")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Soluble common gamma chain exacerbates COPD progress through the regulation of inflammatory T cell response in mice
Authors Lee B, Ko E, Lee J, Jo Y, Hwang H, Goh TS, Joo M, Hong C
Received 28 September 2016
Accepted for publication 20 December 2016
Published 8 March 2017 Volume 2017:12 Pages 817—827
DOI https://doi.org/10.2147/COPD.S123405
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Byunghyuk Lee,1 Eunhee Ko,1 Jiyeon Lee,2 Yuna Jo,1 Hyunju Hwang,1 Tae Sik Goh,1,3 Myungsoo Joo,2 Changwan Hong1
1Department of Anatomy and Cell Biology, Pusan National University School of Medicine, 2Division of Applied Medicine, School of Korean Medicine, Pusan National University, Yangsan, 3Department of Orthopedic Surgery, Medical Research Institute, Pusan National University School of Medicine, Busan, South Korea
Abstract: Cigarette smoking (CS) is a major cause of considerable morbidity and mortality by inducing lung cancer and COPD. COPD, a smoking-related disorder, is closely related to the alteration of immune system and inflammatory processes that are specifically mediated by T cells. Soluble common gamma chain (sγc) has recently been identified as a critical regulator of the development and differentiation of T cells. We examined the effects of sγc in a cigarette smoke extract (CSE) mouse model. The sγc level in CSE mice serum is significantly downregulated, and the cellularity of lymph node (LN) is systemically reduced in the CSE group. Overexpression of sγc enhances the cellularity and IFNγ production of CD8 T cells in LN and also enhances Th1 and Th17 differentiation of CD4 T cells in the respiratory tract. Mechanistically, the downregulation of sγc expression mediated by CSE is required to prevent excessive inflammatory T cell responses. Therefore, our data suggest that sγc may be one of the target molecules for the control of immunopathogenic progresses in COPD.
Keywords: COPD, T cell, soluble common gamma chain, cytokine
Introduction
COPD is a lung disorder defined as a limitation of irreversible airflow that is generally both progressive and associated with enhanced inflammatory responses of the lungs to noxious particles or gases.1 Cigarette smoking (CS) exposure is the primary risk factor for the development of COPD.2 The understanding of how CS alters the immune cells and their responses is important in control of the inflammatory lung disease. Although it has been reported that T cell infiltration is increased in bronchial biopsies of patients with COPD,3 how CS functionally regulates T cell responses is still unclear. It has been presumed that CS promotes Th2 immune response as shown by enhanced IL-4 and IL-13 production from the peripheral blood mononuclear cells (PBMC) of smokers.4,5 Mechanistically, CS induces the production of thymic stromal lymphopoietin (TSLP),6,7 which then allows dendritic cells (DCs) to promote Th2 polarization.8,9 While many reports suggest that CS induces Th2 immune response, other studies suggest that CS induces Th1 immune response. The expression of IFNγ in infiltrated T cells into the peripheral airways was observed in bronchial biopsies of COPD patients.10 Furthermore, the phosphorylation of STAT4, which is activated by IL-12, a primary cytokine in Th1 differentiation,11,12 is enhanced in CD4 T cells of smokers with COPD.10 Accordingly, the induction of phosphor-STAT4 and IFNγ correlates with the degree of airflow limitation in patients with COPD.
The cytotoxic CD8 T cells are also dominantly observed in the respiratory tracts and the lung parenchyma of COPD patients.13–16 This suggests that these cells are involved in airflow obstruction and emphysema with tissue damage. CS triggers innate inflammation that leads to tissue injury and production of antigenic self-substances.17 This chain of events may cause DCs to mature and migrate to the draining lymphoid organs, where T cells are activated.17 Cytolytic CD8 T cells, with the support of helper T cells, kill target cells through secretion of proteolytic enzymes, such as perforin, granulysin, and granzyme, in the lungs of COPD patients.18–20
The common gamma chain (γc) cytokines are essential for the development and homeostasis of immune cells.21 We recently reported that the soluble form of common gamma chain (sγc), generated by alternative splicing, regulates T cell response and survival with an antagonistic effect in γc cytokine signaling.22,23 The inhibitory function of soluble common gamma chain (sγc) in γc cytokine signaling exacerbated the inflammation by promoting the differentiation of pathogenic Th17 cells both in vitro and in vivo.22 Since COPD is developed with T cell-mediated immunopathogenesis by CS,24 sγc would be involved in the progression of diseases such as COPD.
In this study, we identified sγc as one of the key regulators in T cell-mediated immunopathogenesis of COPD and suggest that the downregulation of sγc expression in COPD mouse model could represent a mechanism to prevent excessive T cell responses and then tissue damage in the respiratory tracts. We found that sγc overexpression results in dramatically enhanced IFNγ production of CD8 lymph node T (LNT) cells and skewed Th1 and Th17 differentiation in the respiratory tracts, which are critical in inflammatory response. These data uncover a previously unknown role of sγc in the progression of COPD induced by cigarette smoke extract (CSE) and propose that sγc could be a novel target for the management of COPD development.
Materials and methods
Animals
C57BL/6 mice were obtained from the Orient Bio (Korea). Soluble γc-transgenic mice were described and maintained in our colony. Animal experiments were approved by the Pusan National University Institutional Animal Care and Use Committee (PNU-2014-0620). All mice were cared for in accordance with the guidelines put forth by Pusan National University School of Medicine and National Institutes of Health.
