLI) was measured ex vivo at two time points (5 h and 24 h) in subcutaneous tumors and all organs after carefully washing each organ with physiological serum. The emitted FLI, expressed as average radiant efficiency, measures the amount of accumulated T22-AUR, detected by the fluorescence emission of the GFP domain, in each tissue. FLI from experimental mice was calculated subtracting the FLI auto-fluorescence of control mice. FLI was registered in the IVIS Spectrum 200 Imaging System (Perkin Elmer, Waltham, MA, USA). The area under the curve (AUC) of the emitted FLI during the 0–24 h period was calculated using the GraphPad Prism 6 program.
Finally, tumors were collected, fixed and paraffined to perform immunofluorescence assays.
In vivo Nanoconjugate Antineoplastic Effect
NOD/SCID mice were intravenously injected with 20 million luciferase-transfected U-2932 cells in 200 μL of physiological serum. Three days after cell injection, mice were divided randomly in the control group, administered with buffer (166 mM NaCO3H 333 mM NaCl, pH=8), and the experimental group administered with 100 μg T22-AUR. The administration of buffer or T22-AUR was performed three times per week for twelve doses. Lymphoma dissemination was monitored, once per week, using bioluminescence imaging (BLI, total radiance photons) in the IVIS Spectrum (Perkin-Elmer). Mice were anesthetized with 3% isoflurane in oxygen and BLI was captured 5 min after intraperitoneal injection of firefly D-luciferin (2.25 mg/mouse, Perkin Elmer). Moreover, mouse body weight was registered throughout the course of the study.
All mice were euthanized when the first mouse presented relevant signs of disease such as poor mobility or significant weight loss. At that day, the BLI of lymphoma-infiltrated organs was analyzed ex vivo. Finally, all organs were collected, fixed in 4% formaldehyde and paraffin-embedded for further histopathological or immunohistochemical evaluations.
Immunofluorescence Microscopy
Tumor paraffin sections were used for the simultaneous immunodetection of GFP, CXCR4 and DAPI staining in a confocal microscopy. Tumor slides were heated 15 min at 60°C, dewaxed and rehydrated. Antigen retrieval was performed using pH=9 citrate buffer with DeCloaking Chamber. For the immunodetection, sections were washed 3 times with TBS for 5 min and blocking buffer (TBS1x + 0.5% triton + 3% donkey serum) was added 1 h at RT. Slides were incubated with the GFP chicken IgY 1:250 (AVES, Davis, CA, USA) and the CXCR4 rabbit IgG 1:250 (Abcam) primary antibodies overnight at 4°C and 2 h at RT. Then, sections were washed and the secondary antibodies, an anti-chicken IgY-Cy2 1:50 antibody together with an anti-rabbit IgG-Cy5 1:200 antibody, were added to the tissue sections for 2 h at 37°C. After three additional TBS washes, the sections were stained with DAPI 1:10000 for 10 min at RT and, finally, the mounting medium was added.
Furthermore, to determine whether T22-AUR colocalized with lysosomes, live U-2932 cells were incubated for 5 h with 1 µM T22-AUR. Then, cells were centrifuged and resuspended with 1 mL of cultured medium. LysotrackerTM DeepRed (Thermofisher) and Hoescht bisBenzimide H 33342 trihydrochloride (Sigma-Aldrich) were added at a final concentration of 70 nM (37°C for 1 h) and 1 μg/mL (37°C for 30 min), respectively.
All immunofluorescence pictures were taken with 4.17 or 5.00 zoom using the 63X/1.30 objective in a Confocal TCS SPE microscope (Leica, Wetzlar, Germany) using LAS AF software (Leica).
Histopathology
All organ sections of paraffin-embedded samples were analyzed histopathologically (H&E staining) by two independent observers to assess either the antineoplastic effect in DLBCL infiltrated organs or the possible toxicity in non-DLBCL infiltrated organs. Pictures were taken at 100x or 200x using an Olympus DP73 digital camera and processed with the cellSens Dimension 1.9 software (Olympus).
Immunohistochemistry (IHC)
Anti-CXCR4 (1:300, Abcam) antibody was used for the evaluation of CXCR4 expression in cellular blocks from three different DLBCL cell lines. Anti-H2AX pSer139 (γH2AX, 1:800, Novus, Centennial, CO, USA) and anti-cleaved Poly (ADP-ribose) polymerase (cleaved PARP, 1:300, Promega, Madison, Wisconsin, USA) antibodies were used in cellular blocks from U-2932 cells treated with T22-AUR. Anti-CD20, anti-CD79a, anti-Ki67 (Dako, Santa Clara, CA, USA) and anti-CXCR4 (1:300, Abcam) IHC were performed in mouse paraffin-embedded organs. All IHC analyses were performed in a DAKO Autostainer Link48 following the manufacturer’s instructions. The stained area of positive cells was quantified using the Olympus CellD Imaging 3.3 software in 6-fields for cellular blocks and 12-fields for mouse organs. Data were shown as positive stained area of samples treated with T22-AUR divided by the positive stained area of buffer-treated samples. The pictures were taken at 100x, 200x or 400x. Moreover, for the CXCR4 determination in cellular blocks, pictures were captured with the exact same exposure time and the CXCR4 intensity was measured using the Image J software (1.8.0.172). We used the Colour Deconvolution Plugin with the H DAB vector to split the brown staining adjusting the threshold to 30. After that, the “Analyze particles” plugin was used to detect all stained areas and the mean gray value was obtained combining all selected black areas and using the ROI Manager. The intensity value of each analyzed slide was calculated subtracting 255 to the mean gray value obtained by the Image J analysis.
Statistical Analysis
All results were expressed as mean ± standard error (SE). Differences between groups were analyzed using either the T-Student test or Mann–Whitney U-test after the testing of normality (Shapiro Wilk test). Differences were considered statistically significant at p≤0.05. Statistical calculations were performed using SPSS software v21 (New York, NY, USA).
Results
Characterization of Functionalization of T22-AUR Nanoconjugate
T22-AUR nanoconjugate was generated by the covalent binding of maleimidocaproyl functionalized MMAE molecules (MC-MMAE) through solvent exposed protein lysine-amines of T22-GFP-H6 nanocarrier in a one-pot reaction (Figure 1A). MALDI-TOF mass spectrometry spectra revealed up to 12 peaks with a sequential increment of around 911 Da over the T22-GFP-H6 protein molecular weight (30.6 kDa) (Figure 1B), corresponding each one to the incorporation of an additional MC-MMAE molecule (911 Da). T22-GFP-H6 conjugation did not significantly disturb nanoparticles supramolecular structure, which showed a mild increment in size (+5.5 nm) upon conjugation (Figure 1C). Finally, SEC-MALS analysis showed an average molar mass increment of 207 kDa in T22-AUR nanoconjugate (564 kDa) over parental T22-GFP-H6 nanoparticles (357 kDa). This fact suggested an average incorporation of around 19 MMAE molecules per protein (or around 228 MMAE molecules per nanoparticle) considering that each nanoparticle is composed by approximately 12 monomers of T22-GFP-H6 protein (Figure 1D).
