")
Back to Journals » International Journal of Nanomedicine » Volume 12
Spatiotemporal delivery of nanoformulated liraglutide for cardiac regeneration after myocardial infarction
Authors Qi Q, Lu L, Li H, Yuan Z, Chen G , Lin M, Ruan Z, Ye X, Xiao Z, Zhao Q
Received 10 January 2017
Accepted for publication 8 May 2017
Published 7 July 2017 Volume 2017:12 Pages 4835—4848
DOI https://doi.org/10.2147/IJN.S132064
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
The supplementary video shows nanoformulated liraglutide injection.
Views: 12678
Quan Qi,1,* Lei Lu,2,3,* Haiqing Li,1,* Zhize Yuan,1 Gaoxian Chen,2,3 Miao Lin,2,3 Zhengwen Ruan,4 Xiaofeng Ye,1 Zeyu Xiao,2,3,5 Qiang Zhao1
1Department of Cardiac Surgery, Rui Jin Hospital, 2Department of Pharmacology, Institute of Medical Sciences, 3Translational Medicine Collaborative Innovation Center, Shanghai Jiao Tong University School of Medicine, Shanghai, 4Department of Cardiology, Yuyao People’s Hospital, Yuyao, Zhejiang, 5Collaborative Innovation Center of Systems Biomedicine, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: The local, intramyocardial injection of proteins into the infarcted heart is an attractive option to initiate cardiac regeneration after myocardial infarction (MI). Liraglutide, which was developed as a treatment for type 2 diabetes, has been implicated as one of the most promising protein candidates in cardiac regeneration. A significant challenge to the therapeutic use of this protein is its short half-life in vivo. In this study, we evaluated the therapeutic effects and long-term retention of liraglutide loaded in poly(lactic-co-glycolic acid)–poly(ethylene glycol) (PLGA–PEG) nanoparticles (NP-liraglutide) on experimental MI. PLGA–PEG nanoparticles (NPs) have been shown to efficiently load liraglutide and release bioactive liraglutide in a sustained manner. For in vitro test, the released liraglutide retained bioactivity, as measured by its ability to activate liraglutide signaling pathways. Next, we compared the effects of an intramyocardial injection of saline, empty NPs, free liraglutide and NP-liraglutide in a rat model of MI. NPs were detected in the myocardium for up to 4 weeks. More importantly, an intramyocardial injection of NP-liraglutide was sufficient to improve cardiac function (P<0.05), attenuate the infarct size (P<0.05), preserve wall thickness (P<0.05), promote angiogenesis (P<0.05) and prevent cardiomyocyte apoptosis (P<0.05) at 4 weeks after injection without affecting glucose levels. The local, controlled, intramyocardial delivery of NP-liraglutide represents an effective and promising strategy for the treatment of MI.
Keywords: spatiotemporal delivery, nanoformulated liraglutide, cardiac regeneration
Introduction
Heart failure (HF) after myocardial infarction (MI) is the leading cause of death, with an estimated 7.3 million deaths per year worldwide.1 Despite their clinical importance, current treatment methods are insufficient for the control of HF after MI.2 Some accepted treatment methods for MI still have several limitations.3 Thus, several new therapies have been developed to regenerate the infarcted myocardium, such as the administration of stem cells,4 growth factors or proteins5 or gene therapy.6 Liraglutide has initially been approved for clinical use as a treatment for type 2 diabetes.7 Recently, liraglutide has been suggested to be beneficial for MI, independent of glycemic control.7,8 In fact, the therapeutic benefits of liraglutide have been reported in several studies, including improved cardiac performance,9 an attenuated infarct size,10 increased myocardial blood flow,11 inhibition of myocardial apoptosis12 and activation of myocardial signaling pathways.13 Furthermore, the Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) trial, a randomized, double-blind, placebo-controlled clinical trial with long-term follow-up, confirmed the favorable effects of liraglutide on MI.8
However, like many other proteins, the clinical applicability of liraglutide is limited by its short half-life (13 h) under biological conditions, thereby requiring repeated subcutaneous injections.14 Furthermore, subcutaneous administration of liraglutide is associated with undesired systemic side effects, such as a relatively higher frequency of nausea, diarrhea and vomiting.15 Hypoglycemia is infrequent but is still observed, particularly when liraglutide is used in combination with other hypoglycemics.14 A relatively high percentage of subjects withdrew from the clinical trial due to systemic adverse events (5%).16
In this study, we propose spatiotemporal delivery of liraglutide-loaded poly(ethylene glycol) (PEG)-modified poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NP-liraglutide) into the infarcted myocardium as a more appealing and promising therapeutic approach for treating MI. For spatial delivery, NP-liraglutide is locally injected into the myocardium through a mini-thoracotomy injection. Such a locally intramyocardial delivery of liraglutide leads to minimal tissue exposure, thus reducing the undesired systemic side effects. For temporal delivery, NP-liraglutide delivery systems allow the release behavior of liraglutide to be controlled over a prolonged period of 4 weeks, thus overcoming challenges posed by its short half-life. Our team has proven experience in the development of biodegradable PLGA–PEG nanoparticles (NPs).17–20 We extend this study to a further exploration of the possibility of NP-liraglutide delivery systems to treat MI. We hypothesized that the controlled delivery of liraglutide from PLGA–PEG NPs would maximize its therapeutic effects on MI.
Materials and methods
Preparation of PLGA–PEG NPs containing liraglutide
An overview of the work flow is demonstrated in Figure 1. Liraglutide was purchased from Creative Peptides (Shirley, NY, USA). PLGA (poly-D,L-lactide/glycolide 50/50; molecular weight [MW] =30 kDa) was purchased from Jinan Daigang Biomaterial Co., Ltd. (Jinan, China). PEG (NH2–PEG–COOH, MW =5 kDa) was obtained from JenKem Technology (Tianjin, China). Poly(vinyl alcohol) (PVA-217, degree of polymerization 1,700, degree of hydrolysis 88.5%, MW =72–84 kDa) was purchased from Kuraray (Osaka, Japan). Acetonitrile and potassium dihydrogen phosphate (both in high-pressure liquid chromatography [HPLC] grade) were obtained from Tedia (Cincinnati, OH, USA). We synthesized PLGA–PEG using the classical method.21 NP-liraglutide were prepared with the double emulsion method. First, 3.8 mg of liraglutide was dissolved in 0.5 mL of a 1% (w/v) aqueous sodium acetate solution with 2% PVA, and 25 mg of PLGA–PEG was dissolved in 2.5 mL of dichloromethane (DCM).22 The solution was then emulsified by sonication on ice in 12.5 mL of a 2% PVA solution using a probe sonicator. Then, the emulsion was stirred at 550 rpm for 3 h to evaporate the solvent. The obtained NPs were collected by centrifugation at 4,000 rpm for 25 min and washed three times with water.
