")
Back to Journals » OncoTargets and Therapy » Volume 9
Sophoridinol derivative 05D induces tumor cells apoptosis by topoisomerase1-mediated DNA breakage
Authors Zhao W, Zhang C, Bi C, Ye C, Song D, Liu X, Shao R
Received 5 January 2016
Accepted for publication 8 March 2016
Published 11 May 2016 Volume 2016:9 Pages 2805—2817
DOI https://doi.org/10.2147/OTT.S103671
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Wuli Zhao, Caixia Zhang, Chongwen Bi, Cheng Ye, Danqing Song, Xiujun Liu, Rongguang Shao
Key Laboratory of Antibiotic Bioengineering, Ministry of Health, Laboratory of Oncology, Institute of Medicinal Biotechnology, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, People’s Republic of China
Abstract: Sophoridine is a quinolizidine natural product of Sophora alopecuroides and has been applied for treatment of malignant trophoblastic tumors. Although characterized by low toxicity, the limited-spectrum antitumor activity hinders its further applications. 05D, a derivative of sophoridine, exhibits a better anticancer activity on diverse cancer cells, including solid tumors, and hematologic malignancy. It could inhibit topoisomerase 1 (top1) activity by stabilizing DNA–top1 complex and induce mitochondria-mediated apoptosis by promoting DNA single- and double-strand breakage mediated by top1. Also, 05D induced HCT116 cells arrest at G1 phase by inactivating CDK2/CDK4–Rb–E2F and cyclinD1–CDK4–p21 checkpoint signal pathways. 05D suppressed the ataxia telangiectasia mutated (ATM) and ATM and Rad3-related (ATR) activation and decreased 53BP level, which contributed to DNA damage repair, suggesting that the novel compound 05D might be helpful to improve the antitumor activity of DNA damaging agent by repressing ATM and ATR activation and 53BP level. In addition, the priorities in molecular traits and druggability, such as a simple structure and formulation for oral administration, further prove 05D to be a promising targeting topoisomerase agent.
Keywords: topoisomerase inhibitor, topoisomerase 1, DNA breakage, sophoridinol, anticancer, apoptosis, cell cycle
Introduction
Sophoridine (Figure 1A, left) is a quinolizidine natural product extracted from Sophora alopecuroides and has been applied for its antivirus1,2 and anti-inflammatory3,4 activities and for the improvement of cardiac function.5 Its anticancer activity was first discovered in 1977 and it obtained the clinical study approval in 1993.6,7 Given that it is highly effective on malignant trophoblastic tumors and its lower toxicity, in 2005, sophoridine injection was approved by the Chinese Food and Drug Administration to treat malignant trophoblastic tumors.6 Meanwhile, reports also demonstrated that sophoridine has inhibitory effect on only malignant trophoblastic tumors and has almost no effect on other tumor cells,6,7 which suggest a limited-spectrum antitumor activity. Thus, exploring a novel compound derived from sophoridine that both has low toxicity and possesses a broad antitumor spectrum would be beneficial to the anticancer drug development.
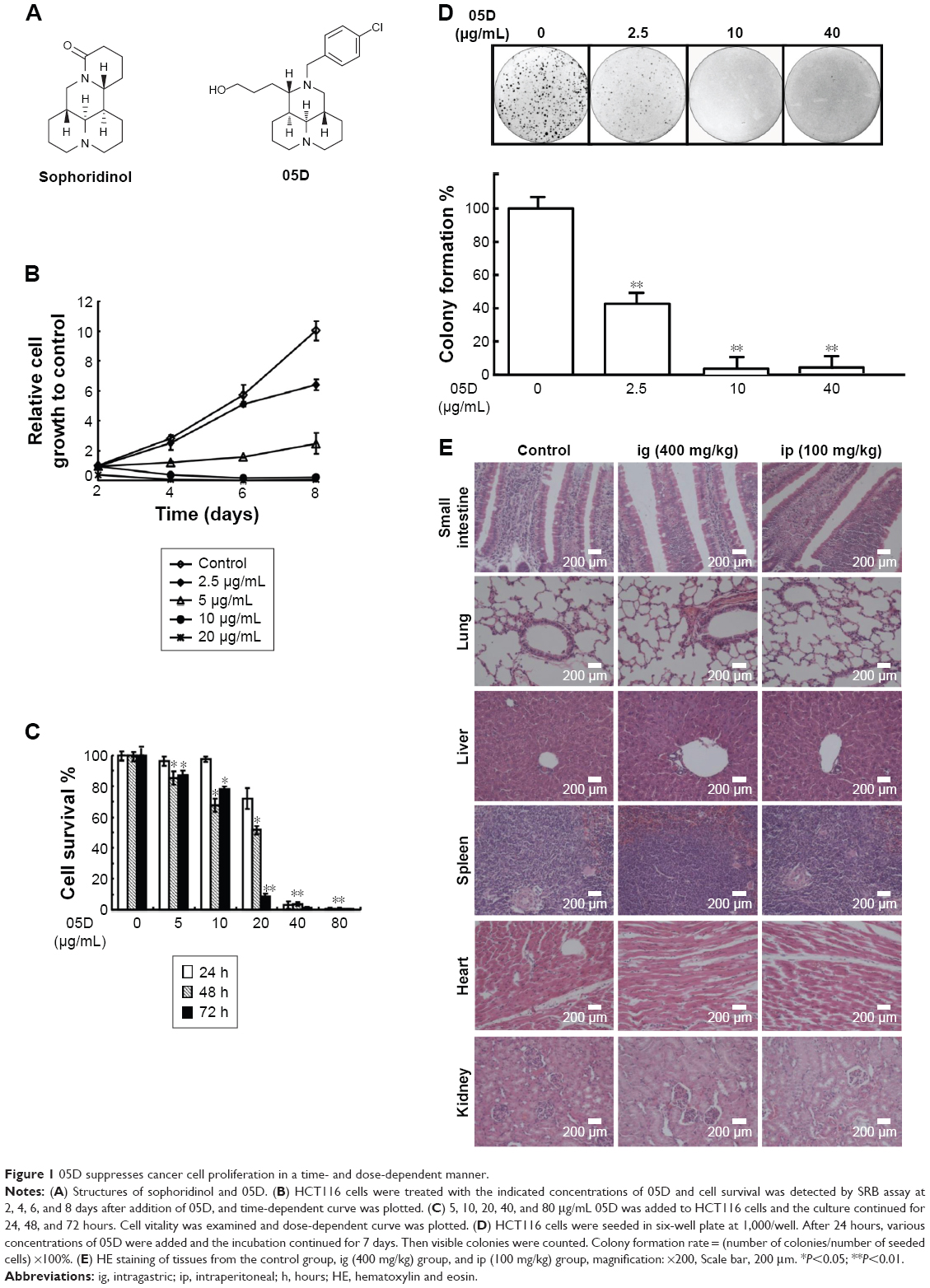 | Figure 1 05D suppresses cancer cell proliferation in a time- and dose-dependent manner. |
Our lab has been engaged in sophoridinic structure modification and has recently found that sophoridinic acid with a three-ring structure scaffold exhibited a better antiproliferative activity compared with its parent sophoridine with four-ring scaffold.8 Based on the earlier results, after a series of structure modifications, we found a novel compound, 05D (Figure 1A, right), that not only has a lower toxicity (Table 1), but can also inhibit the growth of various tumor cells (Table 2), compared with its parental sophoridine. Also, it possesses good water solubility. Although researchers had verified previously that the anticancer mechanism of sophoridine was to target topoisomerase 1 (top1), they did not further elucidate the inhibitory mechanism on top1 and its further effects on cancer cells by which way tumor cell death is induced.