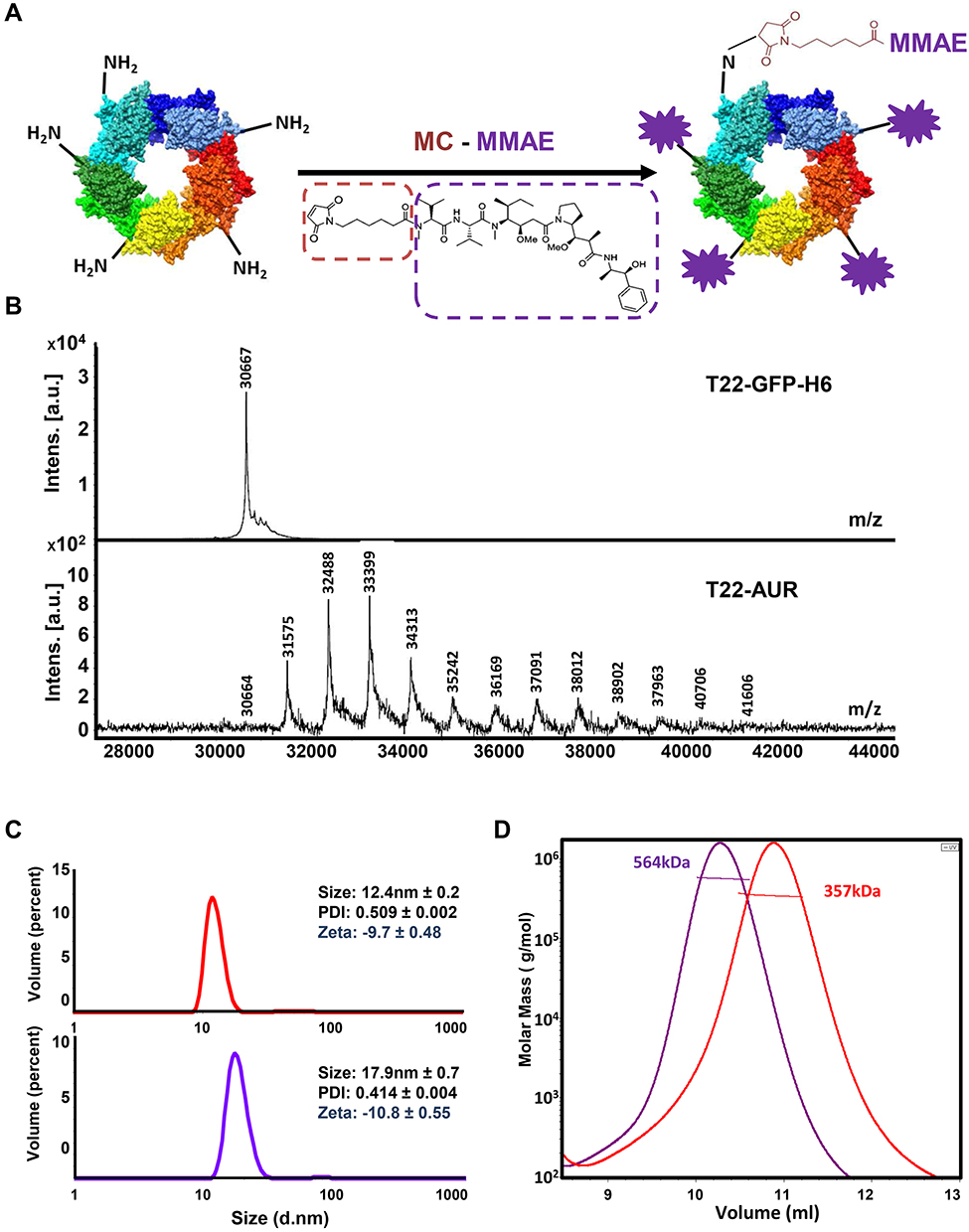 |
Figure 1 T22-AUR nanoconjugate characterization. (A) T22-GFP-H6 nanoparticle conjugation with maleimide functionalized MMAE (MC-MMAE) through protein lysine amines in order to form the T22-AUR nanoconjugate. The number of conjugated MMAE molecules (purple stars) is only illustrative. (B) MALDI-TOF mass spectrometry spectra of parental T22-GFP-H6 and T22-AUR nanoconjugate. Each peak over 30.6 kDa in T22-AUR indicates the covalent incorporation of an additional MMAE molecule (+911Da). (C) Volume size distribution (size) and zeta potential (zeta) of T22-GFP-H6 nanoparticle (red) and T22-AUR nanoconjugate (purple) determined by light scattering. Pdi indicates polydispersion index. Data are presented as mean ± standard error. (D) Average molar mass distribution of parental T22-GFP-H6 nanoparticle (red) and T22-AUR nanoconjugate (purple) determined by size exclusion chromatography coupled to a multi angle light scattering (SEC-MALS). |
Specific T22-AUR Entrance Through the CXCR4 Receptor in DLBCL Cell Lines
Before evaluating the therapeutic effect of the T22-AUR, we first determined the in vitro internalization of the nanoconjugate inside the DLBCL cell lines through the CXCR4 receptor expressed in the cell membrane. Toledo and U-2932 DLBCL cell lines had high levels of CXCR4 receptor (CXCR4+ DLBCL cells), achieving intensity mean values as high as 227.68±0.06 and 226.42±0.05, respectively, over a total of 255. In contrast, the SUDHL-2 cell line did not express the receptor (Figure 2A and B). Then, the T22-AUR internalization was measured, after 1 h exposure, by quantifying the fluorescence emission of the GFP domain incorporated to the nanoconjugate. T22-AUR internalized in CXCR4+ DLBCL cells in a high amount, being the MFI 27.88±3.01 and 14.25±1.64 in Toledo and U-2932 cells, respectively. This internalization was CXCR4-dependent since when CXCR4+ DLBCL cells were pre-incubated 1 h with AMD3100, an antagonist of CXCR4 receptor, the nanoconjugate entrance was blocked and the MFI significantly decreased, reaching levels as low as 5.92±0.22 in Toledo cells and 4.02±0.09 in U-2932 cells. Besides that, the negative-CXCR4 SUDHL-2 cell line showed low MFI levels (3.15±0.13) after 1 h exposure to T22-AUR, which practically did not differ from control cells treated with buffer (2.97±0.12) or cells pre-incubated with AMD3100 before the T22-AUR addition (2.97±0.01) (Figure 2C).
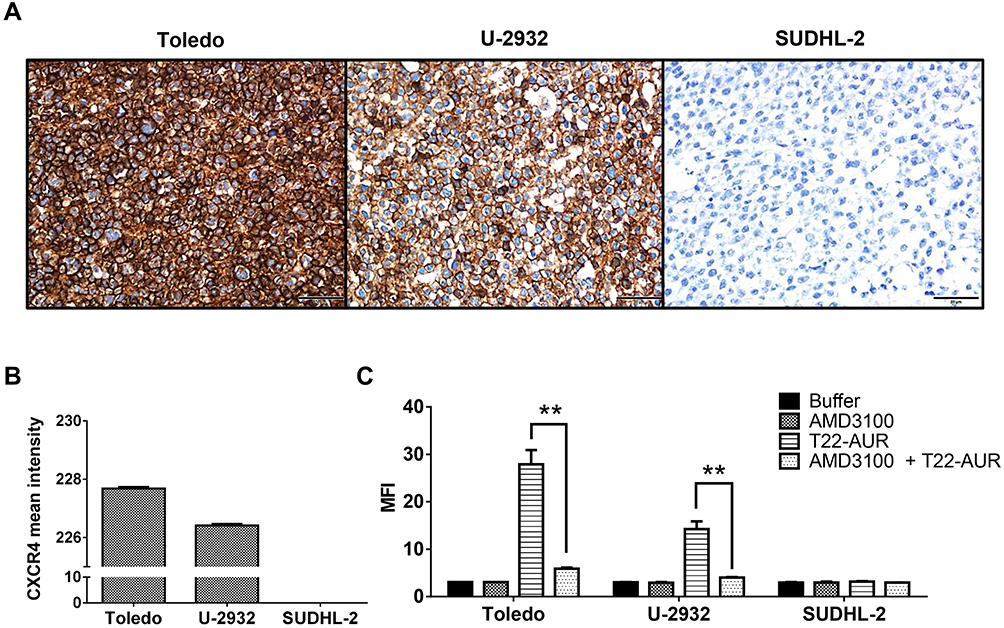 |
Figure 2 Assessment of CXCR4 expression intensity and nanoconjugate internalization in different DLBCL cell lines. (A) CXCR4 IHC of Toledo, U-2932 and SUDHL-2 cellular blocks. Pictures were taken at 400x (scale bars=50 μm). (B) Quantitation of mean CXCR4 expression intensity counting six IHC-stained fields (400x) for Toledo, U-2932 and SUDHL-2 cellular blocks. (C) MFI quantification by flow cytometry in Toledo, U-2932 and SUDHL-2 cells after their incubation in buffer, 100 nM AMD3100 or 10 nM T22-AUR for 1 h, or after pretreatment for 1 h with 100 nM AMD3100 followed by the addition of 10 nM of nanoconjugate for 1 h (AMD3100 + T22-AUR). This experiment was performed in biological triplicates. All data are shown as mean ± standard error. **p≤0.01. Abbreviations: IHC, immunohistochemistry, MFI, mean fluorescence intensity. |
High Tumor Uptake and Internalization of T22-AUR in SC Tumors of CXCR4+ DLBCL Cells
After a single intravenous dose of 325 μg of T22-AUR in a subcutaneous (SC) mouse model bearing CXCR4+ Toledo cells, the nanoconjugate achieved a high tumor uptake in SC tumors and negligible fluorescence levels (FLI) in the normal organs (spleen, pancreas, liver, heart, lungs, kidneys and bone marrow) (Figure 3A). The FLI peak of T22-AUR in SC tumors was at 5 h (1.36±0.51·107 radiant efficiency) and decreased considerably at 24 h (0.61±0.29·107 radiant efficiency). The highest FLI levels in non-DLBCL infiltrated organs were observed in kidneys at 5 h (0.35±0.06·107 radiant efficiency) and liver at 24 h (0.22±0.22·107 radiant efficiency), which showed 3.88-fold and 2.77-fold lower levels than SC tumors, respectively, at each corresponding time point (Figure 3B above). Moreover, the AUC of the emitted FLI revealed that the highest percent of the total T22-AUR injected dose accumulated in SC tumors (57.91 ±5.01%), followed by the uptake in kidneys (15.10±2.32%) and liver (10.74±1.84%) (Figure 3B below).