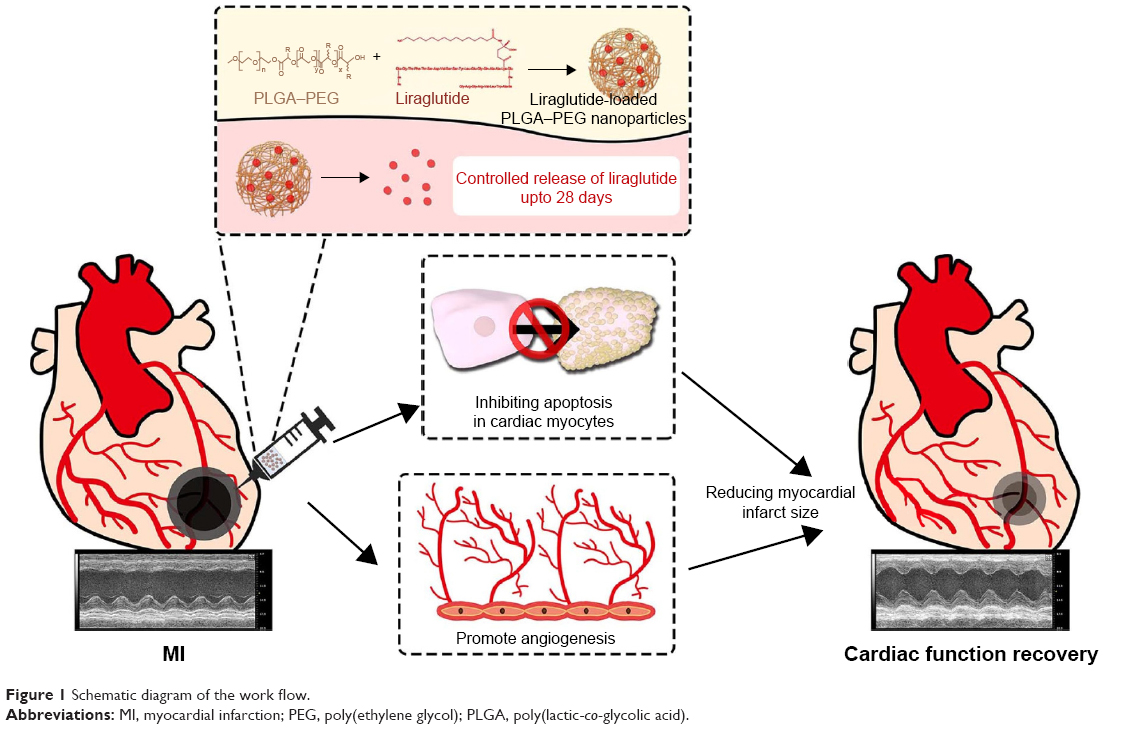 | Figure 1 Schematic diagram of the work flow. |
Characterization of the PLGA–PEG NPs and measurement of liraglutide release
The particle size, size distribution and zeta potential of the NPs were determined by dynamic light scattering (DLS) analysis using a Zetasizer Nano ZS instrument (Malvern, Worcestershire, UK). The morphology of the NPs was determined using transmission electron microscopy (TEM; H-600; Hitachi, Tokyo, Japan) immediately after the samples were negatively stained with sodium phosphotungstate. To determine the drug encapsulation efficiency (EE) of the liraglutide-loaded NPs, a predetermined amount of NPs was dissolved in acetonitrile to release the liraglutide. The resulting mixture was agitated to ensure complete dissolution, then filtered and analyzed by reverse-phase HPLC (L-2000; Hitachi) with a C18 column (dimensions of 250×4.6 mm) to determine the liraglutide concentrations. The mobile phase consisted of acetonitrile (solvent A) and a 0.05-M aqueous KH2PO4 solution (pH 4.0, solvent B). The flow rate was set to 1.0 mL/min. NP samples (100 μL) at 0.02 mg/mL, enclosed in a mini dialysis unit (Thermo Fisher Scientific, Waltham, MA, USA), were incubated in 1 mL PBS (pH 7.4) at 37°C under 100-rpm orbital shaking. Thereafter, 100 μL samples were taken from the incubation medium and analyzed for liraglutide by HPLC as described earlier. After sampling, the incubation medium was replenished with blank PBS. The controlled release of liraglutide from the NPs was measured for 4 weeks.
In vitro bioactivity of the released liraglutide
The bioactivity of the released liraglutide was assessed by Western blotting to measure the phosphorylation level of AKT protein kinase in vitro. The procedure used to isolate the cells in this study was approved by the Institutional Review Committee of Shanghai Jiao Tong University School of Medicine. Neonatal ventricular myocytes were obtained from 0- to 3-day-old Sprague Dawley (SD) rats as previously described.23 The collected cells were pre-plated for 30 min at 37°C. Then, the cardiomyocyte-enriched fraction was plated in six-well plates (1×106 cells/well). The neonatal ventricular myocytes were cultured in cardiac myocyte medium (CMM; ScienCell, Carlsbad, CA, USA), and the culture medium was changed every 2 days. After serum starvation for 24 h, the 7-day-old neonatal cardiomyocytes were washed with fresh medium without serum and treated with the conditioned media from the liraglutide release system in the first 3 days, which has been diluted to 100 nmol/L liraglutide, for 20 min;22 cells cultured with plain medium, free liraglutide (100 nmol/L) or non-loaded NPs were used as the control groups. After the incubation, the medium was removed and the wells containing cells were quickly frozen in liquid nitrogen and stored at −80°C. Western blot analyses were performed using a previously described method.24 After blocking, the membranes were incubated with anti-p-AKT and anti-total AKT antibodies (1:1,000 dilution; Cell Signaling Technology, Beverly, MA, USA), incubated with corresponding horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibodies (1:5,000 dilution; Hua An, Hangzhou, China) and developed using the standard enhanced chemiluminescence (ECL) procedure (Pierce, Rockford, IL, USA). Blots were visualized using Biomax film (Kodak, Rochester, NY, USA). Densitometric analyses were performed using the Image-pro plus6.0 (Media Cybernetics, Silver Spring, MD, USA).
In vivo studies of the rat MI model
SD rats were obtained from the Laboratory Animal Care Facility of Shanghai Jiao Tong University School of Medicine. All the procedures in this study conformed to the “Guide for the Care and Use of Laboratory Animals” (National Institutes of Health, Publication No 85-23, Revised) and were performed under the supervision of the Shanghai Jiao Tong University Institutional Animal Care and Use Committee. Male SD rats (200–250 g) were anesthetized with pentobarbital sodium (30 mg/kg, intraperitoneal), intubated and subjected to left anterior descending coronary artery (LAD) ligation below the atrium (Video S1). The result was confirmed by an increase in the electrocardiogram ST-segment and regional cyanosis. The baseline echocardiographic measurements were collected 2 days following MI.25 After the baseline and screening echocardiograms, 40 rats were randomly divided into four groups. Next, the chests were reopened, and 100 μL of NP-liraglutide (n=8; 0.38 mg of liraglutide), the same amount of liraglutide-loaded saline (liraglutide, n=8; 0.38 mg of liraglutide), non-loaded PLGA–PEG NPs (n=7) or saline solution (n=9) was injected into four points around the infarct areas at a depth of ≈3 mm with a 29-G insulin syringe (25 μL/injection) (Video S2). For the sham group (n=8), a suture was placed around the LAD but was not ligated.
Confirmation of the in vivo visualization of the NPs in the myocardium
A group of 16 infarcted rats was euthanized 1, 3, 5, 7, 14, 21 and 28 days after the injection of the fluorescent-labeled NPs (drug-free; n=2). Rhodamine B was applied as a fluorescent marker for in vivo follow-up. The same amount of rhodamine-labeled NPs was injected into the peri-infarcted areas. The sections were stained with 4-6-diamidino-2-phenylindole (DAPI; 5 μL/section; Sigma-Aldrich, St Louis, MO, USA) to label the nucleus.
Echocardiography for evaluating cardiac function
The left ventricular (LV) performance and structural remodeling were assessed by echocardiography using the Vevo 2100 Imaging System (VisualSonics, Toronto, ON, Canada). Echocardiographic data were collected and analyzed 2 days after ligation (baseline) and 4 weeks after injection.26 The LV internal systolic dimension (LVIDs) and LV internal diastolic dimension (LVIDd) were measured by tracing the endocardial border to calculate the LV ejection fraction (EF) and fractional shortening (FS) using the following equations: EF (LVIDd3 − LVIDs3)/LVIDd3 ×100% and FS (LVIDd − LVIDs)/LVIDd ×100%. All measurements were performed by two independent, qualified echo operators.