CSE preparation and treatment
CSE was prepared as previously described.25 Briefly, Kentucky 1R5F research reference cigarettes (The Tobacco Research Institute, University of Kentucky) were smoked using a peristaltic pump. Each cigarette without filter was smoked for 5 min with a 17-mm butt remaining, which was bubbled through 20 mL of phosphate-buffered saline (PBS) in an impinger. CSE was sterilized with a 0.22-mm filter prior to experiments. Mice (8–10 weeks old) received a single intratracheal injection of 30 μL of CSE for 5 days per week, and CSE was administered for 3 weeks.
Blood collection
After anesthetization, blood was collected from the orbital sinus by inserting the tip of a fine-walled Pasteur pipette into the corner of the eye socket underneath the eyeball.
Lung analyses
The trachea was exposed through midline incision and cannulated with a sterile 24-gauge intravascular catheter. Bilateral bronchoalveolar lavage (BAL) was performed by two consecutive instillations of 1.0 mL of PBS. Total cells in bronchoalveolar lavage fluid (BALF) were counted with hemocytometer. Mouse lungs were perfused with saline. The left lung was inflated with 0.5% low-temperature agarose to pressurize equally over lung fields and fixed with paraformaldehyde solution immediately.26 After paraffin embedding, ten 5 mm sections were cut, placed on charged slides, and stained with standard hematoxylin and eosin (H&E) staining method. Three separate H&E-stained sections were evaluated in 100× microscopic magnifications per mouse. Airspace, the ratio of alveolar wall to parenchyma, and mean linear intercepts were calculated using ImageJ (Bethesda, MD, USA).27,28
Flow cytometry
Single cell suspensions were prepared from the indicated mice. All data were acquired using FACSAria or FACSCanto (BD Biosciences, San Jose, CA, USA) and analyzed using FlowJo (Ashland, OR, USA). The live cells were gated by forward scatter exclusion of dead cells stained with propidium iodide. The following antibodies were used for staining: TCRβ (H57-597) and isotype control antibodies, all from eBioscience (Waltham, MA, USA); CD44 (IM7), CD62L (MEL-14), CD4 (GK1.5 and RM4.5), and CD8α (53-6-7) from BD Biosciences; CXCR3 (CXCR3-173), IFNγ (XMG1.2) and IL-17A (TC11-18H10), all from BioLegend. Anti-mouse CD16/32 antibody (2.4G2; BioLegend [San Diego, CA, USA]) was incubated to block Fcγ receptor. All antibodies were incubated at 4°C for 30 min.
Intracellular cytokine determination
For intracellular cytokine staining, cells were restimulated for 3 h with PMA and ionomycin (all from Sigma-Aldrich Co., St Louis, MO, USA) with the addition of brefeldin A and then fixed and permeabilized with intracellular fixation buffer (eBioscience).
Detection of soluble common gamma chain levels
Serum sγc was detected in a sandwich enzyme-linked immunosorbent assay using polyclonal anti-γc antibodies (R&D Systems Inc., Minneapolis, MN, USA) as capture antibodies and biotin-conjugated monoclonal anti-γc antibodies (4G3; BD Biosciences) as detection antibodies. Recombinant sγc protein was used as a positive control.
Statistical analysis
Statistical differences were analyzed by unpaired two-tailed Student’s t-test. P-values of <0.05 were considered significant. *P<0.05, **P<0.01, and ***P<0.001. All statistical analyses were performed using Graphpad Prism (La Jolla, CA, USA).
Results
CSE reduces sγc expression
To investigate the effects of sγc in a COPD animal model induced by CSE, we first analyzed the sγc level in sera from CSE-treated mice. Interestingly, after 14 days of CSE exposure, the sγc level started to significantly decrease, with further descending tendencies at later time points (Figure 1A). Because T cells are a major source of sγc, we specifically analyzed the T cell profile and response in the peripheral lymph node (LN) of CSE or control mice. CSE significantly reduced the total number of LN cells (Figure 1B), and we also observed that the numbers of naïve (CD44lowCXCR3−) and activated (CD44hiCXCR3+) CD8 T cells were decreased by CSE (Figure 1C and D). Next, we examined the population of CD4 T cells and found that there was a significant reduction in the number of naïve (CD44lowCD62Lhi) CD4 LNT cells, while the number of activated (CD44hiCD62Llow) CD4 LNT cells was unaffected by CSE (Figure 1C and E). This translated into increased percentages of activated CD4 T cells in the CSE group (Figure 1C), since the number of overall LN cells was decreased (Figure 1B). Since sγc production is enhanced in activated T cells,22 these data indicate that a reduction in the sγc level by CSE results from a diminished cell number, specifically activated T cells.