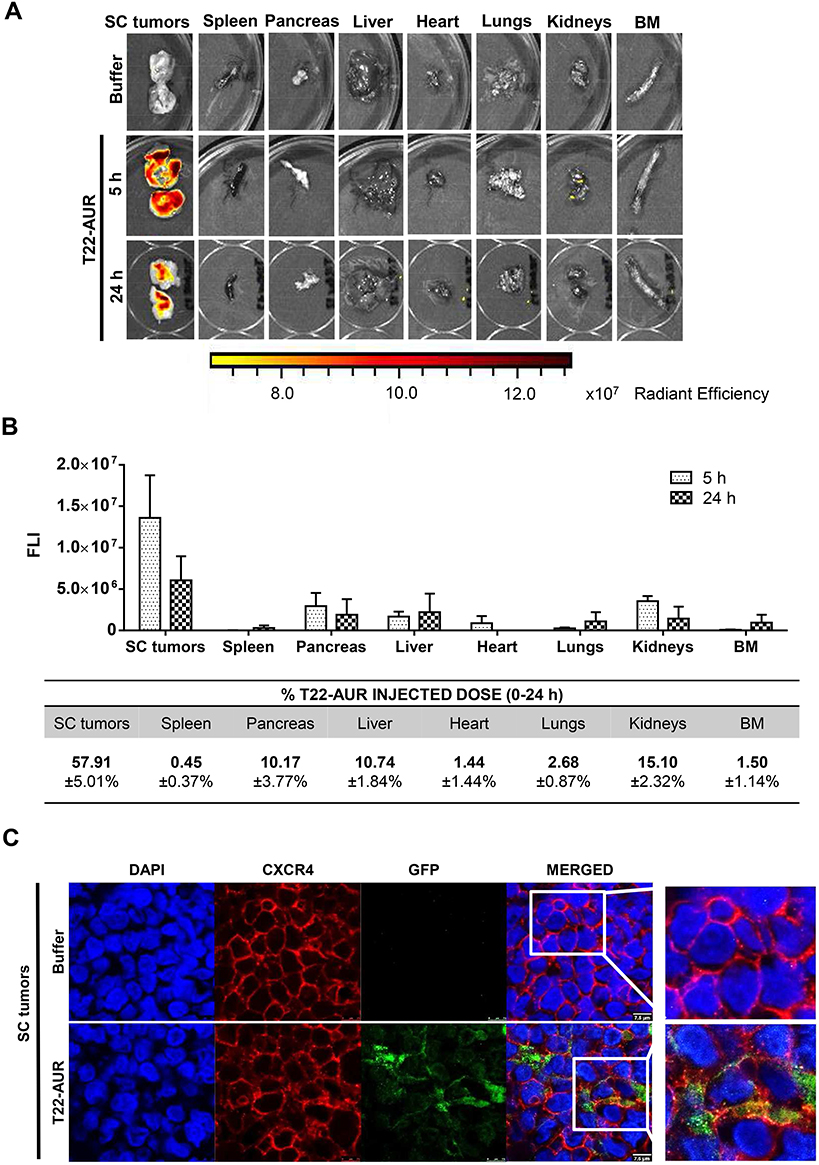 |
Figure 3 T22-AUR tissue biodistribution in a SC mouse model bearing CXCR4+ DLBCL cells. (A) Representative images of the FLI emitted in SC Toledo tumors and normal organs in animals treated either with buffer or the fluorescent nanoconjugate T22-AUR at 5 and 24 h. (B) Above: FLI quantification measured in SC Toledo tumors and normal organs in T22-AUR-treated mice 5 h (n=3) and 24 h (n=3) postinjection. FLI was calculated subtracting the autofluorescence of each tissue from buffer-treated mice (n=3) and represented as radiant efficiency. Below: Quantification of the percentage of T22-AUR injected dose that accumulated in SC tumors and non-infiltrated organs (spleen, pancreas, liver, heart, lungs, kidneys and BM). This percentage was measured using the area under the curve (AUC) of emitted FLI registered in each organ calculated in relation to the total emitted FLI during the 0–24 h period. (C) Confocal images visualizing cell nuclei (DAPI), CXCR4 receptor, GFP protein and the merged of the three staining in the SC Toledo tumors from animals treated with buffer or T22-AUR at 5 h. Scale bars=7.5 μm. All data are shown as mean ± standard error. Abbreviations: BM, bone marrow; FLI, fluorescence intensity; SC, subcutaneous. |
Furthermore, we analyzed the in vivo T22-AUR internalization in the CXCR4+ Toledo cells of SC tumors. By immunofluorescence, we could detect the localization of T22-AUR, with an anti-GFP antibody, observing that it was located in the membrane as well as in the cytosol of Toledo cells in the SC tumors of mice treated with a single bolus of T22-AUR. In the buffer-treated animals, there was no presence of GFP in the SC tumors. Regarding the CXCR4 expression, the receptor was detected predominantly in the membrane of SC tumor cells of animals treated with buffer whereas the CXCR4 expression was more dot-pointed in the group of animals treated with the nanoconjugate, suggesting the endocytosis of the receptor inside the cytosol of lymphoma cells. In the merged images, a co-localization of CXCR4 and T22-AUR was visualized in the membrane and inside of lymphoma cells (Figure 3C).
Cytotoxic Effect of Untargeted Free MMAE and CXCR4-Targeted T22-AUR Nanoconjugate on the DLBCL Cell Lines
Here, we assessed the sensitivity of the three DLBCL cell lines to untargeted free MMAE (the drug conjugated to the T22-AUR nanoconjugate). Our results demonstrated that U-2932 cells were the most sensitive (IC50=0.33±0.14 ng/mL) followed by SUDHL-2 (IC50=0.50±0.08 ng/mL) and finally Toledo (IC50=0.87±0.09 ng/mL). Thus, Toledo cells (with high CXCR4 expression) incubated with free MMAE showed significantly less cell death and, therefore, significantly higher IC50 than U-2932 (with high-intermediate CXCR4 expression) and SUDHL-2 cells (lacking CXCR4 expression) (Figure 4A and B). These data show a lack of relationship of the cytotoxicity of free MMAE with CXCR4 expression in DLBCL cell lines.
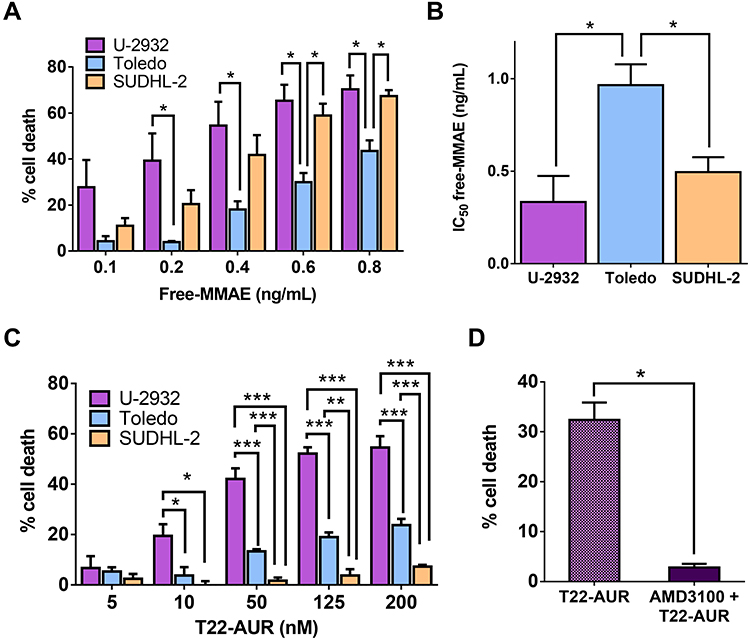 |
Figure 4 Cytotoxic effect of untargeted free MMAE and CXCR4-targeted T22-AUR nanoconjugate on the DLBCL cell lines. (A) Percentage of cell death induced after the incubation of free-MMAE for 48 h in CXCR4+ DLBCL cell lines (U-2932 and Toledo) and CXCR4− DLBCL cell line (SUDHL-2) measured by colorimetric cell proliferation kit (XTT). (B) Quantification of the half-maximal inhibitory concentration (IC50) of free-MMAE in each DLBCL cell line. (C) Percentage of cell death induced by different concentrations of T22-AUR for 48 h in DLBCL cell lines. (D) Competition assays (measured as percentage of cell death) done by 1 h pretreatment of U-2932 cells with 100 nM AMD3100 followed by the addition of 10 nM of nanoconjugate. All experiments were performed in biological triplicates and data are shown as mean ± standard error. *p≤0.05; **p≤0.01 and ***p≤0.005. |
Next step was to assess the cytotoxic effect of T22-AUR as well as its CXCR4-dependent cytotoxicity in the three DLBCL cell lines. The higher cytotoxic effect was shown in U-2932 cells (52.14±2.51% at 125 nM T22-AUR) followed by Toledo cells (18.95±1.93% at 125 nM T22-AUR), and finally the SUDHL-2 cell line, which is the CXCR4-negative DLBCL cell line, only showed barely cell death (3.76±2.44% at 125 nM T22-AUR). Moreover, U-2932 cells underwent a significantly higher level of cell death than Toledo or SUDHL-2 cells at both low and high concentrations, whereas Toledo cell line achieved significant differences with SUDHL-2 at high T22-AUR concentrations (Figure 4C). Thus, despite Toledo cells express higher levels of CXCR4 receptor than U-2932 cells, they are less sensitive to T22-AUR because of their lower sensitivity to the untargeted drug itself (free MMAE) (Figure 4A and B). Moreover, SUDHL-2 cells, which are more sensitive to free MMAE than Toledo cells, but lack CXCR4 expression, show very low levels of cell death induction after T22-AUR treatment demonstrating that the entry of the nanoconjugate in the cells through CXCR4 is essential for the nanoconjugate cytotoxic effect. Additionally, we confirmed the CXCR4-dependence of the T22-AUR cytotoxic effect because after blocking the receptor with AMD3100 the cell death of U-2932 cells decreased significantly from 32.42±2.00% to 2.83±0.40% (Figure 4D).
Mechanism of Action and Cell Death Induction by T22-AUR in CXCR4+ DLBCL Cells
Since T22-AUR included a non-cleavable linker in its conjugation with MMAE, the T22-AUR must be proteolyzed within the lysosomes for the release of the MMAE into the cytoplasm, where it can exert the drug cytotoxic effect in DLBCL cells, as it has been described for ADCs that use non-cleavable linkers to bind and transport MTAs.23 To check this process, we performed an immunofluorescence assay with nuclear (Hoescht dye) and lysosomal (lysotracker Deep Red dye) staining in live U-2932 cells treated with T22-AUR. We detected the presence of T22-AUR (green), by the GFP protein, into lysosomes (red), which both merged (yellow) within U-2932 cells. Moreover, we also observed the presence of the nanoconjugate in the cell membrane (Figure 5A). These results suggested the endocytosis of T22-AUR and its subsequent destination to lysosomes, undergoing complete proteolysis of the protein nanocarrier which releases the attached MMAE to the cytosol where it binds and inhibits tubulin to induce cell death. Thus, T22-AUR lysosomal entry and the subsequent CXCR4-dependent cell death confirm the successful conjugation of MMAE to the nanocarrier.