Histological analysis of the LV cavity areas and scar areas
The rats were euthanized, and the hearts were collected 4 weeks following the injection. The hearts were perfused with 4% formaldehyde for 20 min, rinsed with PBS for approximately 10 min and sectioned into five slices in the transverse orientation below the ligature to the apex at 2-mm intervals. The heart samples were fixed, paraffin-embedded and sectioned into 5-μm slices. The histological analysis was performed on Masson’s trichrome-stained slides from the closest section plane27 The following data were measured using Image J software: ventricular wall thickness, septum thickness (mm), scar thickness (mm), LV cavity area (mm2) and whole LV area (mm2). Each measurement was performed three times. Then, the average of each parameter was calculated. Relative scar thickness was calculated as the ratio of the average thickness of the scar wall to the average thickness of the uninfarcted wall. The infarct expansion index was defined as follows: (LV cavity area/whole LV area)/relative scar thickness.23
Immunohistochemical measurements of neovascularization
An immunofluorescence analysis was carried out to identify blood vessel formation. Blood vessel staining was performed with a mouse monoclonal antibody against CD31 (1:100; Abcam, Cambridge, MA, USA) and a rabbit polyclonal antibody against alpha smooth muscle actin (α-SMA; 1:200; Abcam), with the following secondary antibodies: anti-rabbit Alexa Fluor® 594 (A21207; 1/1,000; Thermo Fisher Scientific) and biotin–streptavidin Alexa Fluor® 488 (BA-2001, 1:200; Thermo Fisher Scientific – 532354, 1:150, Thermo Fisher Scientific). For confocal imaging, an LSM710 Meta confocal microscope (Carl Zeiss, Feldbach, Switzerland) was used. α-SMA and CD31-positive structures were identified and photographed. Arterioles and capillaries bordering the infarct at the mid-infarct level were identified by counting the α-SMA- and CD31-positive structures, subsequently computed using a fluorescence microscope in a blinded manner from eight randomly selected images as the mean within 1-mm2 areas. The maturation index was calculated based on the number of α-SMA-positive vessels relative to the total number of vessels.28 All data analyses were performed in a blinded manner. All measurements were performed using Image J software.
TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) staining to measure apoptosis
TUNEL staining was performed according to the manufacturer’s instructions. The nicked ends of DNA were visualized with Fluorescence (number 11684795910; Hoffman-La Roche Ltd., Basel, Switzerland). Apoptotic nuclei and total nuclei bordering the infarct at the mid-infarct level were identified and subsequently computed using a fluorescence microscope in a blinded manner from three randomly selected fields (200× magnification). The numbers of TUNEL-positive nuclei or total nuclei were calculated using Image J software. The percentage of TUNEL-positive nuclei was defined as the number of apoptotic nuclei compared to the total number of nuclei.29
Blood glucose levels
Blood samples of rats from four groups were collected. Nonfasting blood glucose was randomly measured at 1, 2, 3 and 4 weeks after injection by an Automatic Biochemical Analyzer (Hitachi).
Statistical analysis
All variables are presented as mean ± standard error of the mean (SEM) and were calculated using SPSS19.0 (IBM) statistics software. Comparisons of the densitometry, echocardiography data, morphology, blood vessel analysis, apoptotic cell counts and blood glucose levels among groups were performed using one-way ANOVA with Tukey’s post hoc test. P<0.05 was considered statistically significant.
Results
Characterization and in vitro delivery of the NP-liraglutide
In this study, the double emulsion method was used to synthesize spherical PLGA–PEG NPs loaded with liraglutide. TEM observation showed a rough structure of the NPs (Figure 2A). The size of the PLGA–PEG NPs was 350±1.83 nm (Figure 2B), with a zeta potential of −0.369 mV. The EE of liraglutide was 95% for the PLGA–PEG NPs, which corresponded to a final loading of 95 μg of liraglutide per milligram of NPs. In summary, although the PEG coating neutralized the zeta potential of the NPs and slightly increased the particle size, the NPs maintained their negative zeta potential and shape. Importantly, the EE remained at a high level. The controlled release of liraglutide from the NPs was achieved over 4 weeks (Figure 2C). The large variation in the release behavior at the early stage is likely due to the burst release of liraglutide from PLGA–PEG. Approximately 50% cumulative release was observed on the 7th day; thereafter, a sustained, very slow rate of liraglutide release was observed. On the 28th day, approximately 100% of the liraglutide was released, thereby achieving sustained release. In summary, the PLGA–PEG NPs were useful in controlling the release behavior of liraglutide for prolonged periods.
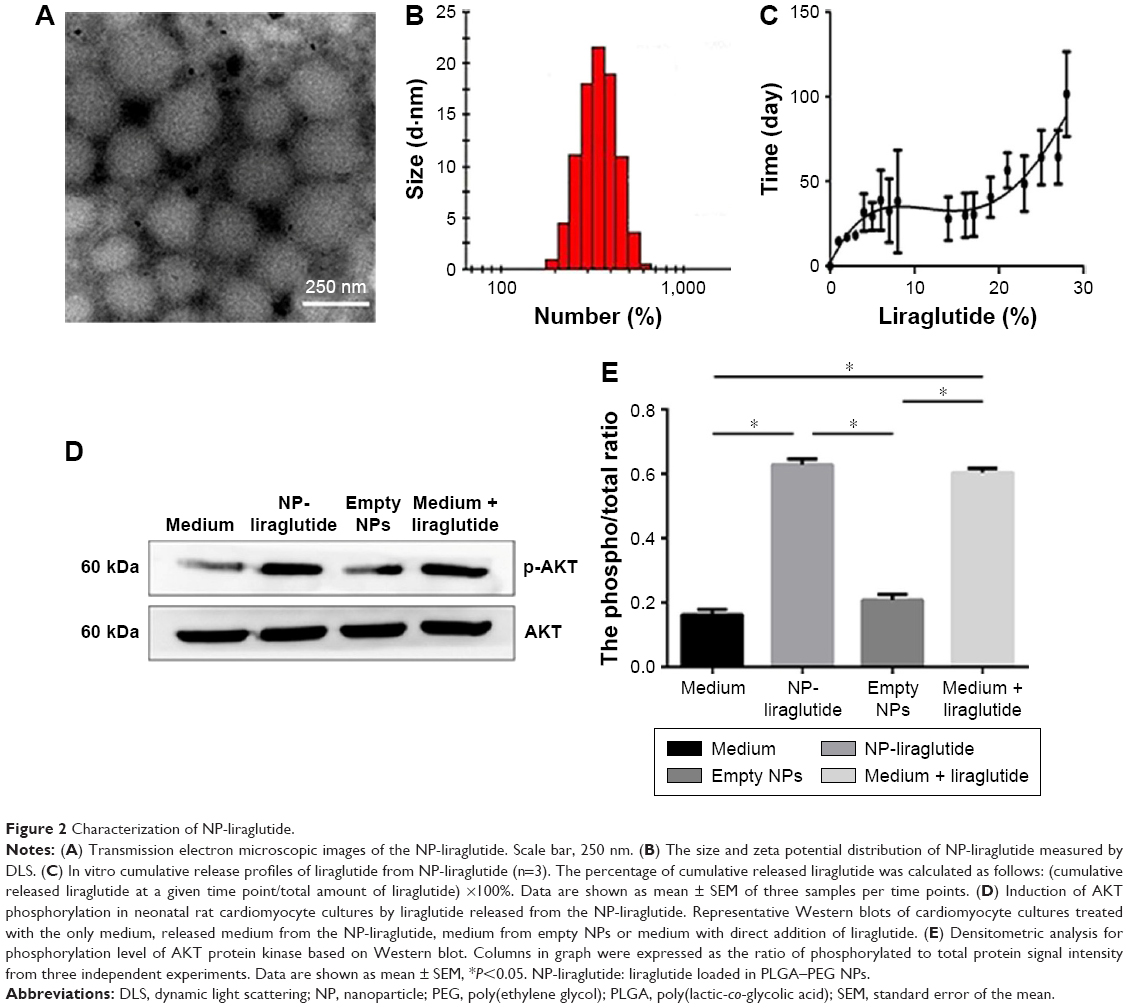 | Figure 2 Characterization of NP-liraglutide. |
The in vitro bioactivity of the NP-liraglutide
Within the organism, liraglutide activates AKT protein kinase, a known downstream target of the liraglutide signaling pathway, to initiate cellular signals.10 Therefore, the bioactivity of the released liraglutide was measured by Western blotting to determine the level of AKT protein kinase phosphorylation. As shown in Figure 2D and E, the released liraglutide induced AKT phosphorylation compared to plain medium or medium containing empty NPs (P<0.05). No differences were observed between the released liraglutide group and the free liraglutide group, suggesting that the released liraglutide retained bioactivity.
