")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 9
Sources and implications of NADH/NAD+ redox imbalance in diabetes and its complications
Authors Wu J, Jin Z, Zheng H, Yan L
Received 9 February 2016
Accepted for publication 16 March 2016
Published 10 May 2016 Volume 2016:9 Pages 145—153
DOI https://doi.org/10.2147/DMSO.S106087
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Jinzi Wu,1Zhen Jin,1Hong Zheng,1,2Liang-Jun Yan1
1Department of Pharmaceutical Sciences, UNT System College of Pharmacy, University of North Texas Health Science Center, Fort Worth, TX, USA; 2Department of Basic Theory of Traditional Chinese Medicine, College of Basic Medicine, Shandong University of Traditional Chinese Medicine, Jinan, People’s Republic of China
Abstract: NAD+ is a fundamental molecule in metabolism and redox signaling. In diabetes and its complications, the balance between NADH and NAD+ can be severely perturbed. On one hand, NADH is overproduced due to influx of hyperglycemia to the glycolytic and Krebs cycle pathways and activation of the polyol pathway. On the other hand, NAD+ can be diminished or depleted by overactivation of poly ADP ribose polymerase that uses NAD+ as its substrate. Moreover, sirtuins, another class of enzymes that also use NAD+ as their substrate for catalyzing protein deacetylation reactions, can also affect cellular content of NAD+. Impairment of NAD+ regeneration enzymes such as lactate dehydrogenase in erythrocytes and complex I in mitochondria can also contribute to NADH accumulation and NAD+ deficiency. The consequence of NADH/NAD+ redox imbalance is initially reductive stress that eventually leads to oxidative stress and oxidative damage to macromolecules, including DNA, lipids, and proteins. Accordingly, redox imbalance-triggered oxidative damage has been thought to be a major factor contributing to the development of diabetes and its complications. Future studies on restoring NADH/NAD+ redox balance could provide further insights into design of novel antidiabetic strategies.
Keywords: mitochondria, complex I, reactive oxygen species, polyol pathway, poly ADP ribosylation, sirtuins, oxidative stress, oxidative damage
Introduction
Chronic elevation of blood glucose, known as diabetic hyperglycemia, is a hallmark of diabetes mellitus.1–4 This persistent hyperglycemia can lead to long term damage to tissues such as the kidney, eyes, nerves, blood vessels, and heart. 3,5,6 For non-insulin-dependent tissues, a high level of blood glucose would mean a high level of glucose metabolism as glucose entry into the cells is not limited by insulin deficiency.7,8 Since one of the major purposes of glucose metabolism is to provide electrons that are stored mainly in NADH and FADH2 for ATP production via the processes of glycolysis and mitochondrial metabolic pathways, NADH would be in an oversupply state when glucose overload occurs. This excess NADH can break the redox balance between NADH and NAD+, and eventually can lead to oxidative stress and a variety of metabolic syndromes.9–13 Hence, it suffices to say that diabetes is a redox imbalance disease.14,15
In this review, we delineate the sources and the pathways that contribute to NADH/NAD+ redox imbalance, and the potential consequences of this redox imbalance in diabetes. Regarding pathways that contribute to NADH/NAD+ redox imbalance, we focus on both the conventional glucose metabolic pathways and polyol pathway that get activated by high level of blood glucose.16–18 We also discuss the pathways that utilize NAD+ as substrates such as sirtuins deacetylation pathways19,20 and poly ADP ribosylation pathway.21,22 Additionally, NADH/NAD+-recycling enzymes such as lactate dehydrogenase (LDH) and mitochondrial complex I (NADH-ubiquinone oxidoreductase23,24) are also discussed. We believe that the consequences triggered by NADH/NAD+ redox imbalance are eventually reflected by oxidative stress and cell death that are known to be involved in the pathogenesis of diabetes and its complications.
NADH production by the conventional glucose metabolic pathways
The pair of NADH and NAD+ plays a crucial role in metabolism and redox signaling.25–30 The central pathways involved in complete glucose breakdown and electron storage in NADH are the glycolytic pathway and the Krebs cycle. As shown in Figure 1, glyceraldehyde 3-phosphate dehydrogenase in the glycolytic pathway makes NADH from NAD+. This is followed by pyruvate dehydrogenase complex that also makes NADH from NAD+, whereby the actual enzyme catalyzing NADH formation is dihydrolipoamide dehydrogenase.31,32 After acetyl-CoA enters into the Krebs cycle, more molecules of NADH are produced, which can be ascribed to the action of isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, and malate dehydrogenase, respectively. Fatty acid β-oxidation fueling the production of acetyl-CoA can also be a significant source of NADH.33 Additionally, glutamate dehydrogenase, a central enzyme involved in α-ketoglutarate formation from glutamate,34 can also make NADH from NAD+.35,36 Under hyperglycemic conditions, both the glycolytic pathway and the Krebs cycle can be intensively fluxed by glucose.37 Therefore, NADH can be overproduced in diabetes via these pathways,38 and excess NADH is known to cause reductive stress.13,39–43
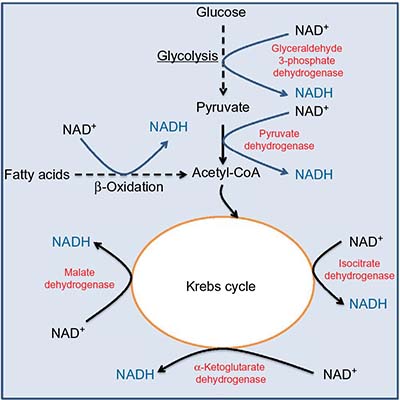 | Figure 1 Metabolic pathways and enzymes involved in NADH production using NAD+ as their cofactor. |
NADH production by polyol pathway