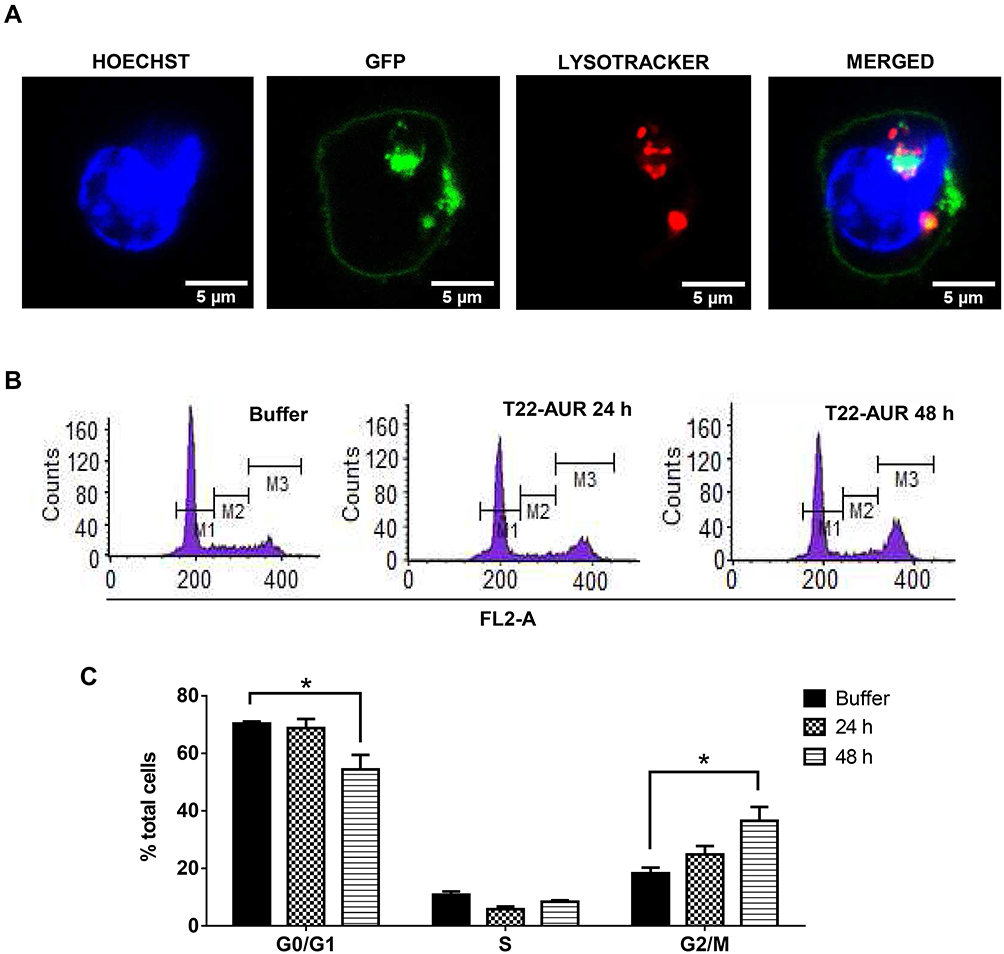 |
Figure 5 T22-AUR-lysosomal colocalization and cell cycle arrest by T22-AUR in CXCR4+ DLBCL cells. (A) Hoechst (blue staining), GFP protein (green staining) and lysotracker (red staining) immunofluorescence detection in live U-2932 cells exposed to 1 μM T22-AUR for 5 h. Scale bars=5 μm. (B) Representative graphics for cell cycle analysis in U-2932 cells treated with buffer or 24 or 48 h with 125 nM T22-AUR. M1: G0/G1 phase, M2: S phase and M3: G2/M phase. (C) Quantification of the percentage of total U-2932 cells in different cell cycle phases (G0/G1, S and G2/M) after incubation with buffer or 125 nM T22-AUR (24 and 48 h). This experiment was performed in biological triplicates and data are shown as mean ± standard error. *p≤0.05. |
In order to further elucidate the cell death mechanisms underlying T22-AUR cytotoxicity, we performed different experiments. We analyzed the cell cycle phase distribution after 24 h and 48 h of 125 nM T22-AUR exposition in U-2932 cells. As time elapsed, the percentage of U-2932 cells in G0/G1 phase decreased from 70.37±0.68% in the buffer-treated cells to 54.44±5.01% after 48 h with the nanoconjugate incubation. This G0/G1 reduction was mainly due to the increase of the G2/M phase, which represented the 18.29±1.96% of total cells in the buffer group and the 36.58±4.80% of T22-AUR treated cells for 48 h (Figure 5B and C).
After performing DAPI staining, we observed a 9.16-fold significant increase of U-2932 cells undergoing mitotic catastrophe (MC) after 24 h of T22-AUR incubation compared to cells treated with buffer. At 48 h, cells undergoing MC diminished compared to the T22-AUR incubation for 24 h, although the increase (2.42-fold) continued being significant with respect to buffer-treated cells. The ratio of apoptosis in cells treated with T22-AUR was significantly higher compared to buffer-treated cells, 17.35-fold and 38.93-fold at 24 h and 48 h, respectively (Figure 6A and B).
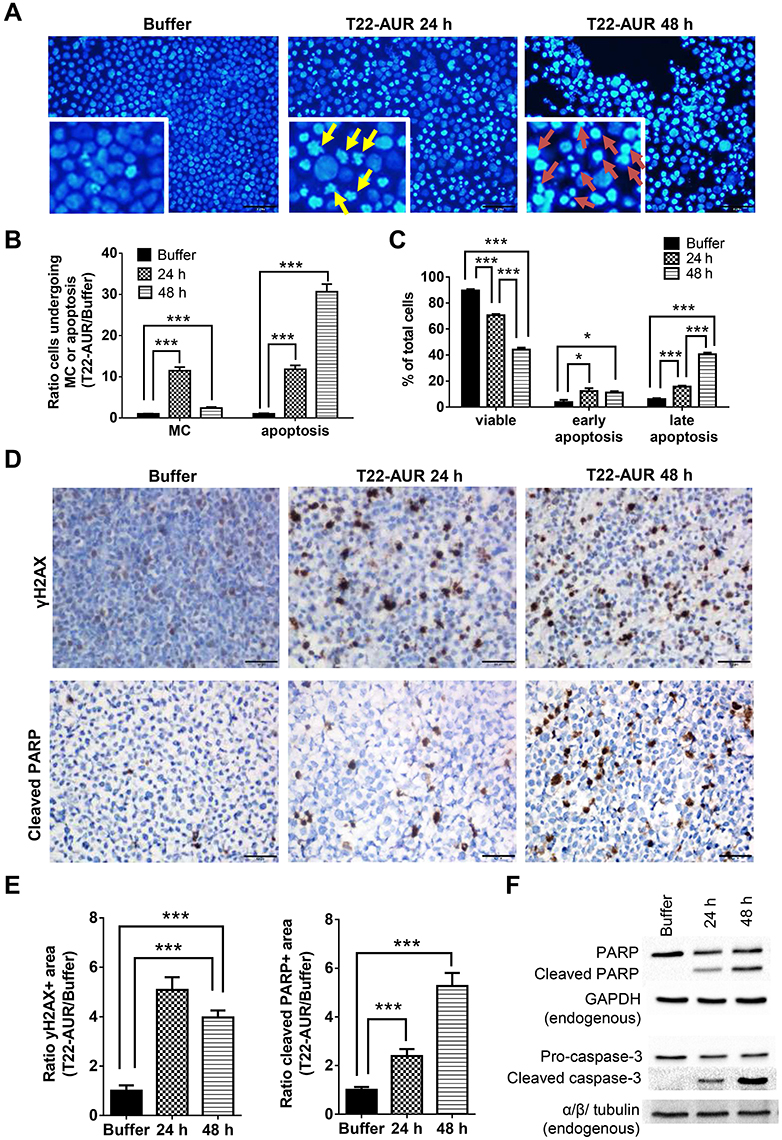 |
Figure 6 Further mechanisms of cell death induction by T22-AUR in CXCR4+ DLBCL cells. (A and B) DAPI staining pictures and ratio quantification of MCs (yellow arrows) or apoptotic bodies (red arrows) in U-2932 cells treated with buffer or 125 nM-T22-AUR (24 and 48 h). Ratio quantification was measured dividing the number of cells undergoing MC or apoptosis in 10 fields of T22-AUR samples (24 and 48 h) by 10 fields of buffer-exposed samples. (C) Annexin/PI assay showing the total percentage of viable, early apoptotic and late apoptotic U-2932 cells treated with buffer or 125 nM T22-AUR (24 h and 48 h). These experiments were performed in biological triplicates. (D) IHC staining using γH2AX and cleaved PARP antibodies after buffer or 125 nM T22-AUR treatment (24 and 48 h) in U-2932 cells. (E) Ratio quantification of the IHC positive stained area marked by γH2AX and cleaved PARP in buffer or T22-AUR-treated samples. Ratio quantification was represented dividing the area of positive cells from 6 fields of T22-AUR-treated samples (24 and 48 h) by 6 fields of buffer-treated samples. (F) Anti-PARP, cleaved PARP, pro-caspase-3 and cleaved caspase-3 detection, by Western blotting, in U-2932 extracts treated with buffer or 125 nM T22-AUR for 24 h and 48 h. GAPDH and α/β tubulin were used as endogenous controls. All pictures were taken at 400x (scale bars= 50 μm). All data are shown as mean ± standard error. *p≤0.05; ***p≤0.005. Abbreviation: MC, mitotic catastrophe. |
Moreover, by performing the Annexin/PI staining in U-2932 cells, we identified a significant decrease in the percentage of viable cells treated with the nanoconjugate, from 89.62±0.97% in cells treated with buffer to 70.54±1.01% after 24 h exposure to T22-AUR and 44.17±1.53% after 48 h exposure. The low viability was associated with a significant increase of the percentage of T22-AUR-treated cells undergoing early apoptosis (from 3.82±1.76% to 12.27±2.30% after 24 h and 11.15±0.99% after 48 h) and late apoptosis (from 6.06±0.72% to 15.68±0.72% at 24 h and 40.55±1.25% at 48 h) (Figure 6C).