In vivo therapeutic efficacy in the rat model of MI
The fate of the NPs was assessed to confirm the extent to which the NPs were retained in the myocardium over prolonged periods (4 weeks). The level of the NPs decreased over time. However, 4 weeks after treatment, some NPs still resided in the myocardium and the rhodamine B marker was observed up to 4 weeks post injection, confirming the suitability of using NPs for the controlled delivery of liraglutide in the heart (Figure 3).
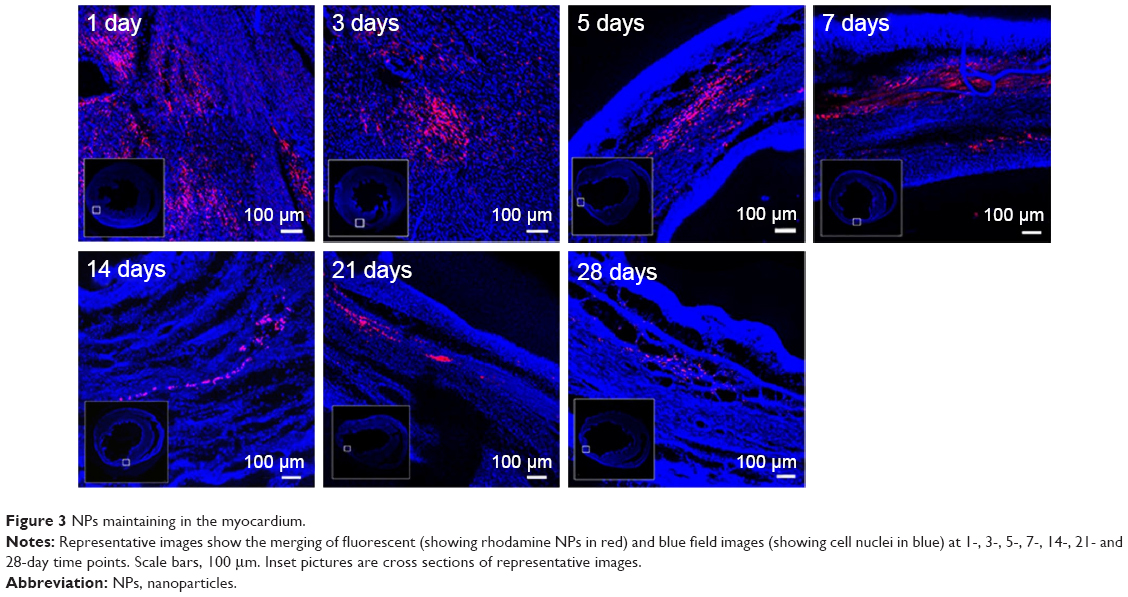 | Figure 3 NPs maintaining in the myocardium. |
Rats underwent echocardiography 2 days (baseline, 2 days after ligation) and 4 weeks post injection to evaluate cardiac function. Differences in the LVEF, LVFS, LVIDs and LVIDd were not observed among the groups at baseline, indicating a comparable degree of initial cardiac injury (Figure S1). The NP-liraglutide group showed markedly improved LVEF (56.1%±1.3%) compared to the administration of saline, NP and liraglutide groups (44.6%±3.0%, 44.4%±2.4% and 47.6%±2.7%, respectively; P<0.05). Similarly, the injection of NP-liraglutide group significantly increased the LVFS (30.5%±0.8%) compared to the administration of saline, NP and liraglutide groups (23.6%±1.9%, 23.1%±1.4% and 25.1%±1.7%, respectively, P<0.05). Moreover, the NP-liraglutide group showed the smallest increase in LVIDs (5.29±0.21) compared to the saline, NP and liraglutide groups (6.62±0.32, 6.49±0.23 and 6.62±0.33 mm, respectively, P<0.05). A similar trend was also noted for the LVIDd, where the NP-liraglutide group showed the smallest increases in the LVIDd (7.60±0.22) compared to the saline, NP and liraglutide groups (8.60±0.25, 8.44±0.20, and 8.82±0.29 mm, respectively, P<0.05; Figure 4).