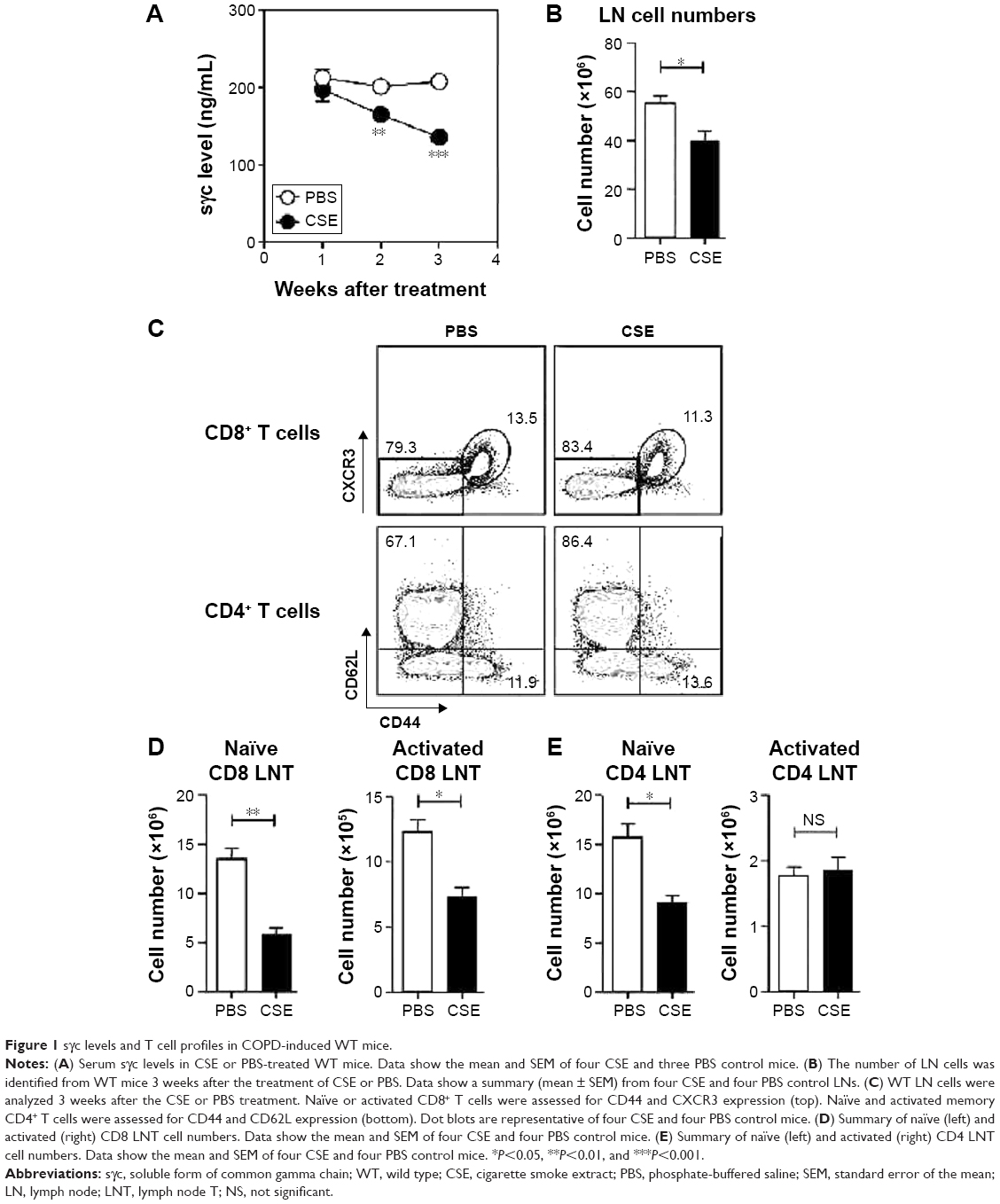 | Figure 1 sγc levels and T cell profiles in COPD-induced WT mice. |
LN cellularity is rescued by sγc overexpression from CSE
To assess the impact of decreased sγc expression in COPD mice, we analyzed sγc transgenic (sγcTg) mice, in which sγc is specifically overexpressed in T lineage cells under the control of a human CD2 promoter.22 Contrary to wild-type (WT) mice with COPD, overexpressed sγc significantly promoted total LN cell numbers (Figure 2A), and we found that there was no decrease in the number of CD8 and CD4 T cells (Figure 2B and C) and an increase in the number of activated CD4 T cells in sγcTg mice with COPD (Figure 2B and D). Collectively, although it is unclear why the sγc levels were reduced by CSE, decreased sγc expression may directly be related to reduced LN cell numbers, as the overexpression of sγc prevents the reduction in the number of LN cells.