The polyol pathway, as shown in Figure 2, involves two consecutive reactions that are catalyzed by aldose reductase and sorbitol dehydrogenase, respectively. This pathway is usually rather inactive under euglycemic condition16 but can become a highly active glucose disposal pathway under diabetic hyperglycemic condition.44,45 The major feature of this pathway is the production of NADH, sorbitol, and fructose.16,46–48 Each of these intermediates or products plays a role in the pathogenesis of diabetes and its complications.16,46–49 For example, sorbitol can accumulate in retinal and renal tissues and causes osmotic stress and cell death,50,51 and fructose can cause nonenzymatic protein glycation or nitration52,53 and contributes to pathogenesis of nonalcoholic fatty liver disease.54 More importantly, a massive NADH production by this pathway is known to perturb redox imbalance between NADH and NAD+, and consumption of NADPH can impair the function of glutathione reductase, leading to accumulation of oxidized form of glutathione and further accentuation of redox imbalance.13,55 As such, inhibition or deletion of aldose reductase, a rate-limiting enzyme in the polyol pathway, has been demonstrated to be antidiabetic.56–60
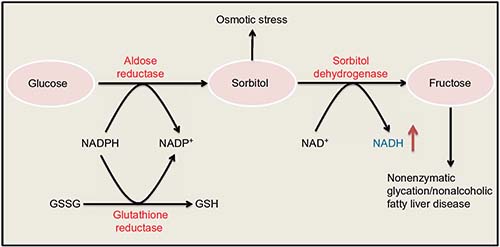 | Figure 2 Polyol pathway. |
NAD+-degradation pathways
NAD+ is not only an electron acceptor but can also serve as a substrate and be degraded during enzyme-catalyzed reactions. Two major enzymatic pathways that use NAD+ as their substrate are sirtuins and poly ADP ribose polymerases (PARPs).27,61 As shown in Figure 3A, sirtuins use NAD+ for their deacetylation reactions, whereby NAD+ is degraded and nicotinamide and 2’-O-acetyl-ADP ribose are formed. Sirtuins are inducible enzymes.62,63 Therefore, if NAD+ level is low, sirtuin protein content would be low.63,64 As acetylated proteins usually exhibit impaired functions,65,66 deacetylation by sirtuins usually improve the function of the target proteins.67 Therefore, sirtuins can be activated by starvation or caloric restriction to safeguard cell survival.68,69 On the other hand, overnutrition such as in diabetes that usually produces excess NADH with diminished NAD+ content can often lead to attenuation of sirtuin protein content.20,70 Therefore, enhancing sirtuin expression in diabetic tissues has been suggested as a therapeutic approach for treating diabetes and its complications.71,72 It should be noted that among the seven members of the sirtuin family,19,73 sirtuin 4 does not possess deacetylation activity but rather exhibits mono- or poly ADP ribosyltransferase activity.74
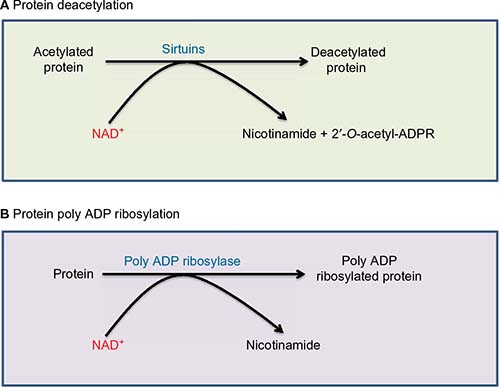 | Figure 3 Two enzyme systems that are involved in NAD+ degradation. |
While numerous studies demonstrate that elevating sirtuin protein content, such as that of sirtuin 3, ameliorates diabetes and its complications,67,75–77 a question arising is that whether it is possible that elevated levels of sirtuins consume more NAD+ and make the redox imbalance situation worse. This seemingly is not the case. It is probable that elevated levels of sirtuins alter the profiles of a given acetylated/deacetylated proteome, rendering metabolic pathways more efficient, which leads to more NADH utilization and thus more NAD+ regeneration.74 It has been reported that deacetylation by sirtuin protein can enhance NADPH production, which may be involved in restoring cellular redox balance.78 Nonetheless, whether elevation of sirtuin levels in diabetes could restore or improve NADH/NAD+ redox balance needs to be further thoroughly investigated.
Another enzyme system that consumes and degrades NAD+ is PARPs, especially PARP-179 that can be activated by DNA damage.22,80 As shown in Figure 3B, the products of PARP-catalyzed reaction are poly ADP ribosylated proteins and nicotinamide derived from NAD+. The problem caused by activation of PARP in diabetes is that the enzyme is often overactivated,81–83 resulting in potential depletion of NAD+, which would further perturb NADH/NAD+ redox balance, leading to cell death.21,79,84,85 PARP has been touted as a promising target for antidiabetic therapy. Indeed, knocking out or knocking down PARP expression can prevent animals from developing diabetes.86–88 Drugs that inhibit PARP activity have also been developed and tested for antidiabetic therapy.89–93 For example, 1,5-isoquinolinediol as a PARP inhibitor has been shown to improve corneal epithelial innervation in diabetic rats,94 and PARP inhibition could improve erectile function in diabetic rodents.95
Regeneration of NAD+ from NADH
For metabolism to continue, NAD+ has to be regenerated from NADH. There are two major pathways that can achieve this task, namely, LDH8 and mitochondrial complex I that is the first electron entry point in the electron transport chain.96–98 In anaerobic metabolism such as in erythrocytes where no mitochondria exist, LDH is responsible for NAD+ regeneration8,99 (Figure 4A). Under aerobic condition, however, mitochondrial complex I is responsible for NAD+ regeneration8,98 (Figure 4B). Hence, it is imaginable that NADH oversupply could overwhelm LDH100 or complex I.101 Indeed, it has been shown that diabetic hyperglycemia increases the enzyme activity of LDH in red blood cells and in small platelets to handle NADH over-influx.102,103 On the other hand, changes in complex I function in diabetes and its complications remain very sketchy. Nonetheless, it has been reported that complex I activity is decreased in diabetic skeletal muscles104 but increased in diabetic kidneys.105 Therefore, it seems that changes in complex I activity are tissue dependent in diabetic subjects. It would be interesting to survey complex I activity from tissue to tissue in diabetic rodents or possibly humans.