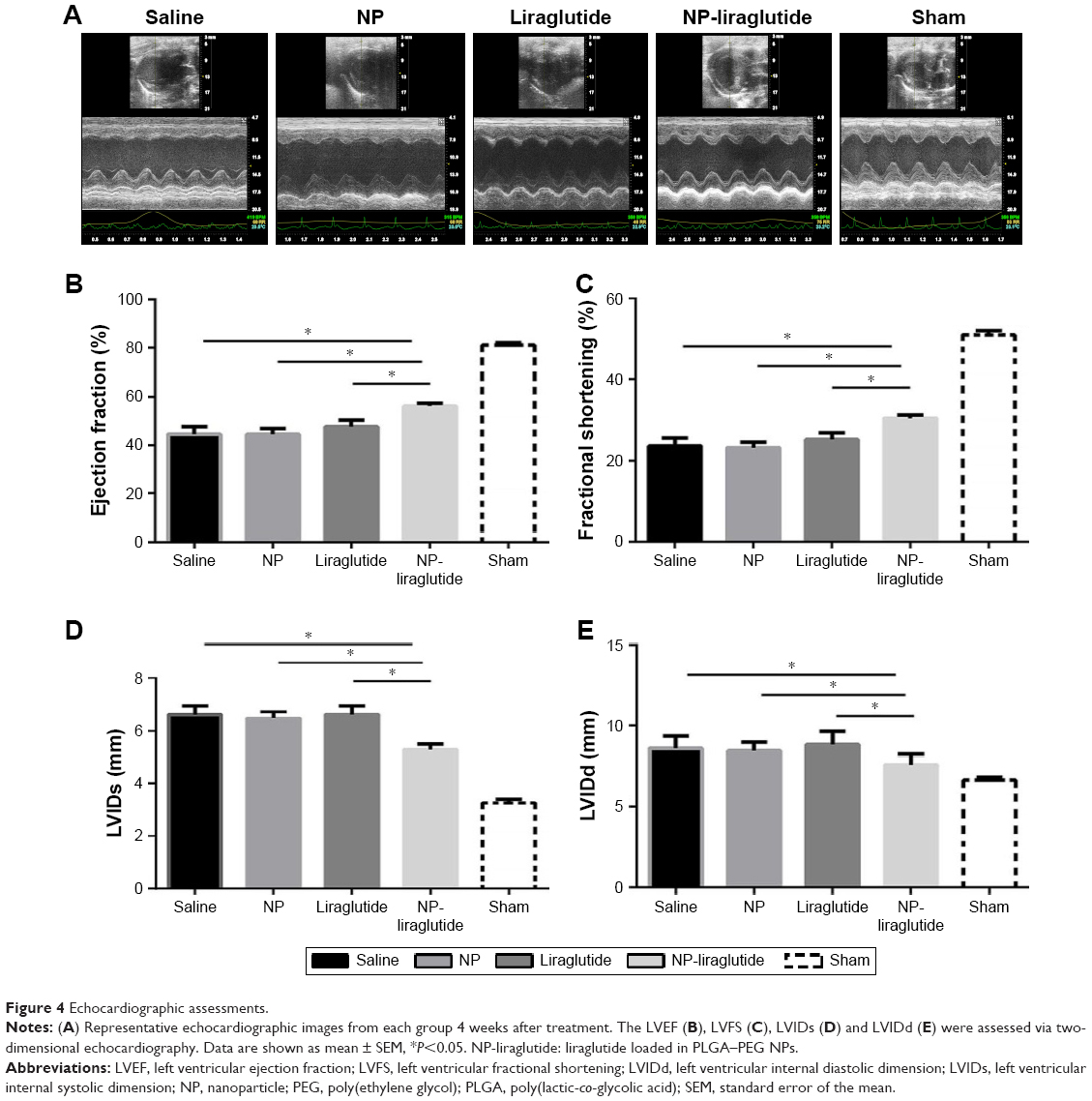 | Figure 4 Echocardiographic assessments. |
H&E staining and Masson’s trichrome staining were performed for the histological analysis of the heart samples from each group (Figure 5). The NP-liraglutide treatment significantly increased the relative scar thickness (0.42±0.01) compared to the saline, NP and liraglutide groups (0.35±0.02, 0.33±0.03 and 0.36±0.02, respectively, P<0.05). A significantly smaller infarct expansion index was also observed in the NP-liraglutide group (0.91±0.03) compared to the saline, NP and liraglutide groups (1.33±0.12, 1.34±0.15 and 1.28±0.14, respectively, P<0.05). The MI-induced decreased scar thickness and cardiac dilation (adverse cardiac remodeling) was significantly attenuated by NP-liraglutide. This finding was further confirmed by the results of the angiogenesis and apoptosis assays.
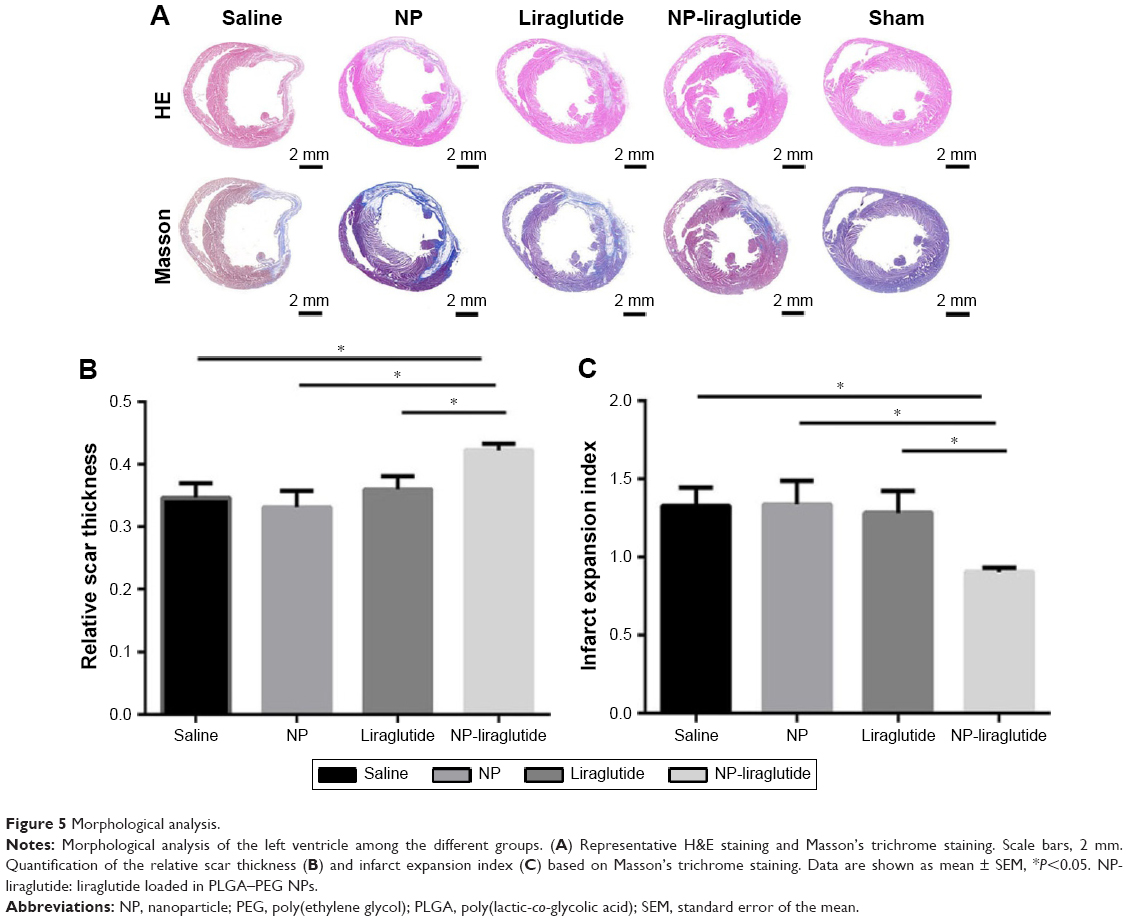 | Figure 5 Morphological analysis. |
We examined whether the delivery of liraglutide would promote neovascularization using immunohistochemical analyses (Figure 6). The NP-liraglutide treatment exerted superior effects on neovascularization compared to the other treatments. NP-liraglutide treatment induced a greater capillary density in the peri-infarct region (60.18±1.96 mm−2) compared to the saline, NP and liraglutide groups (35.91±3.48, 39.84±4.49 and 41.73±4.76 mm−2, respectively, P<0.05; Figure 6B). Furthermore, as shown in Figure 6C, we found co-localization of CD31 and α-SMA, indicating that mature vessels were formed. The maturation index in the NP-liraglutide group (65.5%±1.5%) was increased compared to the saline, NP and liraglutide groups (26.8%±3.4%, 27.9%±4.3% and 34.8%±3.4%, respectively, P<0.05), suggesting that NP-liraglutide induced vessel maturation for myocardial regeneration. These data clearly show that the mechanisms underlying the capacity of NP-liraglutide to improve cardiac function and attenuate cardiac remodeling could be related to enhanced angiogenesis.