Afterwards, we quantified by IHC the phosphorylation of the histone H2AX (γH2AX) in order to know whether DNA double-strand breaks were induced by T22-AUR. U-2932 cells treated with the nanoconjugate achieved a significant 5.09-fold and 3.98-fold increase of γH2AX-positive area at 24 h and 48 h, respectively, compared to cells treated with buffer. Additionally, the positive area of cleaved PARP, a marker of apoptosis, was also significantly higher in these cells treated with T22-AUR, 2.38-fold at 24 h and 5.27-fold at 48 h, compared to buffer group (Figure 6D and E). Finally, we confirmed the activation of the apoptotic pathway, by Western blot, detecting the expression of cleaved PARP as well as cleaved caspase-3 in U-2932 cell extracts treated with T22-AUR for 24 h and 48 h (Figure 6F).
In vivo Antineoplastic Effect of the T22-AUR Nanoconjugate in a CXCR4+ DLBCL Disseminated Mouse Model
Afterwards, we evaluated the in vivo therapeutic effect of T22-AUR in a disseminated mouse model of CXCR4+ U-2932-Luci cells. The lymphoma dissemination, detected by bioluminescence imaging (BLI) emitted by U-2932-Luci cells, was exhibited on CNS and BM (frequently backbone and rarely cranium or jaw). Three days after the intravenous injection of lymphoma cells, we started the administration of buffer or 100 μg T22-AUR (≈60 nmol of MMAE in one dose), three days per week and a total of 12 doses (≈720 nmol of MMAE in the total dosage). The emitted BLI was reduced in the T22-AUR-treated animals compared to buffer-treated animals (Figure 7A). Indeed, the AUC from the whole study showed a significant reduction of BLI in the mice group treated with the therapeutic nanoconjugate (0.53±0.06·108 total radiance photons) compared to the buffer-group (1.71±0.51·108 total radiance photons) (Figure 7B). We ended the experiment when the first buffer-treated mice showed a significant weight loss. Nevertheless, the nanoconjugate-treated mice did not lose weight nor did present any symptoms of illness (Figure 7C). At that time, we analyzed ex vivo the infiltrated lymphoma organs of the U-2932-Luci disseminated mouse model. The organs with highest lymphoma-cell infiltration were bone marrow (BM, backbone) and central nervous system (CNS, brain). Both organs were infiltrated in 7 out of 9 buffer-treated mice. The BM was not infiltrated in any mice treated with T22-AUR and the CNS was infiltrated in only 3 out of 10 T22-AUR-treated mice. Moreover, the levels of BLI in these organs (BM and CNS) were significantly reduced in the T22-AUR-treated compared to buffer-treated animals (Figure 7D).
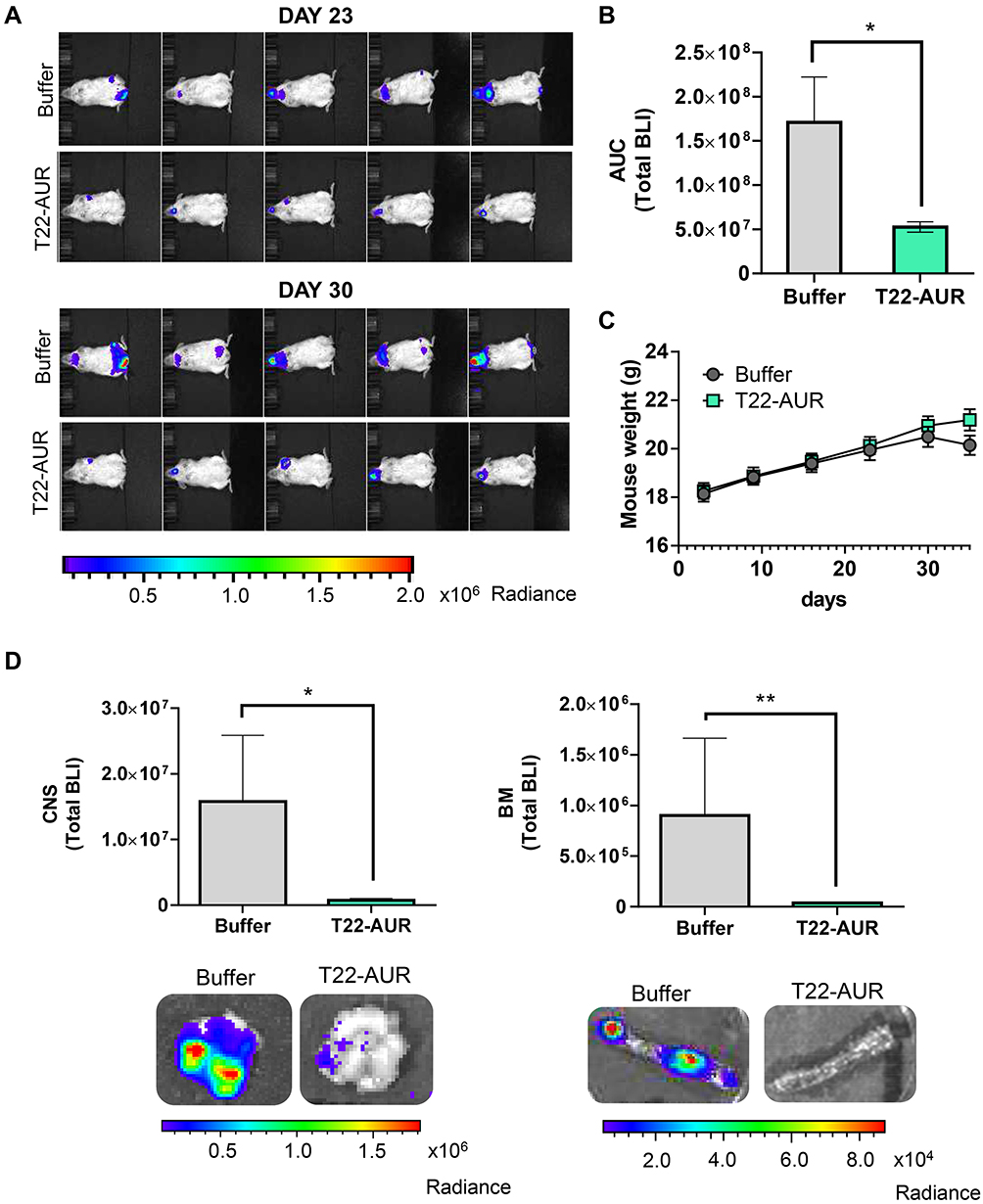 |
Figure 7 In vivo antineoplastic effect of T22-AUR in a disseminated mouse model of CXCR4+ DLBCL-Luci cells. (A) Representative images of lymphoma dissemination, registered by measuring the BLI emitted by U-2932-Luci cells, in animals treated with buffer or T22-AUR at the last days of the experiment (days 23 and 30 postinjection of lymphoma cells). (B) Area under the curve (AUC) of the lymphoma follow-up (from day 1 to day 30) registered by total BLI emission from buffer-treated mice (n=9) and T22-AUR-treated mice (n=10). (C) Body weight of mice treated with buffer (n=9) or T22-AUR (n=10) during the whole experiment. (D) Quantification of the total BLI registered (above) and representative images (below) from CNS and BM in buffer (n=9) and T22-AUR-treated mice (n=10). All data are shown as mean ± standard error. *p≤0.05; **p≤0.01. Abbreviations: BLI, bioluminescence imaging; BM, bone marrow; CNS, central nervous system. |
Lymphoma-Burden Reduction by T22-AUR Nanoconjugate in CXCR4+ DLBCL Infiltrated Organs
By H&E staining, CD20 and CD79a IHC, we confirmed human B cell infiltration in the CNS and BM. Those data showed that buffer treated animals presented a high lymphoma infiltration in the meninges, which covers the CNS parenchyma, and, at many points, this infiltration penetrated to the brain parenchyma. Otherwise, animals treated with T22-AUR presented only residual CD20+/CD79a+ B cells in the meninges, which did not penetrate to the parenchyma in any case. Thus, the quantification of the ratio of CD20/CD79a-positive area showed significant differences between the animals treated with T22-AUR (ratio 0.25±0.03 CD20 and 0.15±0.04 CD79a) and buffer (ratio 1.00±0.14 CD20 and 1.00±0.18 CD79a). Furthermore, we observed that the expression of the CXCR4 receptor was maintained in the human B cells, meaning that we detected also a reduction of the ratio of CXCR4-positive area in the T22-AUR group (0.21±0.04) compared to buffer group (1.00±0.11). Finally, we also demonstrated that DLBCL cells in the CNS of buffer-treated mice expressed Ki67 (1.00±0.22), a marker of proliferation, and the Ki67-positive-cells were significantly reduced in mice treated with T22-AUR (0.11±0.03) (Figure 8A).