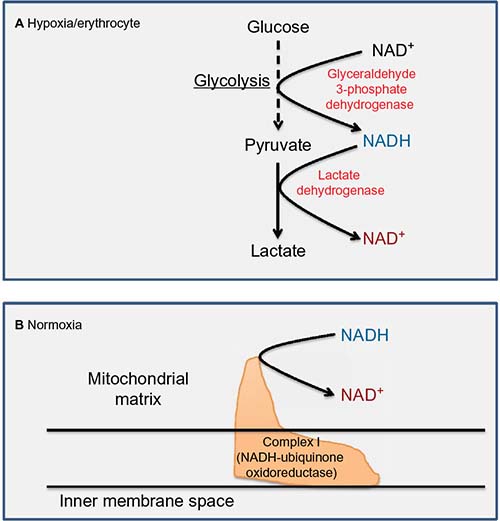 | Figure 4 Major cellular enzymes involved in NAD+ regeneration. |
Detrimental effects of redox imbalance in diabetes
When excess NADH accumulates, the enzymes that produce NADH from NAD+ will be inhibited. For example, both glyceraldehyde 3-phosphate dehydrogenase and dihydrolipoamide dehydrogenase in the pyruvate dehydrogenase complex can be inhibited by NADH,106,107 leading to potential reactive oxygen species (ROS) production.82,108,109 Moreover, mitochondrial electron transport chain can be overloaded by this electron donor.110 The direct pressure of this NADH overload would be on complex I, which is a major site for generation of ROS.111–116 The feature of this 45-subunit complex117 in ROS production is that the more NADH it oxidizes, the more ROS it will produce.112,114,118–120 Therefore, oxidants will overwhelm cellular antioxidant systems, leading to mitochondrial membrane permeability transition pore opening121,122 and mitochondrial dysfunction that are concurrent with extensive oxidative damage to proteins, DNA, and lipids123–127 (Figure 5). These oxidized macromolecules can accumulate over time, manifest diabetic glucotoxicity,128–131 and eventually lead to insulin resistance,132–135 β-cell insulin deficiency,136–138 and global cell death and tissue dysfunction.82,139–144 Indeed, oxidative damage and oxidative stress have been demonstrated to be involved in the pathogenesis of diabetes and its complications.145–148 Relevantly, inhibition of complex I has been shown to activate 5’-AMP-activated protein kinase and improves glucose metabolism in diabetes,149–152 supporting the observation that complex I ROS production plays a role in diabetes.153–157 Therefore, restoring redox balance or attenuating oxidative stress should be a promising approach to treating these chronic age-related diseases. Additionally, roles of antioxidants in antidiabetic therapy should also be highly appreciated as antioxidants usually work by ultimately improving redox balance.158–163
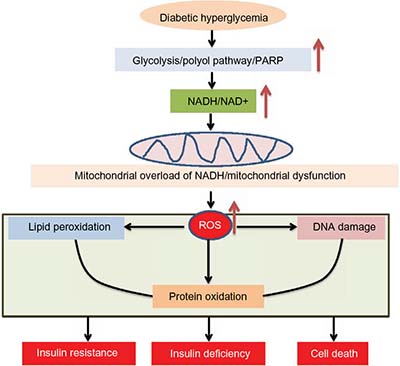 | Figure 5 Consequences of NADH/NAD+ redox imbalance. |
Summary and future perspectives
As has been discussed earlier, there is a severe redox imbalance problem occurring in diabetes and its complications. For cells whose glucose uptake is not dependent on insulin, glucose oversupply can lead to NADH overproduction by both the conventional glucose combustion pathways (Figure 1) and the polyol pathway (Figure 2). On the other hand, overactivation of PARP can diminish or deplete the cellular NAD+ pool (Figure 3B), thereby potentially downregulating sirtuins expression and making the redox imbalance situation worse. While drugs inhibiting aldose reductase in the polyol pathway164,165 or PARP89,90 will continue to remain as active areas of investigation in the future, NAD+ regeneration enzymes such as complex I should also be studied38 to provide insights into how excess NADH can be oxidized under glucose overload conditions. Additionally, administration of NAD+ precursors or analogs166,167 can also serve as an approach to treating diabetes and its complications. The ultimate goals of all these prospective studies are to restore NADH/NAD+ redox balance in diabetes and its complications for therapeutic purposes.
Acknowledgment
LJ Yan was supported in part by the National Institute of Neurological Disorders and Stroke (grant: R01NS079792).
Disclosure
The authors report no conflicts of interest in this work.