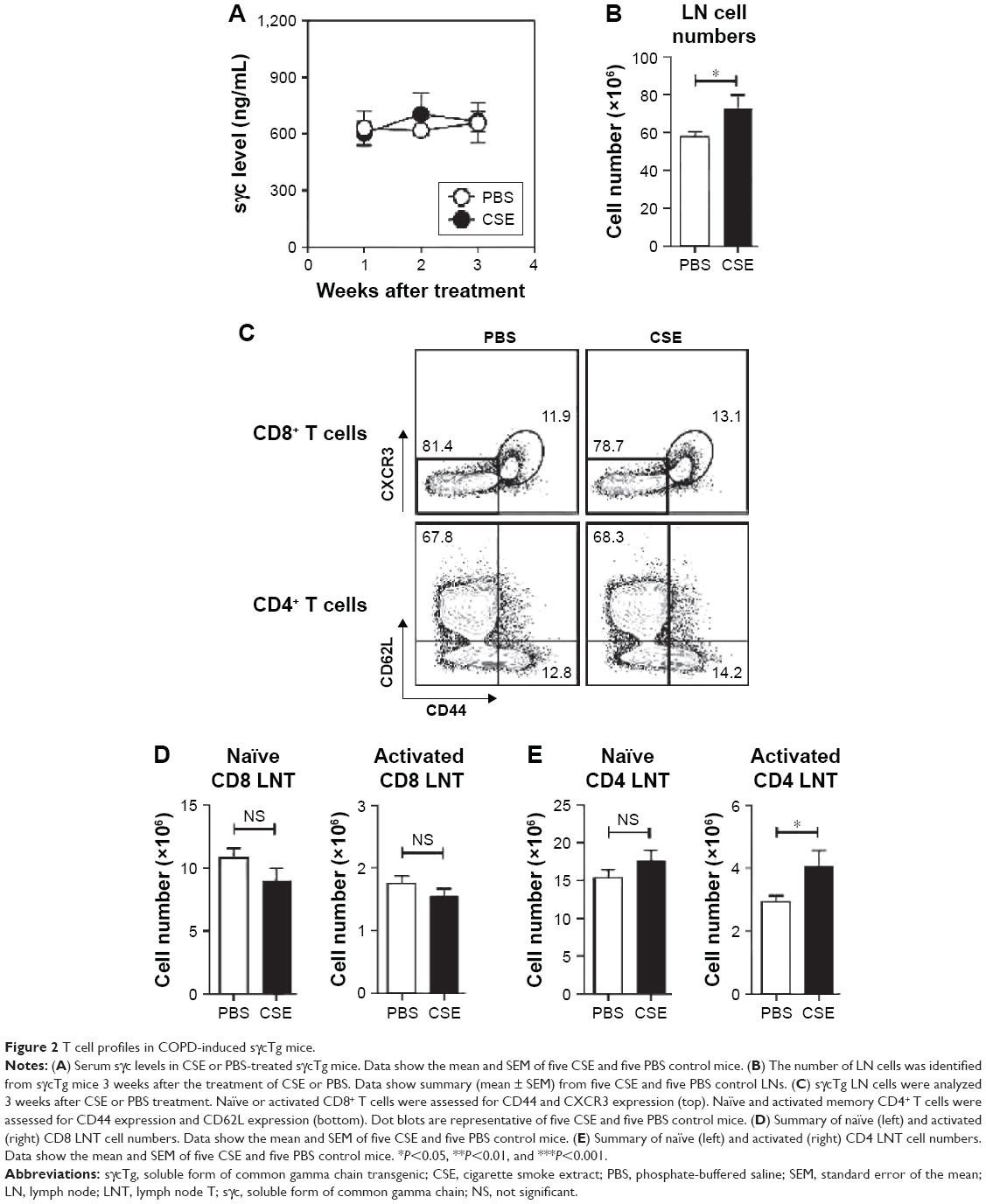 | Figure 2 T cell profiles in COPD-induced sγcTg mice. |
sγc overexpression impairs LNT cell response in CSE
To investigate the functional effect of sγc in LNT cell in response to CSE, LN cells from CSE mice were stimulated with PMA (phorbol 12-myristate 13-acetate)/ionomycin, and the cytokine profiles were analyzed. The percentage of IL-4+ CD4 T cells was reduced, but IFNγ was increased in CD4 LNT cells from WT mice with COPD compared with the PBS control mice, suggesting that CSE acts as an inducer of Th1 (Figure 3A, top and middle). Th17 differentiation was not observed in LN under CSE treatment (Figure 3A bottom). Th1 immunity skewed by CSE was not observed in sγcTg mice compared with CD4 T cell differentiation in WT mice with COPD, implying that sγc overexpression impairs CD4 LNT cell response to CSE. Consequently, CD4 T cells in CSE-treated WT LN were more skewed to Th1 compared to those in PBS-treated WT LN, while Th1 differentiation and Th2 differentiation in PBS- or CSE-treated LNs of sγcTg mice were comparable. Interestingly, reduction in Th17 differentiation was observed in CSE-treated LNs of sγcTg mice. Next, we tested CD8 LNT cell response and found that IFNγ-producing CD8 LNT cells were increased in both WT and sγcTg mice by CSE (Figure 3B). These data suggest that a downregulation of sγc expression in the COPD animal model may be one mechanism to specifically dampen T cell response in LN.