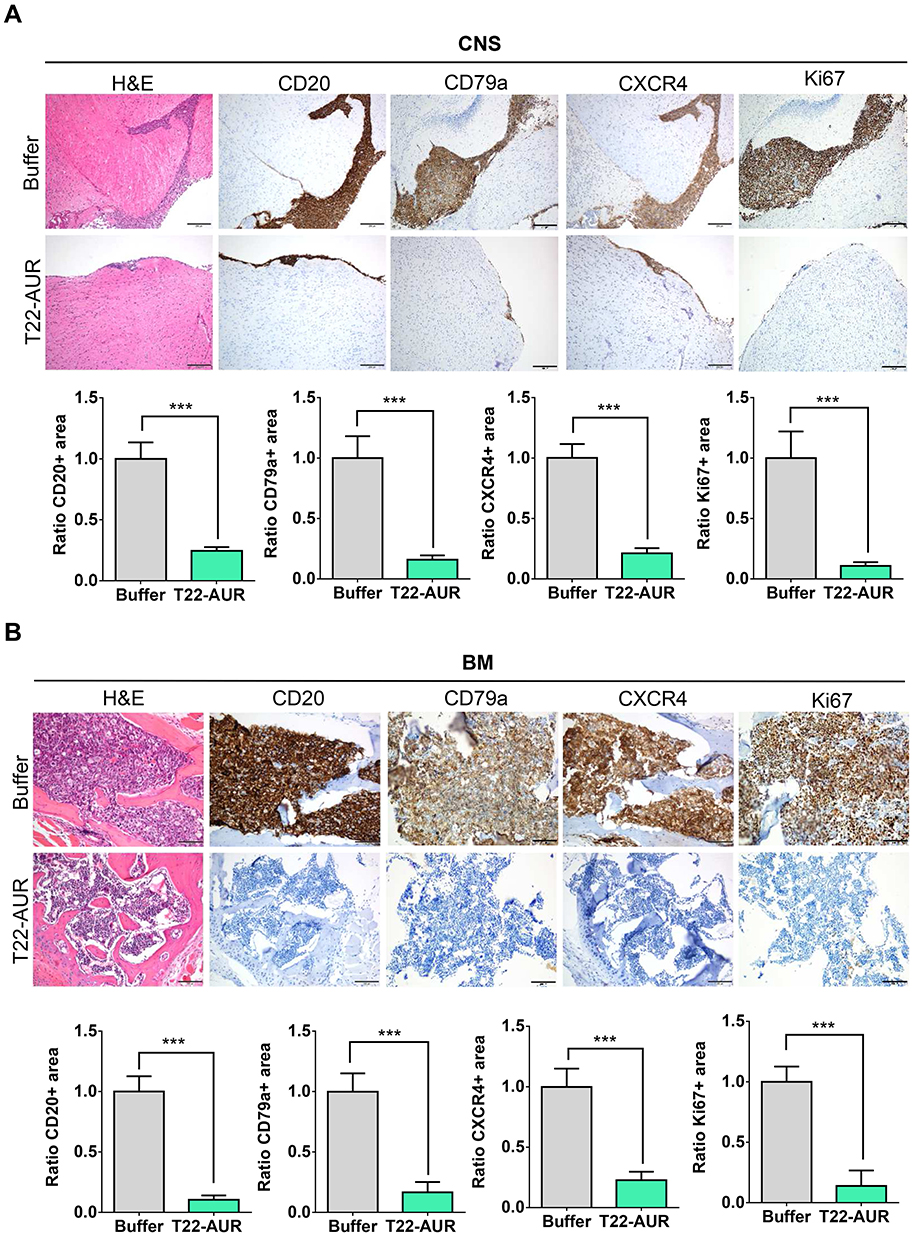 |
Figure 8 T22-AUR effect on lymphoma infiltration burden in the affected organs (CNS and BM). (A and B) Hematoxylin and eosin (H&E) staining, anti-CD20, anti-CD79a, anti-CXCR4 and anti-Ki67 immunohistochemical representative pictures and quantification of the ratio of the positive stained tissue area for each marker in CNS and BM organs from animals treated either with buffer or T22-AUR. Ratio quantification was obtained dividing the area of positive cells in 12 counted fields in samples T22-AUR-treated by the counted positive area in 12 fields of buffer-treated samples. Pictures were taken at 100x (scale bars= 200 μm) for CNS and 200x (scale bars= 100 μm) for BM. All data are shown as mean ± standard error. ***p≤0.005. Abbreviations: BM, bone marrow; CNS, central nervous system. |
Similar findings were observed in the BM, animals treated with buffer had a high lymphoma infiltration (CD20/CD79a-positive cells) which was significantly reduced in T22-AUR-treated mice (ratio 1.00±0.13 CD20-positive area and ratio 1.00±0.15 CD79a-positive area in buffer treated mice vs 0.10±0.04 and 0.17±0.08, respectively, in T22-AUR-treated mice). Moreover, the ratio of the CXCR4-positive area as well as the ratio of Ki67-positive area in the animals treated with T22-AUR was also significantly lower (0.21±0.07 CXCR4-positive area and 0.14±0.13 Ki67-positive area) than in the animals treated with buffer (1.00±0.15 CXCR4-positive area and 1.00±0.13 Ki67-positive area) (Figure 8B).
Absence of Histopathological Alterations in Normal Organs of Mice and Lack of in vitro Cytotoxicity in Human PBMCs After T22-AUR Treatment
The repeated T22-AUR administration in mice did not show any histopathological alteration in the organs non-infiltrated by DLBCL cells, such as liver, spleen, kidneys, heart and lungs (Figure 9A).
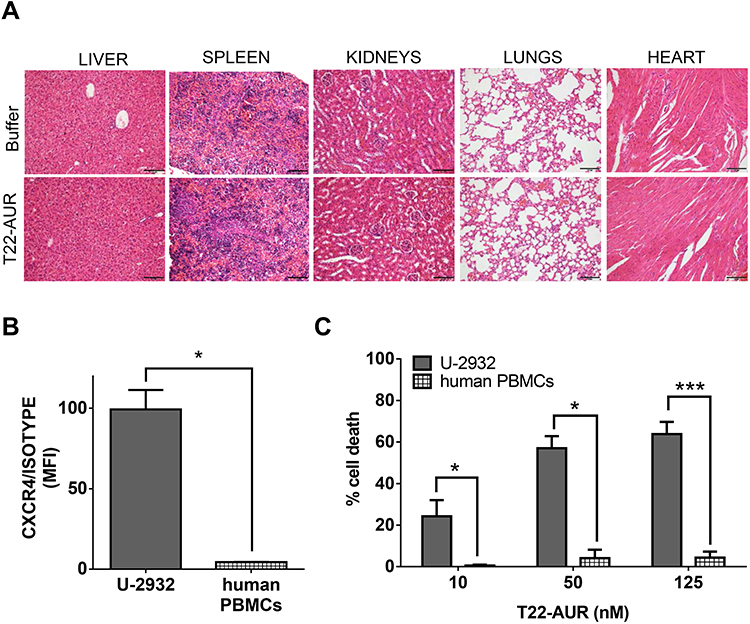 |
Figure 9 T22-AUR does not induce systemic toxicity in CXCR4+ DLBCL-Luci disseminated mice or in vitro cytotoxicity on human PBMCs. (A) Hematoxylin and eosin (H&E) staining of non-DLBCL infiltrated organs (liver, spleen, kidneys, lungs and heart) from buffer and T22-AUR-treated mice. Pictures were taken at 200x (scale bars= 100 μm). (B) Membrane CXCR4 expression measured by cell cytometry in U-2932 cells and human PBMCs from three independent healthy donors. Data were shown as mean fluorescence intensity (MFI) resulted from dividing the CXCR4 MFI by its own isotype MFI. (C) Percentage of cell death measured by colorimetric cell proliferation kit (XTT) in U-2932 and human PBMCs after exposure to T22-AUR in the 10–125 nM range. All experiments were performed in biological triplicates and data are shown as mean ± standard error. *p≤0.05; ***p≤0.005. Abbreviations: MFI, mean fluorescence intensity; PBMCs, peripheral blood mononuclear cells. |
Most importantly, since human hematopoietic cells are reported to express the CXCR4 receptor, we wanted to evaluate the possible hematopoietic toxicity produced by T22-AUR in human PBMCs. Thus, we firstly evaluated the levels of CXCR4 in human PBMCs of three healthy donors, which were significantly lower (4.36±0.14 MFI) than CXCR4+ DLBCL cells (99.32±12.13 MFI) (Figure 9B). Then, we evaluated the cytotoxicity of the T22-AUR to human PBMCs observing almost negligible levels of cell death induction (0.47±0.45% at 10nM; 4.05±4.05% at 50nM and 4.24±2.93% at 125nM) as compared to the cytotoxicity observed in CXCR4+ DLBCL cells (24.17±7.76% at 10 nM; 56.99±5.86% at 50 nM and 63.78±5.93% at 125 nM) (Figure 9C). Therefore, T22-AUR did not produce side toxicity in mice neither in human PBMCs.
Discussion
We have here demonstrated the T22-AUR nanoconjugate capacity to potently block lymphoma dissemination in a CXCR4+ DLBCL mouse model, as well as its induction of mitotic arrest and apoptosis in CXCR4+ DLBCL cells.