References
DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am. 2004;88(4):787–835, ix. | ||
Abdul-Ghani MA, DeFronzo RA. Oxidative stress in type 2 diabetes. In: Miwa S, Beckman KB, Muller FL, editors. Oxidative Stress in Aging. Totowa, New Jersey: Humana Press; 2008:191–212. | ||
Barnett AH. Type 2 Diabetes. 2nd ed. Oxford, UK: Oxford University Press; 2012. | ||
Tuch B, Dunlop M, Proietto J. Diabetes Research: A Guide for Postgraduates. Amsterdam, the Netherlands: Harwood Academic Publishers; 2000. | ||
Dedoussis GV, Kaliora AC, Panagiotakos DB. Genes, diet and type 2 diabetes mellitus: a review. Rev Diabet Stud. 2007;4(1):13–24. | ||
Del Prato S. Role of glucotoxicity and lipotoxicity in the pathophysiology of Type 2 diabetes mellitus and emerging treatment strategies. Diabet Med. 2009;26(12):1185–1192. | ||
Gallwitz B, Kazda C, Kraus P, Nicolay C, Schernthaner G. Contribution of insulin deficiency and insulin resistance to the development of type 2 diabetes: nature of early stage diabetes. Acta Diabetol. 2013;50(1):39–45. | ||
Lieberman M, Marks AD. Marks’ Basic Medical Biochemistry: A Clinical Approach. 4th ed. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins; 2013. | ||
Humphries KM, Szweda PA, Szweda LI. Aging: a shift from redox regulation to oxidative damage. Free Radic Res. 2006;40(12):1239–1243. | ||
Ido Y, Williamson JR. Hyperglycemic cytosolic reductive stress ‘pseudohypoxia’: implications for diabetic retinopathy. Invest Ophthalmol Vis Sci. 1997;38(8):1467–1470. | ||
Teodoro JS, Rolo AP, Palmeira CM. The NAD ratio redox paradox: why does too much reductive power cause oxidative stress? Toxicol Mech Methods. 2013;23(5):297–302. | ||
Lipinski B. Evidence in support of a concept of reductive stress. Br J Nutr. 2002;87(1):93–94; discussion 94. | ||
Yan LJ. Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J Diabetes Res. 2014;2014:137919. | ||
Hayden MR, Tyagi SC. Islet redox stress: the manifold toxicities of insulin resistance, metabolic syndrome and amylin derived islet amyloid in type 2 diabetes mellitus. JOP. 2002;3(4):86–108. | ||
Hayden MR, Sowers JR. Redox imbalance in diabetes. Antioxid Redox Signal. 2007;9(7):865–867. | ||
Dunlop M. Aldose reductase and the role of the polyol pathway in diabetic nephropathy. Kidney Int Suppl. 2000;77:S3–S12. | ||
Hotta N. New concepts and insights on pathogenesis and treatment of diabetic complications: polyol pathway and its inhibition. Nagoya J Med Sci. 1997;60(3–4):89–100. | ||
Lee AY, Chung SS. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 1999;13(1):23–30. | ||
Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med. 2013;56:133–171. | ||
Turkmen K, Karagoz A, Kucuk A. Sirtuins as novel players in the pathogenesis of diabetes mellitus. World J Diabetes. 2014;5(6):894–900. | ||
Pacher P, Szabo C. Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme. Antioxid Redox Signal. 2005;7(11–12):1568–1580. | ||
Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70(3):789–829. | ||
Schapira AH. Human complex I defects in neurodegenerative diseases. Biochim Biophys Acta. 1998;1364(2):261–270. | ||
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem. 1990;54(3):823–827. | ||
Lin SJ, Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol. 2003;15(2):241–246. | ||
Oka S, Hsu CP, Sadoshima J. Regulation of cell survival and death by pyridine nucleotides. Circ Res. 2012;111(5):611–627. | ||
Nikiforov A, Kulikova V, Ziegler M. The human NAD metabolome: functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol. 2015;50(4):284–297. | ||
Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome--a | ||
Mouchiroud L, Houtkooper RH, Auwerx J. NAD(+) metabolism: a therapeutic target for age-related metabolic disease. Crit Rev Biochem Mol Biol. 2013;48(4):397–408. | ||
Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31(2):194–223. | ||
Yan LJ, Yang SH, Shu H, Prokai L, Forster MJ. Histochemical staining and quantification of dihydrolipoamide dehydrogenase diaphorase activity using blue native PAGE. Electrophoresis. 2007;28(7):1036–1045. | ||
Yan LJ, Thangthaeng N, Forster MJ. Changes in dihydrolipoamide dehydrogenase expression and activity during postnatal development and aging in the rat brain. Mech Ageing Dev. 2008;129:282–290. | ||
Ido Y. Pyridine nucleotide redox abnormalities in diabetes. Antioxid Redox Signal. 2007;9(7):931–942. | ||
Li M, Li C, Allen A, Stanley CA, Smith TJ. Glutamate dehydrogenase: structure, allosteric regulation, and role in insulin homeostasis. Neurochem Res. 2014;39(3):433–445. | ||
Gohring I, Mulder H. Glutamate dehydrogenase, insulin secretion, and type 2 diabetes: a new means to protect the pancreatic beta-cell? J Endocrinol. 2012;212(3):239–242. | ||
Otter S, Lammert E. Exciting times for pancreatic islets: glutamate signaling in endocrine cells. Trends Endocrinol Metab. 2016;27(3):177–188. | ||
Jankovic A, Korac A, Buzadzic B, et al. Redox implications in adipose tissue (dys)function-a new look at old acquaintances. Redox Biol. 2015;6:19–32. | ||