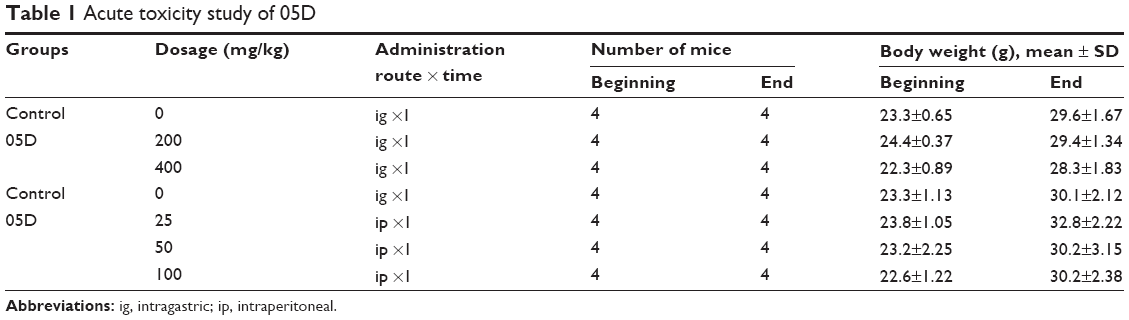 | Table 1 Acute toxicity study of 05D |
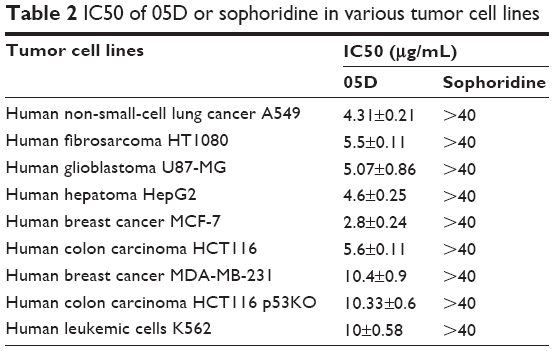 | Table 2 IC50 of 05D or sophoridine in various tumor cell lines |
In the present study, we demonstrated that novel compound 05D could inhibit proliferation of various tumor cells and has a low toxicity. It could inhibit the activity of top1 by stabilizing DNA–top1 complex and exerts its anticancer activity by facilitating DNA breakage mediated by top1 in cells and then triggering mitochondrial pathway of apoptosis.
Materials and methods
Reagents and cells
Hydroxycamp tothecin (HCPT) and etoposide (VP16) were purchased from Sigma-Aldrich (St Louis, MO, USA). Anti-γ-H2AX, anti-caspase 3, anti-caspase 7, anti-PARP, anti-phospho-DNA-PK, anti-DNA-PK, anti-phospho-ataxia telangiectasia mutated (ATM), anti-phospho-ATM and rad3-related (ATR), anti-ATM, anti-ATR, anti-cdc25A, anti-phospho-cdc25A, anti-phospho-cdc2 anti-cdc25A, anti-cdc2, anti-E2F, and anti-cyclinD1 antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA), γ-H2AX antibody conjugated fluorescein isothiocyanate (FITC) was from BD (Franklin Lakes, NJ, USA). Anti-β-actin antibody was obtained from Sigma-Aldrich, and peroxidase-conjugated goat antimouse or goat antirabbit secondary antibodies were purchased from ZSGQ-BIO Company (Beijing, People’s Republic of China). pBR322 DNA and top1 were purchased from Beijing Liuhe Tong Trade Co., Ltd. (Beijing, People’s Republic of China). All animal and cell experiments were approved by the Ethics Committee of Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences (IMBF20060302).
Cell lines
Human non-small-cell lung cancer A549, human fibrosarcoma HT1080, human glioblastoma U87-MG, human hepatoma HepG2, human breast cancer MCF-7, human colon carcinoma HCT116, human breast cancer MDA-MB-231, human colon carcinoma HCT116 p53KO, and human leukemia cells K562 were obtained either from Cell Center of the Institute of Basic Medical Sciences (Beijing, People’s Republic of China) or from American Type Culture Collection (Manassas, VA, USA). A549, HT1080, U87-MG, HepG2, MCF-7, HCT116, MDA-MB-231, and HCT116 p53KO cells were all cultured in Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum in the presence of 5% CO2 at 37°C. K562 cells were cultured in 1640 medium with 10% fetal bovine serum. Anchorage-dependent cells in the exponential growth phase were harvested with 0.25% trypsin–0.02% ethylenediaminetetraacetic acid (EDTA) solution and resuspended in the specified medium. Suspension cells were subcultured regularly. Only single cells with viability over 95% (trypan blue exclusion) were used.
Cell growth inhibition assay
Cell growth inhibition was determined using sulforhodamine B (SRB) assay.9 Cells were seeded in 96-well plates at 4×103–8×103/well, and were treated with increasing concentrations of compound 05D and incubated for 24 or 48 hours. The cells were fixed with 50% trichloroacetic acid (Sigma-Aldrich), and then 0.4% (w/v) SRB in acetic acid (1%) was added. SRB-bound cells were solubilized with 10 mM Trizma base. The absorbance was read at 492 nm. Growth inhibition (%) was calculated at each concentration and the IC50 was calculated by SigmaPlot (Systat Software, Inc., San Jose, CA, USA). Assays were repeated three times and the results are computed as mean and standard deviation.