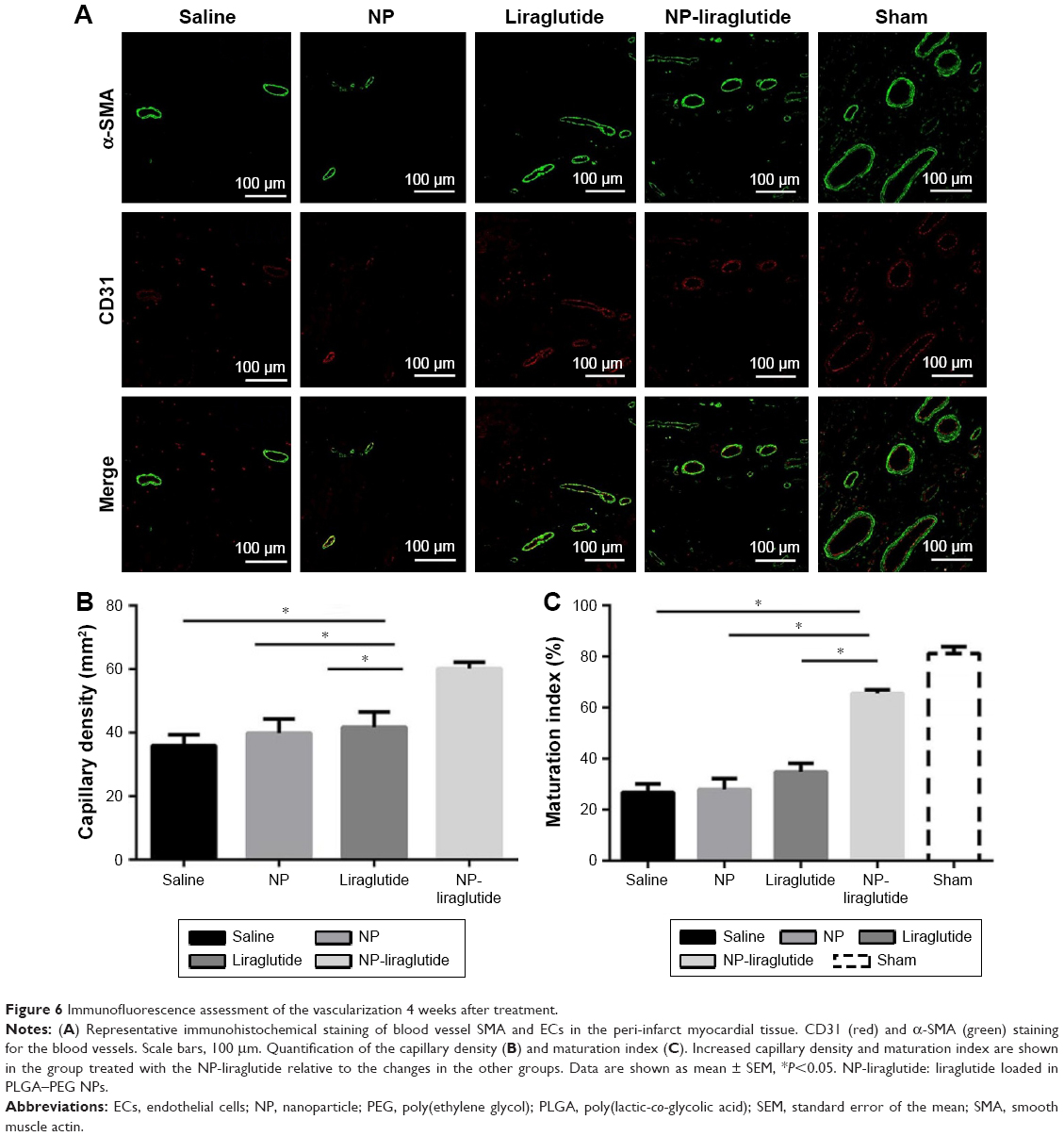 | Figure 6 Immunofluorescence assessment of the vascularization 4 weeks after treatment. |
Cardiomyocyte apoptosis is another leading cause of HF.30 A TUNEL assay (Hoffman-La Roche Ltd) was performed to detect the apoptotic cardiac cells in the peri-infarct region 4 weeks after the injection. As shown in Figure 7, the NP-liraglutide group showed a significant reduction in the number of apoptotic cells (3.0%±0.3%) compared to the saline, NP and liraglutide groups (5.9%±0.7%, 5.1%±0.8% and 5.4%±0.8%, respectively, P<0.05). Thus, cardiomyocyte apoptosis is suppressed by the NP-liraglutide treatment.
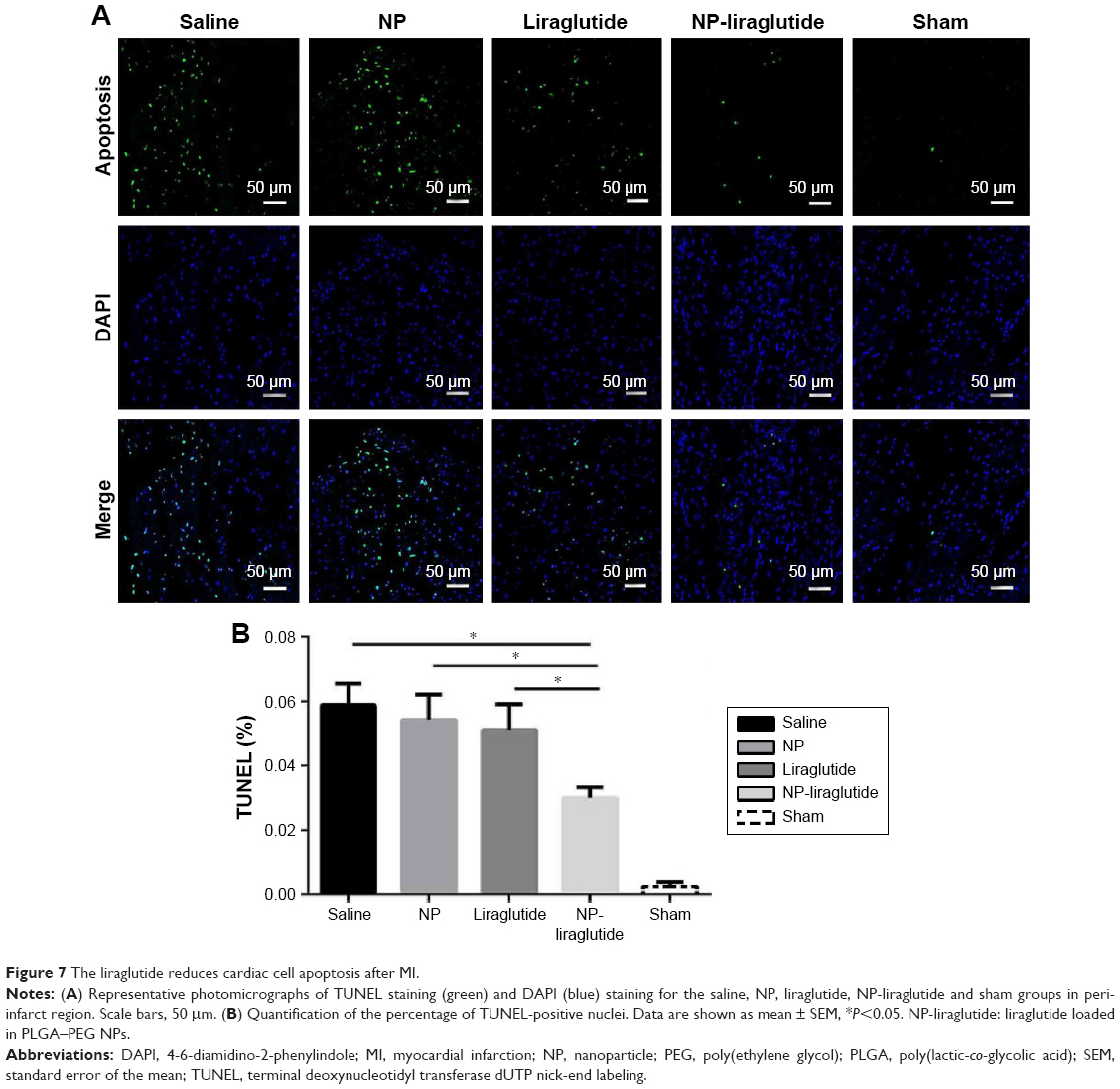 | Figure 7 The liraglutide reduces cardiac cell apoptosis after MI. |
Liraglutide has been approved for the treatment of diabetes. Therefore, we examined whether the delivery of liraglutide in the heart induced hypoglycemia. Significant differences were not detected among the four groups (Figure S2). Therefore, the cardioprotective effects of NP-liraglutide are attainable without affecting the normal blood glucose levels, consistent with the findings of a previous study.10
Discussion
In this study, we confirmed the suitability of using NP-liraglutide for the local, controlled, intramyocardial delivery of liraglutide. In addition, NP-liraglutide may improve cardiac function after MI by relieving adverse cardiac remodeling, inducing angiogenesis and inhibiting cardiac cell apoptosis.