Luo X, Li R, Yan LJ. Roles of pyruvate, NADH, and mitochondrial complex I in redox balance and imbalance in cell function and dysfunction. J Diabetes Res. 2015;2015:512618. | ||
Dawson TL, Gores GJ, Nieminen AL, Herman B, Lemasters JJ. Mitochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes. Am J Physiol. 1993;264(4 Pt 1):C961–C967. | ||
Comporti M, Signorini C, Leoncini S, et al. Ethanol-induced oxidative stress: basic knowledge. Genes Nutr. 2010;5(2):101–109. | ||
Jaeschke H, Kleinwaechter C, Wendel A. NADH-dependent reductive stress and ferritin-bound iron in allyl alcohol-induced lipid peroxidation in vivo: the protective effect of vitamin E. Chem Biol Interact. 1992;81(1–2):57–68. | ||
Yan LJ, Levine RL, Sohal RS. Oxidative damage during aging targets mitochondrial aconitase. Proc Natl Acad Sci U S A. 1997;94:11168–11172. | ||
Valadi H, Valadi A, Ansell R, et al. NADH-reductive stress in Saccharomyces cerevisiae induces the expression of the minor isoform of glyceraldehyde-3-phosphate dehydrogenase (TDH1). Curr Genet. 2004;45(2):90–95. | ||
Fantus IG. The pathogenesis of the chronic complications of the diabetes mellitus. Endocrinol Rounds. 2002;2(4):1–8. | ||
Boesten DM, von Ungern-Sternberg SN, den Hartog GJ, Bast A. Protective pleiotropic effect of flavonoids on NAD(+) levels in endothelial cells exposed to high glucose. Oxid Med Cell Longev. 2015;2015:894597. | ||
Chung SS, Chung SK. Aldose reductase in diabetic microvascular complications. Curr Drug Targets. 2005;6(4):475–486. | ||
Hodgkinson AD, Sondergaard KL, Yang B, Cross DF, Millward BA, Demaine AG. Aldose reductase expression is induced by hyperglycemia in diabetic nephropathy. Kidney Int. 2001;60(1):211–218. | ||
Iwata K, Nishinaka T, Matsuno K, et al. The activity of aldose reductase is elevated in diabetic mouse heart. J Pharmacol Sci. 2007;103(4):408–416. | ||
Kador PF, Kinoshita JH. Role of aldose reductase in the development of diabetes-associated complications. Am J Med. 1985;79(5A):8–12. | ||
Gabbay KH. The sorbitol pathway and the complications of diabetes. N Engl J Med. 1973;288(16):831–836. | ||
Bagnasco SM, Murphy HR, Bedford JJ, Burg MB. Osmoregulation by slow changes in aldose reductase and rapid changes in sorbitol flux. Am J Physiol. 1988;254(6 Pt 1):C788–C792. | ||
Takagi Y, Kashiwagi A, Tanaka Y, Asahina T, Kikkawa R, Shigeta Y. Significance of fructose-induced protein oxidation and formation of advanced glycation end product. J Diabetes Complications. 1995;9(2):87–91. | ||
Prince PD, Lanzi CR, Toblli JE, et al. Dietary (-)-epicatechin mitigates oxidative stress, NO metabolism alterations, and inflammation in renal cortex from fructose-fed rats. Free Radic Biol Med. 2016;90:35–46. | ||
Lirio LM, Forechi L, Zanardo TC, et al. Chronic fructose intake accelerates non-alcoholic fatty liver disease in the presence of essential hypertension. J Diabetes Complications. 2016;30(1):85–92. | ||
Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. 2008;10(2):179–206. | ||
Tang WH, Martin KA, Hwa J. Aldose reductase, oxidative stress, and diabetic mellitus. Front Pharmacol. 2012;3:87. | ||
Ohmura C, Watada H, Azuma K, et al. Aldose reductase inhibitor, epalrestat, reduces lipid hydroperoxides in type 2 diabetes. Endocr J. 2009;56(1):149–156. | ||
Reddy AB, Ramana KV. Aldose reductase inhibition: emerging drug target for the treatment of cardiovascular complications. Recent Pat Cardiovasc Drug Discov. 2010;5(1):25–32. | ||
Suzen S, Buyukbingol E. Recent studies of aldose reductase enzyme inhibition for diabetic complications. Curr Med Chem. 2003;10(15):1329–1352. | ||
Tang J, Du Y, Petrash JM, Sheibani N, Kern TS. Deletion of aldose reductase from mice inhibits diabetes-induced retinal capillary degeneration and superoxide generation. PLoS One. 2013;8(4):e62081. | ||
Massudi H, Grant R, Guillemin GJ, Braidy N. NAD+ metabolism and oxidative stress: the golden nucleotide on a crown of thorns. Redox Rep. 2012;17(1):28–46. | ||
Sauve AA. Sirtuin chemical mechanisms. Biochim Biophys Acta. 2010;1804(8):1591–1603. | ||
Vedantham S, Thiagarajan D, Ananthakrishnan R, et al. Aldose reductase drives hyperacetylation of Egr-1 in hyperglycemia and consequent upregulation of proinflammatory and prothrombotic signals. Diabetes. 2014;63(2):761–774. | ||
Cerutti R, Pirinen E, Lamperti C, et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014;19(6):1042–1049. | ||
Yang T, Sauve AA. NAD metabolism and sirtuins: metabolic regulation of protein deacetylation in stress and toxicity. AAPS J. 2006;8(4):E632–E643. | ||
Kosanam H, Thai K, Zhang Y, et al. Diabetes induces lysine acetylation of intermediary metabolism enzymes in the kidney. Diabetes. 2014;63(7):2432–2439. | ||
Hirschey MD, Shimazu T, Jing E, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44(2):177–190. | ||
Tucci P. Caloric restriction: is mammalian life extension linked to p53? Aging. 2012;4(8):525–534. | ||
Wang Y. Molecular links between caloric restriction and Sir2/SIRT1 activation. Diabetes Metab J. 2014;38(5):321–329. | ||
de Kreutzenberg SV, Ceolotto G, Papparella I, et al. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes. 2010;59(4):1006–1015. | ||