Colony formation assay
Colony formation assay was performed as described in Wang et al.10 Briefly, HCT116 cells were seeded in six-well plate at 1,000/well. After 24 hours, escalating doses of 05D were added and the incubation was continued for 7 days. Then cells were fixed with methanol for 15 minutes and stained with 0.5% crystal violet for 15 minutes at room temperature. A colony is defined to consist of at least 50 cells. Visible colonies were counted. Colony formation rate was calculated as colony formation rate = (number of colonies/number of seeded cells) ×100%.
Acute toxicity study
Single-dose toxicity tests were performed in mice. 05D was given by intragastric (ig) route at a dose of 200 and 400 mg/kg, respectively, or by intraperitoneal (ip) route at a concentration of 25, 50, or 100 mg/kg. Mice survival was observed for 7 days after administration, and body weights were recorded and tissue was isolated at the end of survival observation.
Top1- and Top2α-mediated DNA relaxation assay
The DNA relaxation assay was based on a procedure described previously.11 Briefly, 2 μL of 10x reaction buffer with 1 mM adenosine triphosphate (top2α) or without adenosine triphosphate (top1), 0.5 μg of supercoiled pBR322, 1 unit of top1 or top2α (Topogen, Port Orange, FL, USA) and various concentrations of 05D or HCPT were mixed in a total of 20 μL of reaction buffer. Relaxation was performed at 37°C for 30 minutes and stopped by the addition of 2.5 μL of stop solution (100 mM EDTA, 0.5% sodium dodecyl sulfate [SDS], 50% glycerol, 0.05% bromophenol blue). Electrophoresis was performed in 1% agarose gel (Sigma-Aldrich) in 0.5x Tris-Borate-EDTA (TBE) at 4 V/cm for 1.5 hours. DNA bands were stained with the nucleic acid dye ethidium bromide (EB) and photographed with 300 nm ultraviolet transilluminator.
Top1-mediated DNA cleavage
DNA cleavage assays were performed as described previously.12–14 A 117 base pair (bp) DNA oligonucleotide from Sangon Biotech (Shanghai, People’s Republic of China) encompassing the previously identified top1 cleavage sites identified in the 161 bp fragment from pBluescript SK(-) phagemid DNA was employed. This 117 bp oligonucleotide contains a single 5′-cytosine overhang, which was 3′-end labeled by fill-in reaction with [α-32P]-dGTP in reaction 2 buffer (50 mM Tris-HCl [pH 8.0], 100 mM MgCl2, 50 mM NaCl) with 0.5 U of DNA polymerase I (Klenow fragment; New England BioLabs, Hitchin, UK). Unincorporated 32P-dGTP was removed using mini Quick Spin DNA columns (Hoffman-La Roche Ltd., Basel, Switzerland), and the eluate containing the 3′-end–labeled DNA substrate was collected. Approximately 3 nM of radiolabeled DNA substrate was incubated with recombinant top1 (60 U) in 20 μL of reaction buffer (10 mM Tris-HCl [pH 7.5], 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 15 μg/mL bovine serum albumin [BSA]) at 25°C for 30 minutes in the presence of various drug concentrations. The reactions were terminated by adding SDS (0.5% final concentration), followed by the addition of two volumes of loading dye (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.25% xylene cyanol, and 0.25% bromophenol blue). Aliquots of each reaction mixture were subjected to 16% denaturing polyacrylamide gel electrophoresis. Gels were dried and visualized by using a phosphoimager.
Comet assay
DNA breakage was measured with a neutral comet assay (Trevigen, Gaithersburg, MD, USA), as described in the manufacturer’s procedures and the literature.15 The treated cells were embedded in agarose on a slide and subjected to lysis followed by electrophoresis under neutral conditions. During electrophoresis, the damaged and fragmented negatively charged DNA migrated away from the nucleus toward the anode. The amount of migrated DNA was a measure of the extent of DNA damage. To detect DNA, the slides were stained with SYBR Gold (Thermo Fisher Scientific, Waltham, MA, USA) staining solution. The slides were examined by fluorescence microscopy, and the results were analyzed with the comet analysis software CASP (CASP, Wroclaw, Poland) to quantify DNA damage.16 For each drug concentration, three independent assays were conducted in which comet tails were analyzed in a minimum of 50 randomly selected cells in each assay, and the parameter reflecting the DNA damage was represented as tail moment (TM; percentage of DNA in tail/tail length).17
Western blot
Whole-cell lysates were used for immunoblotting, as described previously.18 The cells were lysed using a lysis buffer (50 mmol/L Tris-HCl [pH 8.0], 150 mmol/L sodium chloride, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, protease inhibitor). Twenty micrograms of protein lysate was resolved by SDS-polyacrylamide gel electrophoresis and analyzed by immunoblotting with the specified antibodies (the dilution ratio of all primary antibodies was 1:1,000 and the secondary antibody was 1:3,000, usually 3 μL of the aforementioned antibodies was used in the assay). The immunoreactive signals were revealed using the enhanced chemiluminescence (EMD Millipore, Billerica, MA, USA) method and visualized with a ChemiImager 5500 imaging system (Alpha Innotech, San Leandro, CA, USA).
Immunofluorescent microscopy
γ-H2AX was detected by immunofluorescent microscopy as described in Rojas et al.17 Cells were treated with different concentrations of 05D and fixed in 4% paraformaldehyde, and then permeabilized with 0.5% Triton X-100 (Sigma-Aldrich) in phosphate-buffered saline containing 5% BSA. Cells were incubated with γ-H2AX (phospho-Ser139) antibody FITC-conjugated at 30-fold dilution in 5% BSA in phosphate-buffered saline overnight at 4°C, and were mounted with mounting medium Vectashield (Vector Laboratories, Peterborough, UK) and sealed with nail polish. γ-H2AX and nuclear staining were viewed with a Olympus FV500 Confocal Laser Scanning Microscope (Olympus Corporation, Tokyo, Japan).
Analysis of cell cycle distribution
Flow cytometric analysis was performed as described in our previous report.11 HCT116 cells were treated with 5, 10, 20, and 40 μg/mL 05D for 24 hours, and were harvested and fixed with 75% ethanol at −20°C overnight. Cells were stained with propidium iodide (25 mg/mL) and RNase A (200 mg/mL) at 37°C for 30 minutes. The DNA content was analyzed with a FACScan flow cytometer (COULTER Epics XL; Beckman Coulter, Inc., Fullerton, CA, USA).