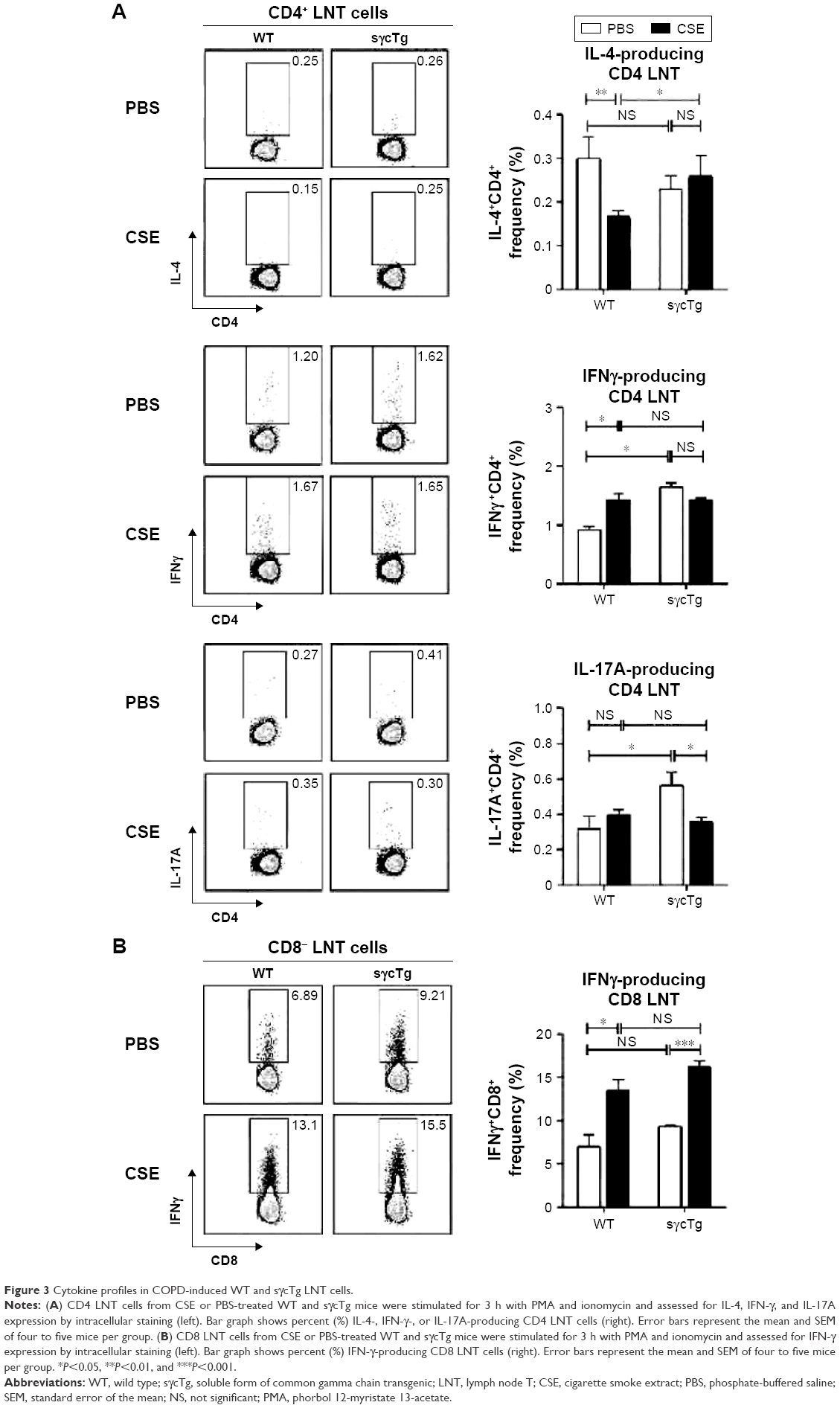 | Figure 3 Cytokine profiles in COPD-induced WT and sγcTg LNT cells. |
sγc overexpression enhances inflammatory response in respiratory tracts
To further determine the biological function of decreased sγc expression in CSE, we assessed the infiltration of inflammatory immune cells into the lung tissue of COPD. We found that there was a significant increase in immune cell infiltration in sγcTg mice (Figure 4A). The numbers of infiltrating TCR+ cells were not shown to be different between WT and sγcTg mice with COPD, while the TCR− cells in sγcTg mice were more infiltrated by CSE compared with WT mice (Figure 4B). To address whether in vivo sγc upregulation had any effect on the infiltrated T cells into the respiratory tract, we stimulated the infiltrated cells with PMA/ionomycin and examined the ex vivo cytokine production profiles of CD8+ and CD4+ T cells. Interestingly, sγcTg CD4+ T cells were more skewed to Th1 and Th17 than were WT CD4+ T cells in CSE (Figure 4C and E); whereas IFNγ and IL-17 productions of CD8 T cells were not affected (Figure 4C and D). Accordingly, decreased sγc level in COPD results from the prevention of a Th1- and Th17-cell-prone proinflammatory environment. Indeed, the lung alveolar sections of CSE-exposed WT and sγcTg mice were stained by H&E, and the data showed that sγcTg mice exhibited more severe interstitial edema, alveolar wall thickening, and inflammatory cell infiltration compared with WT mice, while there were no significant histological differences between PBS-exposed WT versus sγcTg mice (Figure 5). Collectively, these data implicate that IL-17 environment with increased Th17 may recruit more non-T cells such as neutrophils, eosinophils, and macrophages29,30 and it may induce more severe immunopathogenesis of COPD.