Kitada M, Kume S, Kanasaki K, Takeda-Watanabe A, Koya D. Sirtuins as possible drug targets in type 2 diabetes. Curr Drug Targets. 2013;14(6):622–636. | ||
Huynh FK, Hershberger KA, Hirschey MD. Targeting sirtuins for the treatment of diabetes. Diabetes Manag (Lond). 2013;3(3): | ||
Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol. 2008;382(3):790–801. | ||
Pereira CV, Lebiedzinska M, Wieckowski MR, Oliveira PJ. Regulation and protection of mitochondrial physiology by sirtuins. Mitochondrion. 2012;12(1):66–76. | ||
Jing E, Emanuelli B, Hirschey MD, et al. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A. 2011;108(35):14608–14613. | ||
Hou X, Zeng H, He X, Chen JX. Sirt3 is essential for apelin-induced angiogenesis in post-myocardial infarction of diabetes. J Cell Mol Med. 2015;19(1):53–61. | ||
Fritz KS, Galligan JJ, Hirschey MD, Verdin E, Petersen DR. Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. J Proteome Res. 2012;11(3):1633–1643. | ||
Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48(6):749–762. | ||
Puthanveetil P, Zhang D, Wang Y, et al. Diabetes triggers a PARP1 mediated death pathway in the heart through participation of FoxO1. J Mol Cell Cardiol. 2012;53(5):677–686. | ||
Mueller-Dieckmann C, Kernstock S, Lisurek M, et al. The structure of human ADP-ribosylhydrolase 3 (ARH3) provides insights into the reversibility of protein ADP-ribosylation. Proc Natl Acad Sci U S A. 2006;103(41):15026–15031. | ||
Horvath EM, Magenheim R, Kugler E, et al. Nitrative stress and poly(ADP-ribose) polymerase activation in healthy and gestational diabetic pregnancies. Diabetologia. 2009;52(9):1935–1943. | ||
Du X, Matsumura T, Edelstein D, et al. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. 2003;112(7):1049–1057. | ||
Dolle C, Rack JG, Ziegler M. NAD and ADP-ribose metabolism in mitochondria. FEBS J. 2013;280(15):3530–3541. | ||
Obrosova IG, Drel VR, Pacher P, et al. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes. 2005;54(12):3435–3441. | ||
Chiu J, Xu BY, Chen S, Feng B, Chakrabarti S. Oxidative stress-induced, poly(ADP-ribose) polymerase-dependent upregulation of ET-1 expression in chronic diabetic complications. Can J Physiol Pharmacol. 2008;86(6):365–372. | ||
Masutani M, Suzuki H, Kamada N, et al. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999;96(5):2301–2304. | ||
Pieper AA, Brat DJ, Krug DK, et al. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999;96(6):3059–3064. | ||
Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54(3):375–429. | ||
Long CA, Boulom V, Albadawi H, et al. Poly-ADP-ribose-polymerase inhibition ameliorates hind limb ischemia reperfusion injury in a murine model of type 2 diabetes. Ann Surg. 2013;258(6): | ||
Sarras MP Jr, Mason S, McAllister G, Intine RV. Inhibition of poly-ADP ribose polymerase enzyme activity prevents hyperglycemia-induced impairment of angiogenesis during wound healing. Wound Repair Regen. 2014;22(5):666–670. | ||
Szkudelski T. Streptozotocin-nicotinamide-induced diabetes in the rat. Characteristics of the experimental model. Exp Biol Med (Maywood). 2012;237(5):481–490. | ||
Fukaya M, Tamura Y, Chiba Y, et al. Protective effects of a nicotinamide derivative, isonicotinamide, against streptozotocin-induced beta-cell damage and diabetes in mice. Biochem Biophys Res Commun. 2013;442(1–2):92–98. | ||
Obrosova IG, Minchenko AG, Frank RN, et al. Poly(ADP-ribose) polymerase inhibitors counteract diabetes- and hypoxia-induced retinal vascular endothelial growth factor overexpression. Int J Mol Med. 2004;14(1):55–64. | ||
Byun YS, Kang B, Yoo YS, Joo CK. Poly(ADP-ribose) polymerase inhibition improves corneal epithelial innervation and wound healing in diabetic rats. Invest Ophthalmol Vis Sci. 2015;56(3): | ||
Li WJ, Zhou J, Li B, Wang H, Peng YB, Wang Z. PARP inhibition restores erectile function by suppressing corporal smooth muscle apoptosis in diabetic rats. J Sex Med. 2011;8(4):1072–1082. | ||
Hirst J. Mitochondrial complex I. Annu Rev Biochem. 2013;82:551–575. | ||
Andrews B, Carroll J, Ding S, Fearnley IM, Walker JE. Assembly factors for the membrane arm of human complex I. Proc Natl Acad Sci U S A. 2013;110(47):18934–18939. | ||
Papa S, Sardanelli AM, Scacco S, et al. The NADH: ubiquinone oxidoreductase (complex I) of the mammalian respiratory chain and the cAMP cascade. J Bioenerg Biomembr. 2002;34(1):1–10. | ||
Rapoport S. The regulation of glycolysis in mammalian erythrocytes. Essays Biochem. 1968;4:69–103. | ||
Leong HS, Brownsey RW, Kulpa JE, Allard MF. Glycolysis and pyruvate oxidation in cardiac hypertrophy--why so unbalanced? Comp Biochem Physiol A Mol Integr Physiol. 2003;135(4):499–513. | ||
He Q, Wang M, Petucci C, Gardell SJ, Han X. Rotenone induces reductive stress and triacylglycerol deposition in C2C12 cells. Int J Biochem Cell Biol. 2013;45(12):2749–2755. | ||
Lippi G, Mercadanti M, Aloe R, Targher G. Erythrocyte mechanical fragility is increased in patients with type 2 diabetes. Eur J Intern Med. 2012;23(2):150–153. | ||