Cell apoptosis assay
Qualitative analysis of apoptotic cells was accomplished with Annexin V-FITC Apoptosis Detection Kit (Beyotime, Haimen, People’s Republic of China), as described previously.19,20 Briefly, cells were treated with increasing concentrations of 05D and after 48 hours, the cells were collected for flow cytometry analysis by FACScan flow cytometry.
HE staining of tissue sections
HE staining of tissue sections was performed as described in Zhao et al.20
Results
05D suppresses cancer cell proliferation in time- and dose-dependent manner and has a low toxicity
To evaluate the effect of compound 05D on the proliferation of tumor cells, 2.5, 5, 10, and 20 μg/mL 05D were added to the human colon carcinoma cells HCT116 and cell vitality was detected after incubation for 2, 4, 6, and 8 days. Results showed that 05D significantly inhibited cell growth in a time-dependent manner (Figure 1B). At the concentration of 5 μg/mL, cell survival was ~55% of control after 4-day treatment with 05D and ~20% following 8-day treatment. At 20 μg/mL, the number of living cells was only 25% of control after 2-day treatment and decreased to 2% after 8 days. Meanwhile, we also observed its dose-dependent effect with concentrations of 5, 10, 20, 40, and 80 μg/mL at 24, 48, and 72 hours (Figure 1C); for example, at 48 hours, the vital cell counts at 5, 10, 20, 40, and 80 μg/mL were 85.4%, 67.7%, 51.6%, 3.8%, and 0.5%, respectively, of the control.
Given that colony formation analysis is the gold standard of cytotoxic agent effects, we utilized colony formation assay to further assess the inhibitory effect of 05D. HCT116 cells were seeded in six-well plate at 1,000/well and incubated with escalating concentrations of the compound for 7 days prior to colony count. Results showed that 05D could inhibit colony formation of HCT116 cells in a dose-dependent manner, and at 2.5 μg/mL (Figure 1D), a concentration that inhibited cell proliferation only slightly (~10%), it could dramatically inhibit colony formation and the inhibitory rate was ~60%, indicating a higher ability to reduce cancer growth.
Also, we studied the inhibitory effect of 05D on other cancer cells, and results showed that 05D could suppress the proliferation of malignant cells, including lung cancer, fibrosarcoma, glioblastoma, hepatoma, breast cancer, and leukemia (Table 2), indicating a wide anticancer activity. Correspondingly, we also determined the effect of sophoridinol (the parent nucleus of 05D) on these tumor cells and found that there was almost no inhibitory effect at 40 μg/mL.
Acute toxicity study of 05D was performed to assess its toxicity, and results showed that no death of mice was observed either by ig administration (200 or 400 mg/kg) or ip administration (25, 50, 100 mg/kg) (Table 1). In addition, 05D did not influence the body weight of mice also. These results suggest that 05D is considerably safe in vivo with a lower toxicity. HE staining of various tissues showed that there were no obvious pathologic changes observed between control and test groups (Figure 1E).
05D inhibits top1 activity by promoting DNA–top1 complex formation
Then we examined the effect of 05D on top1 activity by unwinding assay. As shown in Figure 2A, 05D inhibited top1 relaxation activity in a dose-dependent manner and at 0.4 mg/mL, 05D could completely inhibit top1 activity. Similarly, we also detected the inhibitory effect of 05D on top2α by relaxation assay, and results revealed that 05D did not affect top2α activity even at a concentration up to 1 mg/mL (Figure 2B). The coculture of DNA and 05D showed that 05D did not influence DNA topology and did not lead to DNA breakage directly (Figure 2C). Meanwhile, we examined the effects of 05D precursor sophoridinol on top1 and top2α and at 2 mg/mL, we did not observe any inhibitory effect on topoisomerase (data not shown). The earlier results indicate that 05D only inhibited top1 activity and had almost no effect on top2α.
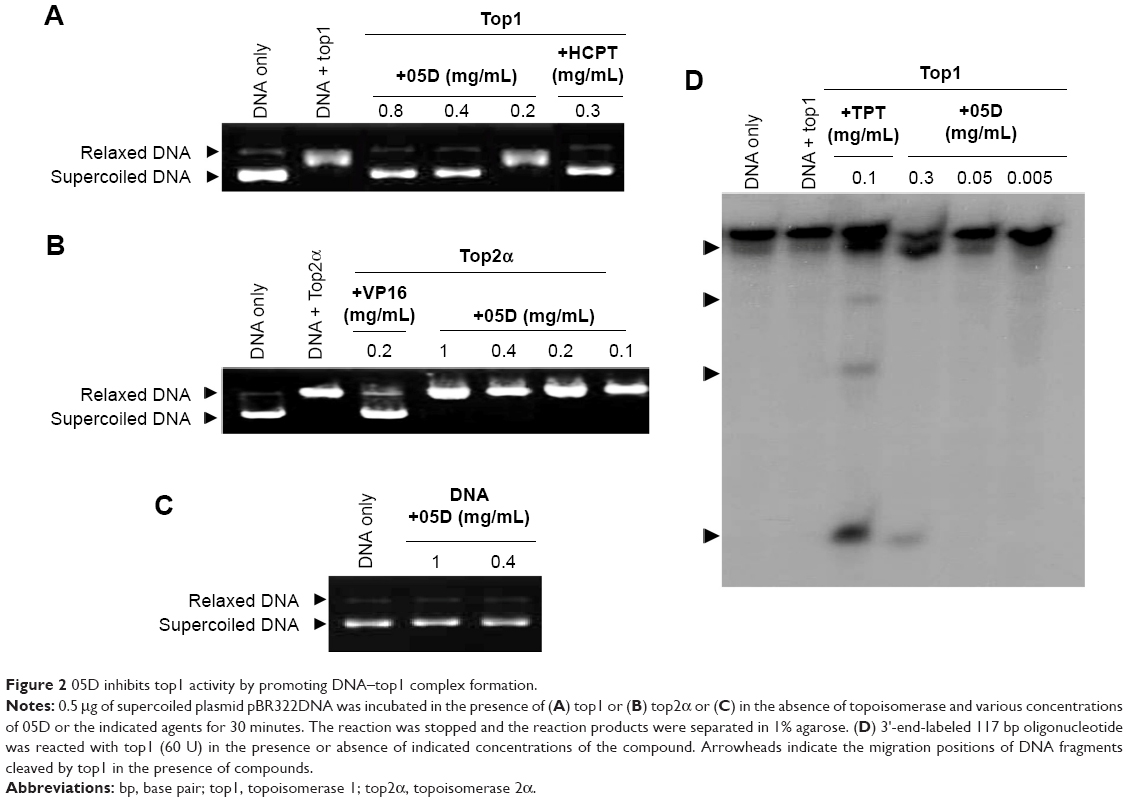 | Figure 2 05D inhibits top1 activity by promoting DNA–top1 complex formation. |
Among top1 inhibitors, most agents act by stabilizing DNA–top1 cleavage complex. So, we continued to perform top1-mediated DNA cleavage assay to detect whether 05D suppressed top1 activity by stabilizing DNA–top1 complexes. As shown in Figure 2D, in the presence of 05D, broken DNA bands that represented the DNA–top1 cleavage complexes were visualized, indicating that 05D could facilitate DNA–top1 cleavage complex formation in vitro.