Adverse cardiac remodeling after MI is a compensatory response that causes an irreversible loss of cardiomyocytes and eventually HF. However, the current therapies are mainly palliative.31 Thus, new therapies must be developed to overcome the poor prognosis of MI. Several alternative strategies based on drug treatments,32 injections of stem cells or proteins,4,5 surgical interventions33 and epicardial restraint devices34 have been used to regenerate the infarcted heart.
The effects of liraglutide have been shown to be beneficial for the treatment of MI.10,35 Several factors may be responsible for the cardioprotective effects of liraglutide. First, glucose metabolism is promoted over fatty acid metabolism, which is a more efficient method for oxygen production36 and may decrease oxygen demand. Second, liraglutide enhances vascularization.37 Finally, liraglutide reduces myocardial apoptosis.10
Protein therapy is suggested to be a promising treatment strategy for MI.38 However, the short half-lives of proteins in their free form under biological conditions limit their successful application.39 Similarly, the clinical application of liraglutide is limited by its short half-life.40
PLGA demonstrated great potential by maintaining the local concentrations of proteins for a prolonged period without the necessity for multiple injections.41 PLGA is a potential vehicle candidate for the sustained release of proteins that show promise as treatments for cardiac diseases.42–45 Moreover, PEG is effective in preventing NP phagocytosis by allowing NPs to evade the immune system.44,46–48 Liraglutide can benefit from the advantages offered by the PLGA–PEG delivery system to overcome the short half-life challenges of protein therapy.
As shown in the in vitro studies, the PLGA–PEG NPs were useful in controlling the release behavior of liraglutide for prolonged periods (Figure 2C). In addition, NPs can remain in the myocardium for at least 4 weeks and elicit long-term benefits without inflammation or fibrosis. After 4 weeks, the NPs are gradually degraded into smaller particles, which are susceptible to phagocytosis.42 Furthermore, the released liraglutide was effective in activating AKT protein kinase. Thus, the NPs are unaffected by the PLGA–PEG manufacturing procedures and maintained the native properties of liraglutide (Figure 2D, E).
As shown in the in vivo study, the local, controlled, intramyocardial delivery of NP-liraglutide attenuated adverse cardiac remodeling and facilitated angiogenesis after MI. Masson’s trichrome staining revealed that an increase in the relative scar thickness and a decrease in the infarct expansion index in the NP-liraglutide group were superior to that in the other three groups (Figure 5). The induction of neovascularization is one of the important elements determining the therapeutic efficacy of treatments for ischemic heart disease, due to its ability to salvage damaged ischemic tissues and initiate self-repair pathways.49 Liraglutide has also been reported to stimulate neovascularization.37 The number of CD31-expressing vessels was significantly increased in the NP-liraglutide group. The NP-liraglutide-induced increases in vascular density would relieve the ischemia in the peri-infarct region. Based on these observations, the improved LV function and geometry may be caused by the increase in revascularization.50 Furthermore, the NP group exhibited the same vessel densities as the saline-only group, suggesting that NPs did not induce angiogenesis (Figure 6). Furthermore, in this study, the NP-liraglutide treatment suppressed cardiomyocyte apoptosis (Figure 7).
Conclusion
Based on these results, the NP-liraglutide was useful in controlling the release behavior of liraglutide for prolonged periods. Furthermore, this strategy significantly improved cardiac function following MI by attenuating adverse cardiac remolding, promoting angiogenesis and suppressing apoptosis. Thus, we identified NP-liraglutide as an ideal system for liraglutide delivery into the infarcted myocardium, and it would serve as a potential treatment to prevent adverse cardiac remodeling after MI.
Acknowledgments
We thank, in particular, Sen li, Mi Zhou, Junfeng Cai, Zhe Wang and Anqing Chen for MI model making. This work was funded by the National Natural Science Foundation of China (Grants 81471779, 31671003, 81671832 and 81571826), Thousand Young Talents Program and the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (No TP2014028).
Disclosure
The authors report no conflicts of interest in this work.
References
Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics – 2011 update: a report from the American Heart Association. Circulation. 2011;123(4):e18–e209. | ||
Lloyd-Jones DM, Huffman MD, Karmali KN, et al. Estimating longitudinal risks and benefits from cardiovascular preventive therapies among Medicare patients: the million hearts longitudinal ASCVD risk assessment tool. J Am Coll Cardiol. 2017;69(12):1617–1636. | ||
Pascual-Gil S, Garbayo E, Diaz-Herraez P, Prosper F, Blanco-Prieto MJ. Heart regeneration after myocardial infarction using synthetic biomaterials. J Control Release. 2015;203:23–38. | ||
Bernstein HS, Srivastava D. Stem cell therapy for cardiac disease. Pediatr Res. 2012;71(4 pt 2):491–499. | ||
Segers VF, Lee RT. Protein therapeutics for cardiac regeneration after myocardial infarction. J Cardiovasc Transl Res. 2010;3(5):469–477. | ||
Gaffney MM, Hynes SO, Barry F, O’Brien T. Cardiovascular gene therapy: current status and therapeutic potential. Br J Pharmacol. 2007;152(2):175–188. | ||
Action to Control Cardiovascular Risk in Diabetes Study Group, Gerstein HC, Miller ME, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358(24):2545–2559. | ||
Marso SP, Poulter NR, Nissen SE, et al. Design of the liraglutide effect and action in diabetes: evaluation of cardiovascular outcome results (LEADER) trial. Am Heart J. 2013;166(5):823.e5–830.e5. | ||
Nikolaidis LA, Mankad S, Sokos GG, et al. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation. 2004;109(8):962–965. | ||
Noyan-Ashraf MH, Momen MA, Ban K, et al. GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes. 2009;58(4):975–983. | ||
Richards P, Parker HE, Adriaenssens AE, et al. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes. 2014;63(4):1224–1233. | ||
Inoue T, Inoguchi T, Sonoda N, et al. GLP-1 analog liraglutide protects against cardiac steatosis, oxidative stress and apoptosis in streptozotocin-induced diabetic rats. Atherosclerosis. 2015;240(1):250–259. | ||
Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. 2014;57(4):660–671. | ||
Marre M, Shaw J, Brandle M, et al. Liraglutide, a once-daily human GLP-1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with Type 2 diabetes (LEAD-1 SU). Diabet Med. 2009;26(3):268–278. | ||