Zappacosta B, De Sole P, Rossi C, Marra G, Ghirlanda G, Giardina B. Lactate dehydrogenase activity of platelets in diabetes mellitus. Eur J Clin Chem Clin Biochem. 1995;33(8):487–489. | ||
Antoun G, McMurray F, Thrush AB, et al. Impaired mitochondrial oxidative phosphorylation and supercomplex assembly in rectus abdominis muscle of diabetic obese individuals. Diabetologia. 2015;58(12):2861–2866. | ||
Wu J, Luo X, Yan LJ. Two dimensional blue native/SDS-PAGE to identify mitochondrial complex I subunits modified by 4-hydroxynonenal (HNE). Front Physiol. 2015;6:98. | ||
Wilkinson KD, Williams CH Jr. NADH inhibition and NAD activation of Escherichia coli lipoamide dehydrogenase catalyzing the NADH-lipoamide reaction. J Biol Chem. 1981;256(5):2307–2314. | ||
Ussher JR, Jaswal JS, Lopaschuk GD. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ Res. 2012;111(5):628–641. | ||
Yan LJ, Thangthaeng N, Sumien N, Forster MJ. Serum dihydrolipoamide dehydrogenase is a labile enzyme. J Biochem Pharmacol Res. 2013;1(1):30–42. | ||
Quinlan CL, Goncalves RL, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J Biol Chem. 2014;289(12):8312–8325. | ||
Galloway CA, Yoon Y. Perspectives on: SGP symposium on mitochondrial physiology and medicine: what comes first, misshape or dysfunction? The view from metabolic excess. J Gen Physiol. 2012;139(6):455–463. | ||
Bridges HR, Jones AJ, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J. 2014;462(3):475–487. | ||
Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochem Soc Trans. 2008;36(Pt 5):976–980. | ||
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. | ||
Cooper JM, Mann VM, Krige D, Schapira AH. Human mitochondrial complex I dysfunction. Biochim Biophys Acta. 1992;1101(2):198–203. | ||
Santidrian AF, Matsuno-Yagi A, Ritland M, et al. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013;123(3):1068–1081. | ||
Pryde KR, Hirst J. Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J Biol Chem. 2011;286(20):18056–18065. | ||
Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE. Bovine complex I is a complex of 45 different subunits. J Biol Chem. 2006;281(43):32724–32727. | ||
Treberg JR, Quinlan CL, Brand MD. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J Biol Chem. 2011;286(31):27103–27110. | ||
Esposti MD, Ngo A, Myers MA. Inhibition of mitochondrial complex I may account for IDDM induced by intoxication with the rodenticide Vacor. Diabetes. 1996;45(11):1531–1534. | ||
Fassone E, Rahman S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J Med Genet. 2012;49(9): | ||
Yan LJ, Rajasekaran NS, Sathyanarayanan S, Benjamin IJ. Mouse HSF1 disruption perturbs redox state and increases mitochondrial oxidative stress in kidney. Antioxid Redox Signal. 2005;7(3–4): | ||
Yan LJ, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J. 2002;21(19):5164–5172. | ||
Ames BN, Shigenaga MK. Oxidants are a major contributor to aging. Ann N Y Acad Sci. 1992;663:85–96. | ||
Helbock HJ, Beckman KB, Shigenaga MK, et al. DNA oxidation matters: the HPLC-electrochemical detection assay of 8-oxo-deoxyguanosine and 8-oxo-guanine. Proc Natl Acad Sci U S A. 1998;95(1):288–293. | ||
Anderson EJ, Katunga LA, Willis MS. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin Exp Pharmacol Physiol. 2012;39(2):179–193. | ||
Yan LJ, Lodge JK, Traber MG, Packer L. Apolipoprotein B carbonyl formation is enhanced by lipid peroxidation during copper-mediated oxidation of human low-density lipoproteins. Arch Biochem Biophys. 1997;339(1):165–171. | ||
Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272(33):20313–20316. | ||
Luo X, Wu J, Jing S, Yan LJ. Hyperglycemic stress and carbon stress in diabetic glucotoxicity. Aging Dis. 2016;7(1):90–110. | ||
Kaiser N, Leibowitz G, Nesher R. Glucotoxicity and beta-cell failure in type 2 diabetes mellitus. J Pediatr Endocrinol Metab. 2003;16(1): | ||
Brunner Y, Schvartz D, Priego-Capote F, Coute Y, Sanchez JC. Glucotoxicity and pancreatic proteomics. J Proteomics. 2009;71(6):576–591. | ||
Wu J, Yan LJ. Streptozotocin-induced type 1 diabetes in rodents as a model for studying mitochondrial mechanisms of diabetic beta cell glucotoxicity. Diabetes Metab Syndr Obes. 2015;8:181–188. | ||
Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102(4):401–414. | ||
Kaneki M, Shimizu N, Yamada D, Chang K. Nitrosative stress and pathogenesis of insulin resistance. Antioxid Redox Signal. 2007;9(3):319–329. | ||
Henriksen EJ, Diamond-Stanic MK, Marchionne EM. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic Biol Med. 2011;51(5):993–999. | ||
Abel ED. Free fatty acid oxidation in insulin resistance and obesity. Heart Metab. 2010;48:5–10. | ||
Guo S, Dai C, Guo M, et al. Inactivation of specific beta cell transcription factors in type 2 diabetes. J Clin Invest. 2013;123(8):3305–3316. | ||
Halban PA, Polonsky KS, Bowden DW, et al. Beta-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care. 2014;37(6):1751–1758. | ||
Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest. 2006;116(7):1802–1812. | ||