05D leads to cell DNA breakage, γ-H2AX foci formation, and represses ATM and Rad3-related (ATR) and ataxia telangiectasia mutated (ATM) activation
The earlier results show that 05D could induce DNA breakage mediated by top1 in vitro. We evaluated its in vivo effect by observing whether it could induce DNA breakage in cells, since DNA strand breakage in cells, especially double-strand breakage, was the initial mechanism of top1 inhibitor inducing apoptosis.21,22 Alkaline single-cell gel electrophoresis (for single-strand breakage detection) and neutral single-cell gel electrophoresis (for double-strand breakage detection) were used to evaluate its effect on DNA breakage. Results showed that comet cells with comet tail were all visualized either in alkaline or neutral condition (Figure 3A), indicating that 05D could induce DNA single- and double-strand breakage in cells. The quantitative data revealed that 05D caused increase of TM in a dose-dependent manner, either in alkaline or neutral electrophoresis. In alkaline conditions, the TM was approximately 3.8-fold of the control at the highest concentration (40 μg/mL) and in neutral conditions, the TM was up to 40-fold of the control, indicating an extensive DNA double-strand breakage.
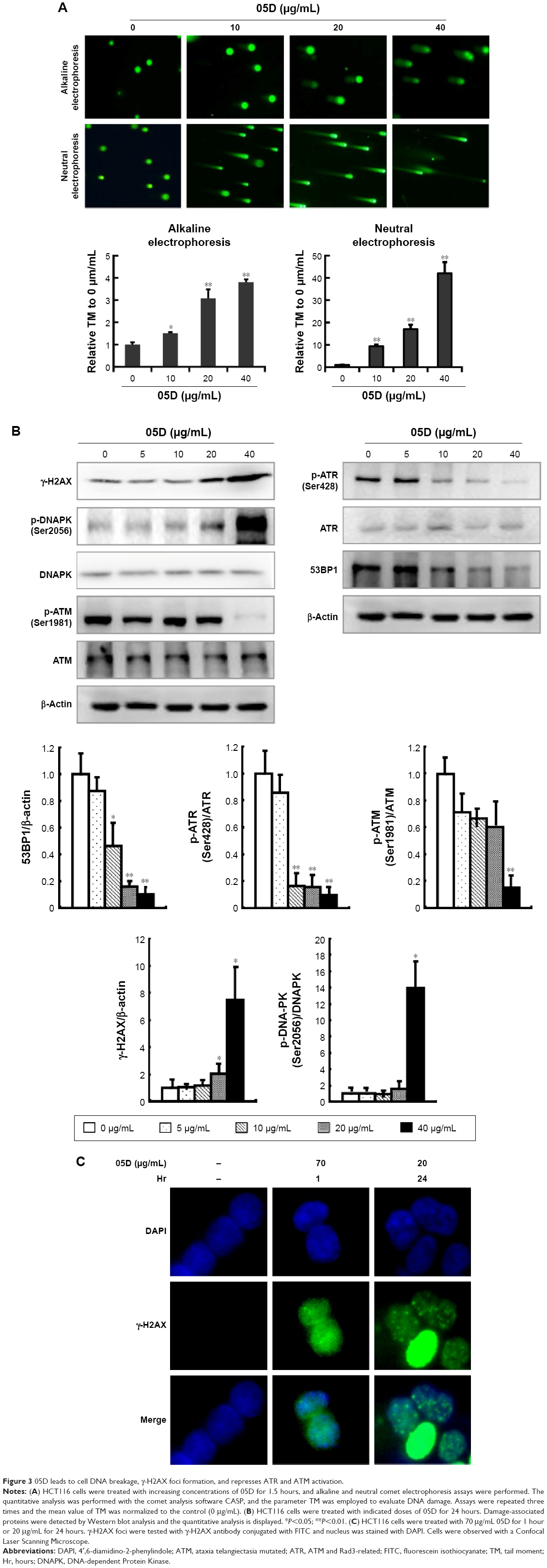 | Figure 3 05D leads to cell DNA breakage, γ-H2AX foci formation, and represses ATR and ATM activation. |
Given that the histone variant H2AX is phosphorylated rapidly on Ser139 (γ-H2AX) in response to DNA double-strand breakage23 in cells, we examined the γ-H2AX level and results showed that the γ-H2AX expression significantly increased after the addition of 05D and its elevation was in a dose-dependent manner (Figure 3B). Then we utilized immunofluorescent analysis to examine whether γ-H2AX foci were formed (a γ-H2AX focus represents a DNA damage site in nucleas)24 in the presence of 05D, and results showed that 05D could cause the formation of γ-H2AX foci in the nuclei of HCT116 cells after 24-hour treatment and these foci were also observed after 1-hour incubation (Figure 3C), indicating that not only could 05D lead to DNA breakage in a short time, but also this damage was persistent in response to 05D.
DNA damage usually evokes repair-associated pathways and their activation is contributed to the repair of broken DNA. So, we measured the repair-associated proteins and results showed that DNA damaging sensor DNA-PK was activated in the presence of 05D, but the phosphorylated levels of ATM and ATR attenuated, although their activation was the key to repairing the damage. Meanwhile, we also observed that the level of 53BP1 decreased (Figure 3C).
05D causes G1 phase arrest by suppressing CDK2/CDK4–Rb–E2F and cyclinD1–CDK4–p21 checkpoint signaling pathways
The impact of 05D on cell cycle progression was evaluated by flow cytometric analysis. HCT116 cells were treated with various concentrations of 05D (5, 10, 20, and 40 μg/mL) for 24 hours prior to cell cycle analysis, and results showed that HCT116 cells were blocked at G1 phase in a dose-dependent manner. At the concentration of 40 μg/mL, ~80% cells were blocked at G1 phase that was significantly higher than control (40% cells in G1 phase) (Figure 4A).