Importantly, we have proved that MMAE conjugation to the nanocarrier, in order to generate the T22-AUR nanoconjugate, does not alter the nanocarrier targeting and specific internalization ability through the CXCR4 receptor in DLBCL cell lines nor the tumor and non-tumor tissue distribution pattern in the mouse model. Thus, T22-AUR displayed a CXCR4-dependent targeting, as we had previously demonstrated for the unconjugated T22-GFP-H6 nanoparticle. In fact, T22-AUR nanoconjugate achieved a high tumor uptake and internalization within CXCR4+ DLCBL tumor cells in a SC mouse model reaching its peak at 5 h post-injection, exactly the same tumor accumulation time we have reported for the nanocarrier.15
The pharmacokinetics of our nanoconjugate may be similar to that reported for ADCs, in which the antibody rather than the drug determines its half-life in blood.24 Moreover, the size of T22-AUR (17.9 nm ± 0.7 nm), which is higher than the renal filtration cut-off (7 nm), avoids its clearance by renal filtration, as also reported by ADCs.25
The MMAE, which belongs to the vinkaloid group and dolastatins family of MTAs, is increasingly used for targeted drug delivery approaches in cancer therapy. Microtubules are actively involved in essential processes, both in dividing and non-dividing cells, which cannot be replaced by other cellular systems. One of the main reasons for the increasing interest in the clinical use of MTAs is because they can kill mitotic, interphase and quiescent cancer cells.13,14
Regarding the mechanism of action of the MMAE delivered payload drug, our results showed that the T22-AUR cytotoxic effect in CXCR4+ DLBCL cells involves G2/M cell cycle arrest, MC and DNA damage (as measured by γH2AX), followed by PARP cleavage and induction of apoptotic bodies. There is an ongoing debate on whether MC is a specific cell death program by itself or a previous event process leading to apoptosis or necrosis.26,27 We observed that MC appeared earlier (ie, 24 h) than apoptosis; however, at a later time point (ie, 48 h), the proportion of cells involved in apoptotic cell death was extremely higher than the proportion of cells undergoing MC at an earlier time point (ie, 24 h). It is likely that the MMAE-induced apoptosis in our model may also be activated independently of MC in non-cycling cells. Therefore, our data support the notion that MMAE may act by two different apoptotic mechanisms, with and without previous MC trigger, in CXCR4+ DLBCL cells. Actually, MTAs, such as MMAE, are mechanistically different from DNA-directed drugs used in chemotherapy and mitosis-specific drugs that kill only cancer cells that are actively dividing.28
For the evaluation of the in vivo T22-AUR antineoplastic effect, we used an extranodal DLBCL disseminated mouse model, with CXCR4+ DLBCL infiltration in the CNS and BM, which we consider a relevant model to test novel therapies since extranodal DLBCL patients involving more than one site present worse overall as well as progression-free survival than nodal DLBCL patients.29–31 In this DLBCL model, repeated administration of the T22-AUR nanoconjugate induces a decrease of lymphoma dissemination during the whole experimental follow-up. This cytotoxic effect was observed in the CXCR4+ DLBCL-infiltrated CNS and especially in the BM, where a nearly complete elimination of CXCR4+ DLBCL cells was achieved in all nanoconjugate-treated animals. Therefore, the use of MMAE as a payload drug is especially relevant to eliminate quiescent resistant cells in R/R DLBCL patients, which remain in the hypoxic niches, including bulky necrotic lymphoma masses and the BM.32 Moreover, the hypoxic tumor BM microenvironment enhances cancer cell survival and reduces the sensitivity to therapy.33–35
Furthermore, the absence of toxicity, either on non-DLBCL infiltrated mouse organs or human PBMCs, could be explained by the “super-selectivity” reported in multivalent systems, such as nanoparticles or nanoconjugates, which consists in the ability of interacting with their target receptor only when its density in the cell surface is above a certain threshold.36,37
Finally, a successful preclinical development of our T22-AUR nanoconjugate could add a therapeutic option for transplantation-ineligible R/R DLBCL patients. In this sense, it must be necessarily compared with the performance of polatuzumab vedotin ADC, which has been recently approved for clinical use in R/R DLBCL patients together with rituximab and bendamustine.38,39 Despite both polatuzumab vedotin and T22-AUR incorporate the same payload drug (MMAE), we believe that, as compared to this ADC, there are five main reasons to further develop our nanoconjugate:
- T22-AUR could be more efficient than polatuzumab vedotin in selectively delivering the MMAE payload to target cells, and in subsequently inducing their elimination, because each nanoconjugate shows a multivalent ligand display of around 12 molecules to internalize through CXCR4-mediated internalization, as compared to the only two epitope-binding domains from two antigen-binding fragment (Fab) present in the antibody of ADCs.16,40
- Each T22-AUR nanoconjugate displays about 228 molecules of MMAE linked to the nanocarrier, as compared to a mean of 3.65 MMAE molecules added to polatuzumab vedotin.41 Thus, lower doses of T22-AUR should be more effective than polatuzumab vedotin.
- Our nanoconjugate is mostly uptaken by cancer target cells in the tumor tissue, while only low or negligible uptake is seen in non-lymphoma infiltrated tissues, whereas ADCs accumulate less than 1% of the administered dose in cancer tissues.21,22
- Whereas gene expression of CD79b, the target of this ADC, is around a median of 7.5 fold-higher in DLBCL patients than non-neoplastic controls, the CXCR4 gene expression, targeted by the T22-AUR nanoconjugate in DLBCL samples is on average 140-fold higher than in normal controls.10,38 Thus, our nanoconjugate can be targeted more specifically due to the huge CXCR4 overexpression existing in DLBCL patients.
- Our nanoconjugate achieves a potent block of lymphoma dissemination with complete lack of associated toxicity in normal organs. In contrast, polatuzumab vedotin is effective at a dosage that induces severe side effects and life-threatening toxicities, such as grade 3–4 cytopenias, peripheral neuropathy, progressive multifocal leukoencephalopathy or tumor lysis syndrome.38
Conclusions
The association of the overexpression of CXCR4 in DLBCL cells with therapy resistance, the specific CXCR4 targeting of the nanoconjugate and the use of the payload MMAE, which is capable to kill quiescent cancer cells, make the T22-AUR a promising therapeutic approach in CXCR4+ transplantation-ineligible R/R DLBCL patients.
Data Sharing Statement
Data available on request from the authors: The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
Acknowledgments
This work was supported by Instituto de Salud Carlos III (ISCIII, Co-funding from FEDER) [PI18/00650, PIE15/00028, PI15/00378 and EU COST Action CA 17140 to R.M., FIS PI17/01246 and RD16/0011/0028 and FIS PI14/00450 to J.S., and FIS PI15/00272 to E.V.]; Agencia Estatal de Investigación (AEI) and Fondo Europeo de Desarrollo Regional (FEDER) [grant BIO2016-76063-R, AEI/FEDER, UE to A.V. and grant PID2019-105416RB-I00/AEI/10.13039/501100011033 to E.V.]; CIBER-BBN [CB06/01/1031 and 4NanoMets to R.M., VENOM4CANCER to A.V. and NANOREMOTE to E.V.]; AGAUR [2017 FI_B 00680 to A.F., 2017- SGR-865 to R.M., 2017-SGR-1395 to J.S. and 2017SGR-229 to A.V.]; Josep Carreras Leukemia Research Institute [P/AG to R.M.] La Marató TV3 [201941-30-31-32 to J.S. and A.V.]; a grant from the Cellex Foundation, Barcelona to J.S.; a grant from the Generalitat de Catalunya (PERIS) [SLT002/16/00433 to J.S.] and a grant from the Generalitat de Catalunya CERCA Programme. A.V. received an Icrea Academia Award. U.U was also supported by Grant PERIS SLT006/17/00093 from la Generalitat de Catalunya and Miguel Servet fellowship (CP19/00028) from Instituto de Salud Carlos III co-funded by European Social Fund (ESF investing in your future). The bioluminescent follow-up of cancer cells and toxicity studies has been performed in the ICTS-141007 Nanbiosis Platform, using its CIBER-BBN Nanotoxicology Unit (http://www.nanbiosis.es/portfolio/u18-nanotoxicology-unit/). Protein production has been partially performed by the ICTS “NANBIOSIS”, more specifically by the Protein Production Platform of CIBER-BBN/IBB (http://www.nanbiosis.es/unit/u1-protein-production-platform-ppp/).
The authors would like to thank Annabel García-León (IIB-Sant Pau, Barcelona) and Lola Mulero Pérez (histology unit from CMRB, Barcelona) for their technical support.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work. Conception and design: AF, EV, RM and IC; Development of methodology: AF, VP, UU, LAC, MAM, PA and IC; Acquisition of data: AF, VP and YN. Histopathological analysis: AF and AG; Analysis and interpretation of data: AF, JS, AV, EV, RM and IC. Drafted the manuscript: AF; Review and revision of manuscript: AV, UU, EV, RM and IC. Study supervision: RM, IC and EV.
Disclosure
AV, EV, UU, RM, IC are cited as inventors in three patents: PCT/EP2018/061732, covering Nanostructured Proteins and uses thereof; PCT/EP2018/069303, covering Therapeutic Nanoconjugates and uses thereof; and PCT/EP2012/050513 (WO2012/095527), covering Methods and reagents for efficient and targeted delivery of therapeutic molecules to CXCR4 cells, all licensed to Nanoligent. AV, EV, RM are scientific advisors and cofounders of Nanoligent. The authors report no other conflicts of interest in this work.