Buse JB, Nauck M, Forst T, et al. Exenatide once weekly versus liraglutide once daily in patients with type 2 diabetes (DURATION-6): a randomised, open-label study. Lancet. 2013;381(9861):117–124. | ||
Nauck M, Frid A, Hermansen K, et al. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care. 2009;32(1):84–90. | ||
Wu J, Kamaly N, Shi J, et al. Development of multinuclear polymeric nanoparticles as robust protein nanocarriers. Angew Chem Int Ed Engl. 2014;53(34):8975–8979. | ||
Xiao Z, Farokhzad OC. Aptamer-functionalized nanoparticles for medical applications: challenges and opportunities. ACS Nano. 2012;6(5):3670–3676. | ||
Xiao Z. CuS nanoparticles: clinically favorable materials for photothermal applications? Nanomedicine. 2014;9(3):373–375. | ||
Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41(7):2971–3010. | ||
Gu F, Zhang L, Teply BA, et al. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc Natl Acad Sci U S A. 2008;105(7):2586–2591. | ||
Noyan-Ashraf MH, Shikatani EA, Schuiki I, et al. A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation. 2013;127(1):74–85. | ||
Ruvinov E, Leor J, Cohen S. The promotion of myocardial repair by the sequential delivery of IGF-1 and HGF from an injectable alginate biomaterial in a model of acute myocardial infarction. Biomaterials. 2011;32(2):565–578. | ||
Ruvinov E, Leor J, Cohen S. The effects of controlled HGF delivery from an affinity-binding alginate biomaterial on angiogenesis and blood perfusion in a hindlimb ischemia model. Biomaterials. 2010;31(16):4573–4582. | ||
Yoshizumi T, Zhu Y, Jiang H, et al. Timing effect of intramyocardial hydrogel injection for positively impacting left ventricular remodeling after myocardial infarction. Biomaterials. 2016;83:182–193. | ||
Paul A, Hasan A, Kindi HA, et al. Injectable graphene oxide/hydrogel-based angiogenic gene delivery system for vasculogenesis and cardiac repair. ACS Nano. 2014;8(8):8050–8062. | ||
Wang Q, Yang H, Bai A, et al. Functional engineered human cardiac patches prepared from nature’s platform improve heart function after acute myocardial infarction. Biomaterials. 2016;105:52–65. | ||
Song M, Jang H, Lee J, et al. Regeneration of chronic myocardial infarction by injectable hydrogels containing stem cell homing factor SDF-1 and angiogenic peptide Ac-SDKP. Biomaterials. 2014;35(8):2436–2445. | ||
Kim JH, Jung Y, Kim SH, et al. The enhancement of mature vessel formation and cardiac function in infarcted hearts using dual growth factor delivery with self-assembling peptides. Biomaterials. 2011;32(26):6080–6088. | ||
Santos-Gallego CG, Vahl TP, Goliasch G, et al. Sphingosine-1-phosphate receptor agonist fingolimod increases myocardial salvage and decreases adverse postinfarction left ventricular remodeling in a porcine model of ischemia/reperfusion. Circulation. 2016;133(10):954–966. | ||
Chang MY, Yang YJ, Chang CH, et al. Functionalized nanoparticles provide early cardioprotection after acute myocardial infarction. J Control Release. 2013;170(2):287–294. | ||
Sabbah HN, Shimoyama H, Kono T, et al. Effects of long-term monotherapy with enalapril, metoprolol, and digoxin on the progression of left ventricular dysfunction and dilation in dogs with reduced ejection fraction. Circulation. 1994;89(6):2852–2859. | ||
Dor V, Sabatier M, Di Donato M, Montiglio F, Toso A, Maioli M. Efficacy of endoventricular patch plasty in large postinfarction akinetic scar and severe left ventricular dysfunction: comparison with a series of large dyskinetic scars. J Thorac Cardiovasc Surg. 1998;116(1):50–59. | ||
D’Amore A, Yoshizumi T, Luketich SK, et al. Bi-layered polyurethane – extracellular matrix cardiac patch improves ischemic ventricular wall remodeling in a rat model. Biomaterials. 2016;107:1–14. | ||
Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375(4):311–322. | ||
Kavianipour M, Ehlers MR, Malmberg K, et al. Glucagon-like peptide-1 (7–36) amide prevents the accumulation of pyruvate and lactate in the ischemic and non-ischemic porcine myocardium. Peptides. 2003;24(4):569–578. | ||
Sato K, Kameda M, Yasuhara T, et al. Neuroprotective effects of liraglutide for stroke model of rats. Int J Mol Sci. 2013;14(11):21513–21524. | ||
Lee J, Tan CY, Lee SK, Kim YH, Lee KY. Controlled delivery of heat shock protein using an injectable microsphere/hydrogel combination system for the treatment of myocardial infarction. J Control Release. 2009;137(3):196–202. | ||
Wang L, Liu Y, Zhang W, Chen X, Yang T, Ma G. Microspheres and microcapsules for protein delivery: strategies of drug activity retention. Curr Pharm Des. 2013;19(35):6340–6352. | ||
Wu J, Williams GR, Branford-White C, Li H, Li Y, Zhu LM. Liraglutide-loaded poly(lactic-co-glycolic acid) microspheres: preparation and in vivo evaluation. Eur J Pharm Sci. 2016;92:28–38. | ||
Formiga FR, Pelacho B, Garbayo E, et al. Sustained release of VEGF through PLGA microparticles improves vasculogenesis and tissue remodeling in an acute myocardial ischemia-reperfusion model. J Control Release. 2010;147(1):30–37. | ||
Pascual-Gil S, Simon-Yarza T, Garbayo E, Prosper F, Blanco-Prieto MJ. Tracking the in vivo release of bioactive NRG from PLGA and PEG-PLGA microparticles in infarcted hearts. J Control Release. 2015;220(pt A):388–396. | ||
Qiao S, Zhao Y, Geng S, et al. A novel double-targeted nondrug delivery system for targeting cancer stem cells. Int J Nanomedicine. 2016;11:6667–6678. | ||
Bi C, Wang A, Chu Y, et al. Intranasal delivery of rotigotine to the brain with lactoferrin-modified PEG-PLGA nanoparticles for Parkinson’s disease treatment. Int J Nanomedicine. 2016;11:6547–6559. | ||
Jahan ST, Haddadi A. Investigation and optimization of formulation parameters on preparation of targeted anti-CD205 tailored PLGA nanoparticles. Int J Nanomedicine. 2015;10:7371–7384. | ||
Simon-Yarza T, Formiga FR, Tamayo E, Pelacho B, Prosper F, Blanco-Prieto MJ. PEGylated-PLGA microparticles containing VEGF for long term drug delivery. Int J Pharm. 2013;440(1):13–18. | ||
Gref R, Luck M, Quellec P, et al. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces. 2000;18(3–4):301–313. | ||
Wattendorf U, Merkle HP. PEGylation as a tool for the biomedical engineering of surface modified microparticles. J Pharm Sci. 2008;97(11):4655–4669. | ||
Haider H, Akbar SA, Ashraf M. Angiomyogenesis for myocardial repair. Antioxid Redox Signal. 2009;11(8):1929–1944. | ||
Gyongyosi M, Khorsand A, Zamini S, et al. NOGA-guided analysis of regional myocardial perfusion abnormalities treated with intramyocardial injections of plasmid encoding vascular endothelial growth factor A-165 in patients with chronic myocardial ischemia – subanalysis of the EUROINJECT-ONE multicenter double-blind randomized study. Circulation. 2005;112(9):I157–I165. |
Supplementary materials
 | Figure S1 Echocardiographic assessments. |
 | Figure S2 Blood glucose levels. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.