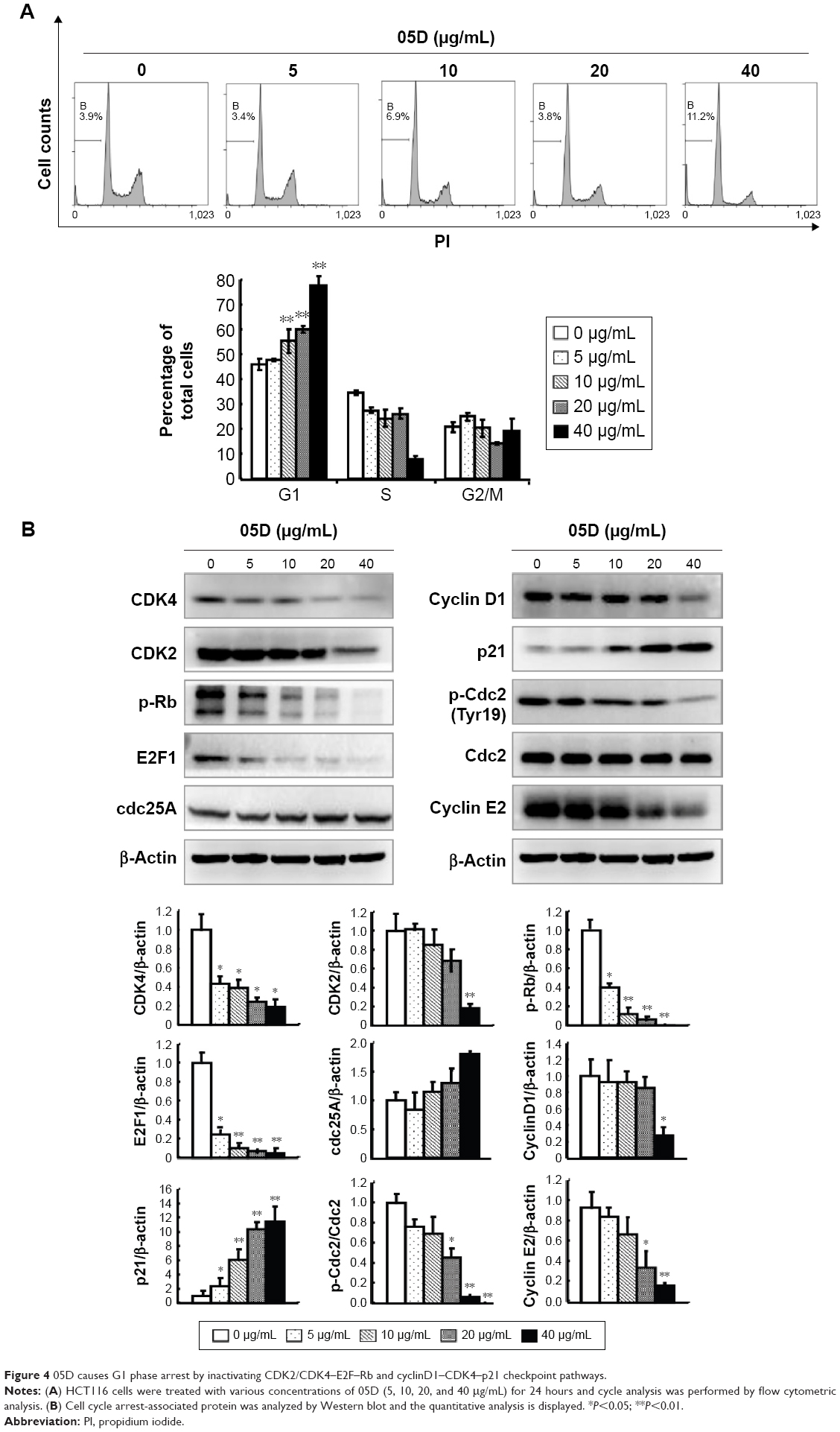 | Figure 4 05D causes G1 phase arrest by inactivating CDK2/CDK4–E2F–Rb and cyclinD1–CDK4–p21 checkpoint pathways. |
Then we assessed two checkpoint signal pathways, CDK2/CDK4–Rb–E2F and cyclinD1–CDK4–p21, that play a key role in the regulation of G1–G2/M transition and whose inactivation would induce cells arrested at G1 phase,22,25,26 and results showed that 05D could strongly repress the activation of CDK2/CDK4–Rb–E2F and cyclinD1–CDK4–p21 pathways (Figure 4B). Meanwhile, the levels of phosphorylated cdc2 and cyclin E2 decreased and Cdc25A level increased; these changes also contributed to G1 phase arrest (Figure 4B).
05D induces cell apoptosis through mitochondria-mediated apoptotic pathway
Cell apoptosis was assessed by flow cytometric analysis and results showed that 05D could extensively induce HCT116 cell apoptosis in a dose-dependent manner and at lower concentrations of 3 and 9 μg/mL, the apoptotic cells were 6.3% and 13% of the total cells, respectively, and at the higher concentration of 27 μg/mL, the apoptotic cells were up to 80.5% (Figure 5A), indicating that 05D plays its anticancer role by inducing apoptosis. Then we examined the corresponding apoptosis signaling pathways and results showed that 05D could lead to the activation of final apoptotic signal proteins caspase 3 and caspase 7 and mitochondria-mediated apoptotic signal protein caspase 9, but the signal proteins of nonmitochondrial-mediated apoptosis, such as caspase 2, caspase 8, and caspase 10, were not activated (Figure 5B), suggesting that 05D induced cancer cell apoptosis by mitochondrial-mediated apoptotic pathway.