References
1. Smith A, Crouch S, Lax S, et al. Lymphoma incidence, survival and prevalence 2004–2014: sub-type analyses from the UK’s haematological malignancy research network. Br J Cancer. 2015;112(9):1575–1584. doi:10.1038/bjc.2015.94
2. Coiffier B, Sarkozy C. Diffuse large B-cell lymphoma: r-CHOP failure—what to do? Hematol Am Soc Hematol Educ Program. 2016;2016(1):366–378. doi:10.1182/asheducation-2016.1.366
3. Raut LS, Chakrabarti PP. Management of relapsed-refractory diffuse large B cell lymphoma. South Asian J Cancer. 2014;3(1):66–70. doi:10.4103/2278-330X.126531
4. González-Barca E, Coronado M, Martín A, et al. Spanish lymphoma group (GELTAMO) guidelines for the diagnosis, staging, treatment, and follow-up of diffuse large B-cell lymphoma. Oncotarget. 2018;9(64):32383–32399. doi:10.18632/oncotarget.25892
5. Fridrik MA, Jaeger U, Petzer A, et al. Cardiotoxicity with rituximab, cyclophosphamide, non-pegylated liposomal doxorubicin, vincristine and prednisolone compared to rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone in frontline treatment of patients with diffuse large B-cell lymphoma: a randomised phase-III study from the Austrian cancer drug therapy working group [Arbeitsgemeinschaft Medikamentöse Tumortherapie AGMT] (NHL-14). Eur J Cancer. 2016;58:112–121. doi:10.1016/j.ejca.2016.02.004
6. Unzueta U, Céspedes MV, Vázquez E, Ferrer-Miralles N, Mangues R, Villaverde A. Towards protein-based viral mimetics for cancer therapies. Trends Biotechnol. 2015;33(5):253–258. doi:10.1016/j.tibtech.2015.02.007
7. Bamrungsap S, Zhao Z, Chen T, et al. Nanotechnology in therapeutics: a focus on nanoparticles as a drug delivery system. Nanomedicine (Lond). 2012;7(8):1253–1271. doi:10.2217/nnm.12.87
8. Chen J, Xu-Monette ZY, Deng L, et al. Dysregulated CXCR4 expression promotes lymphoma cell survival and independently predicts disease progression in germinal center B-cell-like diffuse large B-cell lymphoma. Oncotarget. 2015;6(8):5597–5614. doi:10.18632/oncotarget.3343
9. Moreno MJ, Bosch R, Dieguez-Gonzalez R, et al. CXCR4 expression enhances diffuse large B cell lymphoma dissemination and decreases patient survival. J Pathol. 2015;235(3):445–455. doi:10.1002/path.4446
10. Pansy K, Feichtinger J, Ehall B, et al. The CXCR4-CXCL12-axis is of prognostic relevance in DLBCL and its antagonists exert pro-apoptotic effects in vitro. Int J Mol Sci. 2019;20(19):4740. doi:10.3390/ijms20194740
11. Laursen MB, Reinholdt L, Schönherz AA, et al. High CXCR4 expression impairs rituximab response and the prognosis of R-CHOP-treated diffuse large B-cell lymphoma patients. Oncotarget. 2019;10(7):717–731. doi:10.18632/oncotarget.26588
12. Loong HH, Yeo W. Microtubule-targeting agents in oncology and therapeutic potential in hepatocellular carcinoma. OncoTargets Ther. 2014;7:575–585.
13. Bates D, Eastman A. Microtubule destabilising agents: far more than just antimitotic anticancer drugs. Br J Clin Pharmacol. 2017;83(2):255–268. doi:10.1111/bcp.13126
14. Field JJ, Kanakkanthara A, Miller JH. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg Med Chem. 2014;22(18):5050–5059. doi:10.1016/j.bmc.2014.02.035
15. Falgàs A, Pallarès V, Unzueta U, et al. A CXCR4-targeted nanocarrier achieves highly selective tumor uptake in diffuse large B-cell lymphoma mouse models. Haematologica. 2020;105(3):741–753. doi:10.3324/haematol.2018.211490
16. Pallarès V, Unzueta U, Falgàs A, et al. An Auristatin nanoconjugate targeting CXCR4+ leukemic cells blocks acute myeloid leukemia dissemination. J Hematol Oncol. 2020;13(1):36. doi:10.1186/s13045-020-00863-9
17. Li C, Zhang C, Li Z, et al. Clinical pharmacology of vc-MMAE antibody–drug conjugates in cancer patients: learning from eight first-in-human Phase 1 studies. mAbs. 2020;12(1):1699768. doi:10.1080/19420862.2019.1699768
18. Zhao P, Zhang Y, Li W, Jeanty C, Xiang G, Dong Y. Recent advances of antibody drug conjugates for clinical applications. Acta Pharm Sin B. 2020;10(9):1589–1600. doi:10.1016/j.apsb.2020.04.012
19. Ma H, Sawas A. Combining biology and chemistry for a new take on chemotherapy: antibody-drug conjugates in hematologic malignancies. Curr Hematol Malig Rep. 2018;13(6):555–569. doi:10.1007/s11899-018-0485-3
20. Bourbon E, Salles G. Polatuzumab vedotin: an investigational anti-CD79b antibody drug conjugate for the treatment of diffuse large B-cell lymphoma. Expert Opin Investig Drugs. 2020;29(10):1079–1088. doi:10.1080/13543784.2020.1800638
21. Gerber H-P, Senter PD, Grewal IS. Antibody drug-conjugates targeting the tumor vasculature. mAbs. 2009;1(3):247–253. doi:10.4161/mabs.1.3.8515
22. Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. mAbs. 2016;8(4):659–671. doi:10.1080/19420862.2016.1156829
23. Erickson HK, Lambert JM. ADME of antibody–maytansinoid conjugates. AAPS J. 2012;14(4):799–805. doi:10.1208/s12248-012-9386-x
24. Han TH, Zhao B. Absorption, distribution, metabolism, and excretion considerations for the development of antibody-drug conjugates. Drug Metab Dispos. 2014;42(11):1914–1920. doi:10.1124/dmd.114.058586
25. Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomed. 2008;3(5):703–717. doi:10.2217/17435889.3.5.703
26. Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15(7):1153–1162. doi:10.1038/cdd.2008.47
27. Souza RD, Ayub LC, Yip K. Mitotic catastrophe. In: Apoptosis and Beyond. John Wiley & Sons, Ltd; 2018:475–510.
28. Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell. 2012;23(1):1–6. doi:10.1091/mbc.e10-04-0335
29. Shi Y, Han Y, Yang J, et al. Clinical features and outcomes of diffuse large B-cell lymphoma based on nodal or extranodal primary sites of origin: analysis of 1,085 WHO classified cases in a single institution in China. Chin J Cancer Res. 2019;31(1):152–161. doi:10.21147/j.issn.1000-9604.2019.01.10
30. Yao S, Li J, Yao Z, et al. Extranodal involvement in young patients with diffuse large B-cell lymphoma: distribution, prognostic value and treatment options. Chin J Cancer Res. 2017;29(1):57–65. doi:10.21147/j.issn.1000-9604.2017.01.07
31. Hashwah H, Bertram K, Stirm K, et al. The IL-6 signaling complex is a critical driver, negative prognostic factor, and therapeutic target in diffuse large B-cell lymphoma. EMBO Mol Med. 2019;11(10):e10576. doi:10.15252/emmm.201910576
32. Klener P, Klanova M. Drug resistance in non-hodgkin lymphomas. Int J Mol Sci. 2020;21(6):2081. doi:10.3390/ijms21062081
33. Cavnar SP, Rickelmann AD, Meguiar KF, et al. Modeling selective elimination of quiescent cancer cells from bone marrow. Neoplasia. 2015;17(8):625–633. doi:10.1016/j.neo.2015.08.001
34. Cavnar SP, Xiao A, Gibbons AE, et al. Imaging sensitivity of quiescent cancer cells to metabolic perturbations in bone marrow spheroids. Tomography. 2016;2(2):146–157.
35. Sircar A, Chowdhury SM, Hart A, et al. Impact and intricacies of bone marrow microenvironment in B-cell lymphomas: from biology to therapy. Int J Mol Sci. 2020;21(3):904. doi:10.3390/ijms21030904
36. Martinez-Veracoechea FJ, Frenkel D. Designing super selectivity in multivalent nano-particle binding. Proc Natl Acad Sci. 2011;108(27):10963–10968. doi:10.1073/pnas.1105351108
37. Liu M, Apriceno A, Sipin M, et al. Combinatorial entropy behaviour leads to range selective binding in ligand-receptor interactions. Nat Commun. 2020;11(1):4836. doi:10.1038/s41467-020-18603-5
38. Sehn LH, Herrera AF, Flowers CR, et al. Polatuzumab vedotin in relapsed or refractory diffuse large B-cell lymphoma. J Clin Oncol. 2020;38(2):155–165. doi:10.1200/JCO.19.00172
39. Malecek M-K, Watkins MP, Bartlett NL. Polatuzumab vedotin for the treatment of adults with relapsed or refractory diffuse large B-cell lymphoma. Expert Opin Biol Ther. 2020;
40. Sanchez-Trincado JL, Gomez-Perosanz M, Reche PA. Fundamentals and methods for T- and B-cell epitope prediction. J Immunol Res. 2017;2017:2680160. doi:10.1155/2017/2680160
41. Lu D, Lu T, Gibiansky L, et al. Integrated two‐analyte population pharmacokinetic model of polatuzumab vedotin in patients with non‐hodgkin lymphoma. CPT Pharmacomet Syst Pharmacol. 2020;9(1):48–59. doi:10.1002/psp4.12482
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.