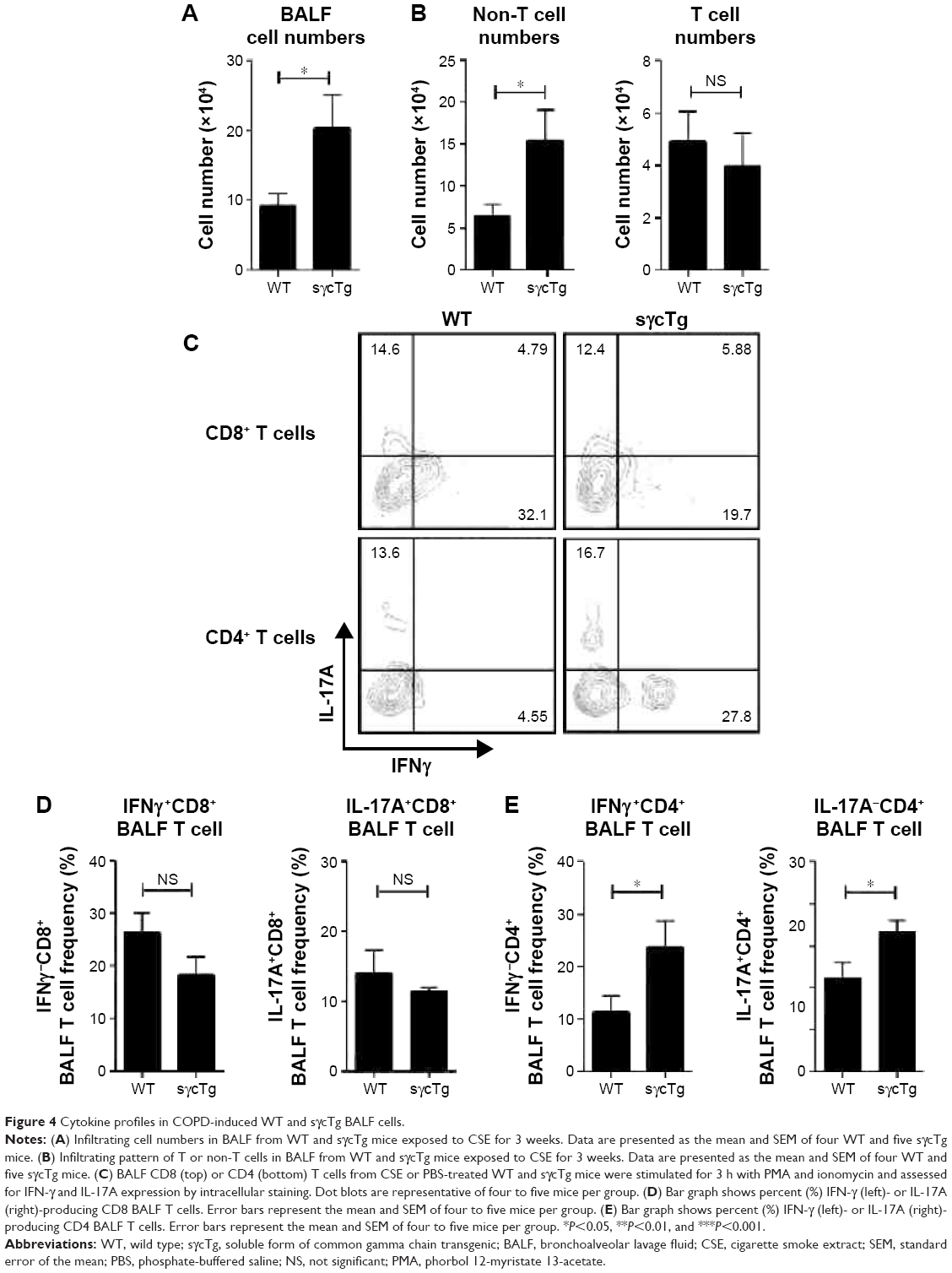 | Figure 4 Cytokine profiles in COPD-induced WT and sγcTg BALF cells. |
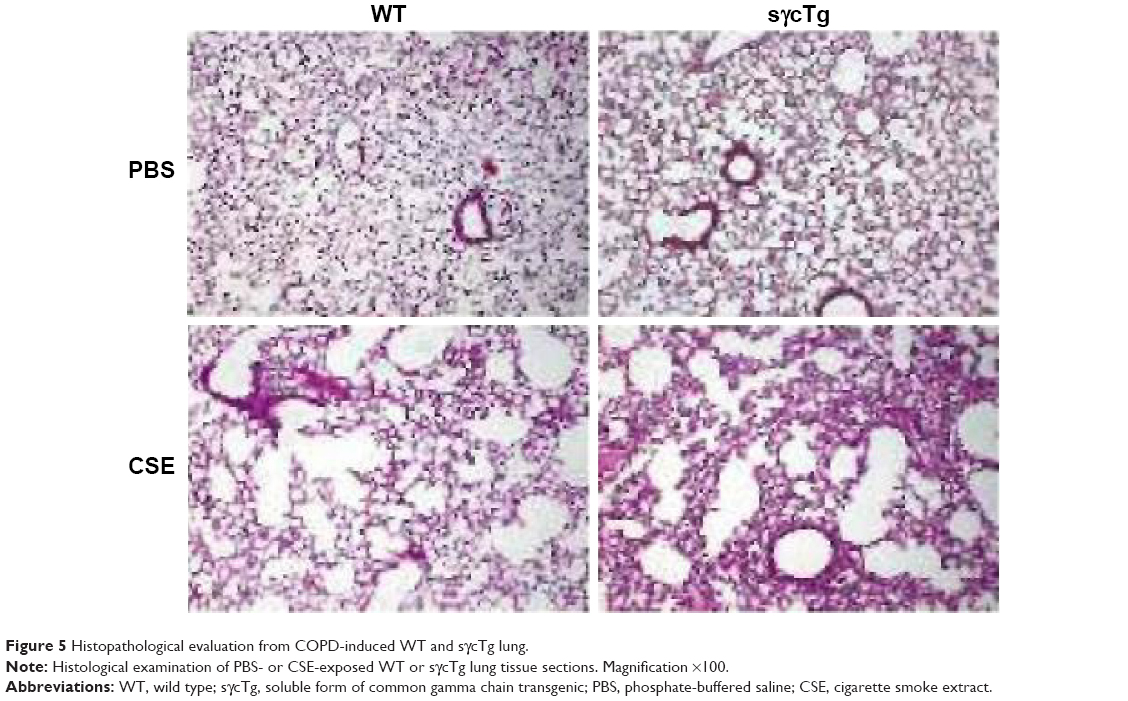 | Figure 5 Histopathological evaluation from COPD-induced WT and sγcTg lung. |
Discussion
COPD is characterized by septal tissue damage, remodeling of small airways, airway obstruction, and subsequent decline in lung function.1 It has been conventionally known that inflammatory responses by innate and adaptive inflammatory immune cells contribute to lung tissue damage.1,31,32 Previous studies have reported that the levels of Th1- and Th17-related cytokines in lung tissues of COPD patients are increased and play a pivotal role in the progression of COPD.33,34 Since sγc was identified as regulators of Th1 and Th17 differentiation,22 it is logical that sγc may closely be related to the immunopathogenesis of COPD. Here, we assessed the pattern of sγc expression on COPD development and found that the expression of sγc was dramatically reduced in a CSE-induced COPD mouse model. To elucidate the biological role of its reduction, we analyzed sγcTg mice and showed that an increased sγc production more skewed CD4 T cells to Th1 and Th17, which resulted in more severe lung tissue damage in COPD mice. These findings propose a new regulatory mechanism in COPD immunopathogenesis that the downregulation of sγc expression is one of the defense mechanisms from excessive inflammation by CSE. Inflammatory immune response is more induced in the sγcTg animal model in which the regulatory mechanism in sγc expression is compromised. Collectively, this study reports a new regulatory role for sγc in enhancing the progression of COPD, and it implicates that sγc can be a novel target to control COPD immunopathogenesis caused by CS.
CS changes a broad range of immunological functions, including innate and adaptive immune responses.24 It has been surmised that many of the health consequences of CS are due to its negative effects on the immune system. One of the adverse effects is an autoreactive CD8 T cell response.35 CS triggers tissue cell death and the release of self-antigens. These events induce DC maturation and generate self-antigen specific cytotoxic CD8 T cells that induces more deterioration of tissue injury.24 Thus, regulatory mechanisms are operated to control self-reactive T cells at the thymic36 and peripheral levels.37,38 The autoreactive T cells are negatively selected in the thymus and controlled by Treg cells in the periphery. IL-2, which plays a critical role in the development and homeostasis of