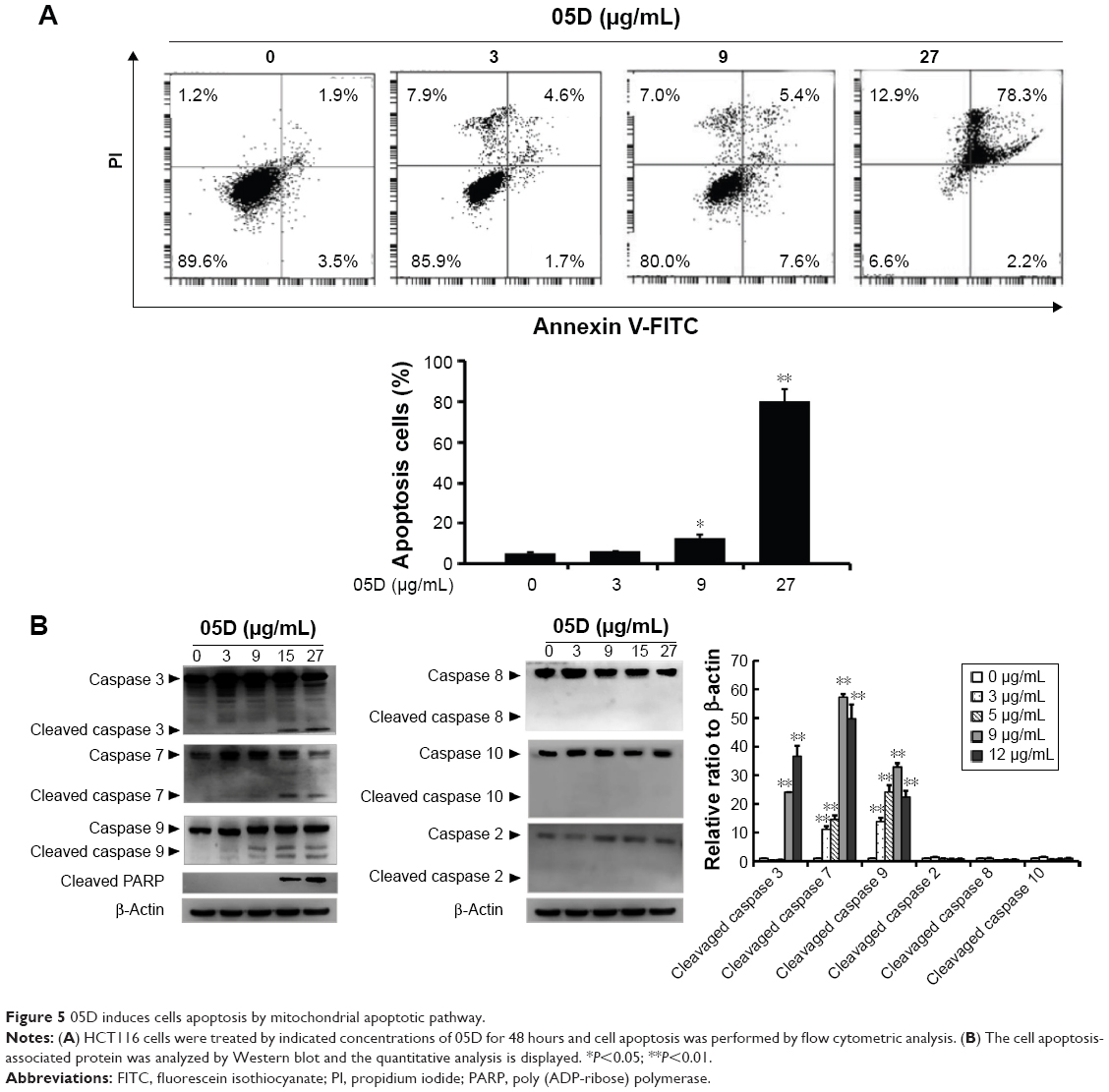 | Figure 5 05D induces cells apoptosis by mitochondrial apoptotic pathway. |
Discussion
Although top1 inhibitors such as camptothecin and hydroxycamptothecin have been widely used clinically for tumor treatment for several decades, the dose-dependent toxicity always disturbs their applications.27 Also, weak water solubility and the difficulties in de novo synthesis due to their large molecular structure also limited their further employment and exploration.28,29 Our reported novel structure compound 05D, compared with hydroxycamptothecin, has a couple of priorities in molecular traits and druggability, such as a simple structure and special scaffold, providing considerable opportunities of formulation for oral administration, thus indicating that it belongs to a novel family of antitumor agents with ideal druggability characteristics.8
Anticancer activity of 05D suggested that the novel compound possessed wide spectrum of anticancer effects compared to its nucleus, that is, 05D could not only inhibit proliferation of solid tumors, including non-small-cell lung cancer, hepatoma, colon carcinoma, ovarian carcinoma, glioblastoma, and so on, but also suppress the growth of hematologic malignancies such as erythroleukemia. Toxicity assay showed 05D did not lead to death of mice either by ip or ig administration, suggesting it was considerably safe in vivo.
As a derivative of sophoridinol, 05D could still depress top1 activity like its parent and its inhibitory activity on top1 was higher than sophoridinol. Further results of top1-mediated DNA cleavage showed that 05D could induce DNA–top1 cleavage complex formation. Meanwhile, we found that there were four DNA cleavage bands in topotecan (TPT)20 (a derivate of camptothecin)-treated group and only two DNA cleavage bands observed in high concentration group of 05D, suggesting that intercalative sites of TPT and 05D were distinct in top1-cleavaged DNA. This difference in intercalative style might be helpful for the combined application of TPT and 05D to improve the antitumor efficacy. Then, we demonstrated that 05D could sustain irreversible DNA breakage, although it did not directly damage DNA, and this nonreversible broken DNA usually initiates the apoptosis, as evidenced in our study.
Generally, DNA damage activates DNA repair pathways. Among them, homologous recombination and nonhomologous end joining repair for broken DNA are the two major repair styles in response to DNA breakage.29 Activation of ATM and ATR and an increase in 53BP1 level are usually the keys in the repair of homologous recombination and nonhomologous end joining, and their activation usually attenuated the effect of DNA damage that was critical to the efficacy of DNA damaging compound.30,31 In our study, 05D could decrease or even eliminate phosphorylated level of ATR and ATM and 53BP1 level, suggesting that 05D might have a potential to enhance the effect of ATM- or ATR-activated DNA damaging drugs by repressing DNA breakage repair pathway.
Conclusion
Compound 05D exhibits antiproliferative activity in a variety of tumor cell lines, including liver, colon, lung, and breast hematopoietic malignancy. It could inhibit top1 activity by stabilizing DNA–top1 complex and induce cell DNA breakage and apoptosis, but depress ATM and ATR activation and 53BP level, which might be attributed to the improvement of therapeutic effectiveness of DNA damaging agents. In addition, the priorities in molecular traits and druggability indicate sophoridinol derivative 05D is a novel and promising antitumor agent with ideal druggability characteristics.