Seo K, Ki SH, Shin SM. Methylglyoxal induces mitochondrial dysfunction and cell death in liver. Toxicol Res. 2014;30(3):193–198. | ||
Berg D, Youdim MB, Riederer P. Redox imbalance. Cell Tissue Res. 2004;318(1):201–213. | ||
Stadtman ER. Protein oxidation and aging. Science. 1992;257(5074):1220–1224. | ||
Stadtman ER. Protein oxidation in aging and age-related diseases. Ann N Y Acad Sci. 2001;928:22–38. | ||
Stadtman ER. Protein oxidation and aging. Free Radic Res. 2006;40(12):1250–1258. | ||
Stadler K. Oxidative stress in diabetes. Adv Exp Med Biol. 2012;771:272–287. | ||
Weaver JR, Grzesik W, Taylor-Fishwick DA. Inhibition of NADPH oxidase-1 preserves beta cell function. Diabetologia. 2015;58(1): | ||
Robertson RP, Tanaka Y, Takahashi H, Tran PO, Harmon JS. Prevention of oxidative stress by adenoviral overexpression of glutathione-related enzymes in pancreatic islets. Ann N Y Acad Sci. 2005;1043: | ||
Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004;279(41):42351–42354. | ||
Karunakaran U, Park KG. A systematic review of oxidative stress and safety of antioxidants in diabetes: focus on islets and their defense. Diabetes Metab J. 2013;37(2):106–112. | ||
Srivastava RA, Pinkosky SL, Filippov S, Hanselman JC, Cramer CT, Newton RS. AMP-activated protein kinase: an emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat cardio-metabolic diseases. J Lipid Res. 2012;53(12):2490–2514. | ||
Huang SL, Yu RT, Gong J, et al. Arctigenin, a natural compound, activates AMP-activated protein kinase via inhibition of mitochondria complex I and ameliorates metabolic disorders in ob/ob mice. Diabetologia. 2012;55(5):1469–1481. | ||
Zheng T, Yang X, Wu D, et al. Salidroside ameliorates insulin resistance through activation of a mitochondria-associated AMPK/PI3K/Akt/GSK3beta pathway. Br J Pharmacol. 2015;172(13):3284–3301. | ||
Jenkins Y, Sun TQ, Markovtsov V, et al. AMPK activation through mitochondrial regulation results in increased substrate oxidation and improved metabolic parameters in models of diabetes. PLoS One. 2013;8(12):e81870. | ||
Lefort N, Glancy B, Bowen B, et al. Increased reactive oxygen species production and lower abundance of complex I subunits and carnitine palmitoyltransferase 1B protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes. 2010;59(10):2444–2452. | ||
Raza H, Prabu SK, John A, Avadhani NG. Impaired mitochondrial respiratory functions and oxidative stress in streptozotocin-induced diabetic rats. Int J Mol Sci. 2011;12(5):3133–3147. | ||
Victor VM, Rocha M, Banuls C, et al. Mitochondrial complex I impairment in leukocytes from polycystic ovary syndrome patients with insulin resistance. J Clin Endocrinol Metab. 2009;94(9): | ||
Batandier C, Guigas B, Detaille D, et al. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J Bioenerg Biomembr. 2006;38(1):33–42. | ||
Hernandez-Mijares A, Rocha M, Apostolova N, et al. Mitochondrial complex I impairment in leukocytes from type 2 diabetic patients. Free Radic Biol Med. 2011;50(10):1215–1221. | ||
Saini AS, Taliyan R, Sharma PL. Protective effect and mechanism of Ginkgo biloba extract-EGb 761 on STZ-induced diabetic cardiomyopathy in rats. Pharmacogn Mag. 2014;10(38):172–178. | ||
El-Awdan SA, Abdel Jaleel GA, Saleh AO. Grape seed extract attenuates hyperglycemia-induced in rats by streptozotocin. Bull Fac Pharm Cairo Univ. 2013;51:203–209. | ||
Niture NT, Ansari AA, Naik SR. Anti-hyperglycemic activity of rutin in streptozotocin-induced diabetic rats: an effect mediated through cytokines, antioxidants and lipid biomarkers. Indian J Exp Biol. 2014;52(7):720–727. | ||
Maritim A, Dene BA, Sanders RA, Watkins JB 3rd. Effects of pycnogenol treatment on oxidative stress in streptozotocin-induced diabetic rats. J Biochem Mol Toxicol. 2003;17(3):193–199. | ||
Parveen K, Khan MR, Mujeeb M, Siddiqui WA. Protective effects of Pycnogenol on hyperglycemia-induced oxidative damage in the liver of type 2 diabetic rats. Chem Biol Interact. 2010;186(2):219–227. | ||
Acharya JD, Pande AJ, Joshi SM, Yajnik CS, Ghaskadbi SS. Treatment of hyperglycaemia in newly diagnosed diabetic patients is associated with a reduction in oxidative stress and improvement in beta-cell function. Diabetes Metab Res Rev. 2014;30(7):590–598. | ||
Yasunari K, Kohno M, Kano H, Minami M, Yoshikawa J. Aldose reductase inhibitor improves insulin-mediated glucose uptake and prevents migration of human coronary artery smooth muscle cells induced by high glucose. Hypertension. 2000;35(5):1092–1098. | ||
Yasunari K, Kohno M, Kano H, Yokokawa K, Horio T, Yoshikawa J. Aldose reductase inhibitor prevents hyperproliferation and hypertrophy of cultured rat vascular smooth muscle cells induced by high glucose. Arterioscler Thromb Vasc Biol. 1995;15(12):2207–2212. | ||
Ying W, Wei G, Wang D, et al. Intranasal administration with NAD+ profoundly decreases brain injury in a rat model of transient focal ischemia. Front Biosci. 2007;12:2728–2734. | ||
Pittelli M, Felici R, Pitozzi V, et al. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol Pharmacol. 2011;80(6):1136–1146. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.