Treg cells,39 is elevated in COPD patients who show disease stability,40 inducing dominant upregulation of Treg cells in smokers with preserved lung function compared with COPD patients.41 As our previous studies demonstrated that sγc inhibits IL-2 signaling,22 a high level of sγc leads to impaired IL-2 signaling, resulting in the inhibition of Treg cell function and survival.39 This suggests that the low level of sγc in a CSE animal model may result in the prevention of COPD progression by restricting excessive T cell response with IL-2-induced Treg cells. On the other hand, Treg development depends on IL-2 and TGFβ, which is also linked to Th17 differentiation.42,43 The function of Th17 in COPD is less known; an enhancement of Th17 differentiation was observed in peripheral blood and bronchial biopsies of COPD patients compared to those of healthy control.44,45 We found that CD4 T cells in sγcTg mice were much more skewed to Th17 compared to WT mice with CSE treatment, indicating that more IL-17 production from increased Th17 cell differentiation may recruit more inflammatory responsible cells in COPD. Indeed, IL-17 may induce chemokine secretion46–48 and promote the infiltration of neutrophils and macrophages to peripheral airway.29,30 It may partially explain the reason for the increased numbers of non-T cells in BALF from sγcTg mice with CSE treatment in our study. If the expression of sγc is regulated and maintained at low levels in smokers, CS-mediated disease stability may be maintained over a long period or it is possible for disease severity to be ameliorated. Whether this is indeed the case remains to be tested.
COPD is a typical disease induced by chronic inflammation.24 It has been known that Th1 and Th17 cells act as helper T cells to robustly induce the recruitment of inflammatory cells and tissue damage in chronic inflammation. As shown in sγcTg mice with COPD that exhibited the enhanced Th1 and Th17 differentiation, sγc may play a critical role in chronic inflammation. Although the downregulation of sγc expression as part of physiological defense mechanism is induced to prevent severe COPD progress, Th1 and Th17 differentiation is induced in a COPD animal model. Therefore, we speculate that a complete block of sγc expression or function may inhibit the progression of COPD or maintain disease stability. In sum, the regulatory role of sγc on the progression and exacerbation of COPD in sγcTg mice put forward a model of inflammation regulatory mechanism that requires the integration of a role of sγc in controlling inflammation.