Acknowledgment
This work was supported by the National Science Fund (81102464 and 81321004) and National Mega-project for Innovative Drugs (2009ZX09103-698 and 2014ZX09201042).
Disclosure
The authors report no conflicts of interest in this work.
References
Yang ZW, Zhou Y, Cao XQ. The effect of sophora alopecuroides on CV B3 in vitro. J Sichuan Traditional Chinese Med. 2003;21:14–16. | ||
Nie HM, Chen JJ, Gao YQ, Jin SG, Wang LT. The anti-HBV study of sophoridine in vitro. Beijing J TCM. 2007;26:678–680. | ||
Hang XM, Li B. Effects of six alkaloids of sophora alopecuroides on LTC4 and LTB4. Chinese Traditional Patent Med. 2003;25(10):824–826. | ||
Hang XM, Li B, Shen L, Wang J. Inhibitory effects of four alkaloids of Sophora alopecuroides on TNFα production by murine peritoneal macrophages. Pharmacology Clin Chinese Mater Med. 2001;17(3):12–14. | ||
Lu H, Shen YF, Cao M, Song XW, Tang Y, Yang YJ. Effects of sophoridine on cardiac function and myocardial ultrastructure of rats with myocardial infarction. AJSMMU. 2012;33(005):470–473. | ||
Li XM, Wu YG, Pan DX, et al. Sophoridine is a new antitumor medicine with new molecular structure. Chinese J New Drugs. 2012;33(005):470–473. | ||
Li XM, Wu YG, Chen SL. The effect of sophoridine on tumor cells. Acta Pharm Sinic. 1987;8(2):153–l58. | ||
Bi CW, Zhang CX, Li YH, et al. Novel N-substituted sophoridinol derivatives as anticancer agents. Eur J Med Chem. 2014;81:95–105. | ||
Zhao WL, He HW, Ren KH, Zhang H, Chen Y, Shao RG. Myofibrillogenesis regulator-1 promotes cell adhesion and migration in human hepatoma cells. Chin Sci Bull. 2013;58(24):3007–3014. | ||
Wang L, Gao C, Yao S, Xie B. Blocking autophagic flux enhances matrine-induced apoptosis in human hepatoma cells. Int J Mol Sci. 2013;14(12):23212–23230. | ||
Li YB, Zhao WL, Wang YX, et al. Discovery, synthesis and biological evaluation of cycloprotoberberine derivatives as potential antitumor agents. Eur J Med Chem. 2013;68:463–472. | ||
Dexheimer TS, Pommier Y. DNA cleavage assay for the identification of topoisomerase I inhibitors. Nat Protoc. 2008;3(11):1736–1750. | ||
Sooryakumar D, Dexheimer TS, Teicher BA, Pommier Y. Molecular and cellular pharmacology of the novel noncamptothecin topoisomerase I inhibitor Genz-644282. Mol Cancer Ther. 2011;10(8):1490–1499. | ||
Beck DE, Abdelmalak M, Lv W, et al. Discovery of potent indenoisoquinoline topoisomerase I poisons lacking the 3-nitro toxicophore. J Med Chem. 2015;58(9):3997–4015. | ||
Znojek P, Willmore E, Curtin NJ. Preferential potentiation of topoisomerase I poison cytotoxicity by PARP inhibition in S phase. Br J Cancer. 2014;111(7):1319–1326. | ||
Konca K, Lankoff A, Banasik A, et al. A cross-platform public domain PC image-analysis program for the comet assay. Mutat Res. 2003;534(1–2):15–20. | ||
Rojas E, Lopez MC, Valverde M. Single cell gel electrophoresis assay: methodology and applications. J Chromatogr B Biomed Sci Appl. 1999;722(1–2):225–254. | ||
Zhao W, He H, Ren K, et al. MR-1 blocks the megakaryocytic differentiation and transition of CML from chronic phase to blast crisis through MEK dephosphorylation. Blood Cancer J. 2013;3:e107. | ||
Pan Y, Ren KH, He HW, Shao RG. Knockdown of Chk1 sensitizes human colon carcinoma HCT116 cells in a p53-dependent manner to lidamycin through abrogation of a G2/M checkpoint and induction of apoptosis. Cancer Biol Ther. 2009;8(16):1559–1566. | ||
Zhao WL, Jiang GH, Bi CW, et al. The dual topoisomerase inhibitor A35 preferentially and specially targets topoisomerase 2alpha by enhancing pre-strand and post-strand cleavage and inhibiting DNA religation. Oncotarget. 2015;35(6):37871–3794. | ||
Furuta T, Takemura H, Liao ZY, et al. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem. 2003;278(22):20303–20312. | ||
Obeyesekere MN, Knudsen ES, Wang JY, Zimmerman SO. A mathematical model of the regulation of the G1 phase of Rb+/+ and Rb−/− mouse embryonic fibroblasts and an osteosarcoma cell line. Cell Prolif. 1997;30(3–4):171–194. | ||
Guo L, Liu X, Nishikawa K, Plunkett W. Inhibition of topoisomerase IIalpha and G2 cell cycle arrest by NK314, a novel benzo[c]phenanthridine currently in clinical trials. Mol Cancer Ther. 2007;6(5):1501–1508. | ||
Kuo LJ, Yang LX. Gamma-H2AX – a novel biomarker for DNA double-strand breaks. In Vivo. 2008;22(3):305–309. | ||
Berthet C, Kaldis P. Cdk2 and Cdk4 cooperatively control the expression of Cdc2. Cell Div. 2006;1:10. | ||
Masamha CP, Benbrook DM. Cyclin D1 degradation is sufficient to induce G1 cell cycle arrest despite constitutive expression of cyclin E2 in ovarian cancer cells. Cancer Res. 2009;69(16):6565–6572. | ||
Rowinsky EK, Kaufmann SH, Baker SD, et al. A phase I and pharmacological study of topotecan infused over 30 minutes for five days in patients with refractory acute leukemia. Clin Cancer Res. 1996;2(12):1921–1930. | ||
Bornkessel B. [Topoisomerase I inhibitors]. Med Monatsschr Pharm. 1997;20(5):114–116. | ||
Hang M, Ding J. Developments of topoisomerase I inhibitors in cancer chemotherapy. Chinese J New Drugs. 2007;16(13):990–1000. | ||
Beucher A, Birraux J, Tchouandong L, et al. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009;28(21):3413–3427. | ||
Rodrigues AS, Gomes BC, Martins C, et al. DNA repair and resistance to cancer therapy. InTech. 2013:489–528. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.