Acknowledgment
This work was supported by a 2-year research grant of Pusan National University.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Pauwels RA, Buist AS, Ma P, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: National Heart, Lung, and Blood Institute and World Health Organization Global Initiative for Chronic Obstructive Lung Disease (GOLD): executive summary. Respir Care. 2001;46(8): 798–825. | ||
Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343(4):269–280. | ||
Saetta M, Turato G, Maestrelli P, Mapp CE, Fabbri LM. Cellular and structural bases of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163(6):1304–1309. | ||
Byron KA, Varigos GA, Wootton AM. IL-4 production is increased in cigarette smokers. Clin Exp Immunol. 1994;95(2):333–336. | ||
de Heens GL, van der Velden U, Loos BG. Cigarette smoking enhances T cell activation and a Th2 immune response; an aspect of the pathophysiology in periodontal disease. Cytokine. 2009;47(3):157–161. | ||
Nakamura Y, Miyata M, Ohba T, et al. Cigarette smoke extract induces thymic stromal lymphopoietin expression, leading to T(H)2-type immune responses and airway inflammation. J Allergy Clin Immunol. 2008;122(6):1208–1214. | ||
Smelter DF, Sathish V, Thompson MA, Pabelick CM, Vassallo R, Prakash YS. Thymic stromal lymphopoietin in cigarette smoke-exposed human airway smooth muscle. J Immunol. 2010;185(5):3035–3040. | ||
Liu YJ, Soumelis V, Watanabe N, et al. TSLP: an epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu Rev Immunol. 2007;25:193–219. | ||
Vassallo R, Tamada K, Lau JS, Kroening PR, Chen L. Cigarette smoke extract suppresses human dendritic cell function leading to preferential induction of Th-2 priming. J Immunol. 2005;175(4):2684–2691. | ||
Di Stefano A, Caramori G, Capelli A, et al. STAT4 activation in smokers and patients with chronic obstructive pulmonary disease. Eur Respir J. 2004;24(1):78–85. | ||
Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100(6):655–669. | ||
Agnello D, Lankford CS, Bream J, et al. Cytokines and transcription factors that regulate T helper cell differentiation: new players and new insights. J Clin Immunol. 2003;23(3):147–161. | ||
Majo J, Ghezzo H, Cosio MG. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur Respir J. 2001;17(5):946–953. | ||
O’Shaughnessy TC, Ansari TW, Barnes NC, Jeffery PK. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Am J Respir Crit Care Med. 1997;155(3):852–857. | ||
Saetta M, Baraldo S, Corbino L, et al. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160(2):711–717. | ||
Saetta M, Di Stefano A, Turato G, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(3 pt 1):822–826. | ||
Steinman L. State of the art. Four easy pieces: interconnections between tissue injury, intermediary metabolism, autoimmunity, and chronic degeneration. Proc Am Thorac Soc. 2006;3(6):484–486. | ||
Chrysofakis G, Tzanakis N, Kyriakoy D, et al. Perforin expression and cytotoxic activity of sputum CD8+ lymphocytes in patients with COPD. Chest. 2004;125(1):71–76. | ||
Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol. 2003;3(5):361–370. | ||
Vernooy JH, Moller GM, van Suylen RJ, et al. Increased granzyme A expression in type II pneumocytes of patients with severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(5):464–472. | ||
Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9(7):480–490. | ||
Hong C, Luckey MA, Ligons DL, et al. Activated T cells secrete an alternatively spliced form of common gamma-chain that inhibits cytokine signaling and exacerbates inflammation. Immunity. 2014;40(6):910–923. | ||
Park JY, Jo Y, Ko E, et al. Soluble gammac cytokine receptor suppresses IL-15 signaling and impairs iNKT cell development in the thymus. Sci Rep. 2016;6:36962. | ||
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23): 2445–2454. | ||
van der Toorn M, Slebos DJ, de Bruin HG, et al. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1211–L1218. | ||
Halbower AC, Mason RJ, Abman SH, Tuder RM. Agarose infiltration improves morphology of cryostat sections of lung. Lab Invest. 1994;71(1):149–153. | ||
DeVoss JJ, Anderson MS. Lessons on immune tolerance from the monogenic disease APS1. Curr Opin Genet Dev. 2007;17(3):193–200. | ||
Chen J, Petrus M, Bryant BR, et al. Induction of the IL-9 gene by HTLV-I Tax stimulates the spontaneous proliferation of primary adult T-cell leukemia cells by a paracrine mechanism. Blood. 2008;111(10):5163–5172. | ||
Curtis JL, Freeman CM, Hogg JC. The immunopathogenesis of chronic obstructive pulmonary disease: insights from recent research. Proc Am Thorac Soc. 2007;4(7):512–521. | ||
Hoshino H, Laan M, Sjöstrand M, Lötvall J, Skoogh BE, Linden A. Increased elastase and myeloperoxidase activity associated with neutrophil recruitment by IL-17 in airways in vivo. J Allergy Clin Immunol. 2000;105(1 pt 1):143–149. | ||
Parker LC, Prince LR, Sabroe I. Translational mini-review series on Toll-like receptors: networks regulated by Toll-like receptors mediate innate and adaptive immunity. Clin Exp Immunol. 2007;147(2):199–207. | ||
Pasare C, Medzhitov R. Toll-like receptors and acquired immunity. Semin Immunol. 2004;16(1):23–26. | ||
Shan M, Cheng HF, Song LZ, et al. Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Transl Med. 2009;1(4):4ra10. | ||
Zivadinov R, Weinstock-Guttman B, Hashmi K, et al. Smoking is associated with increased lesion volumes and brain atrophy in multiple sclerosis. Neurology. 2009;73(7):504–510. | ||
Lee SH, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13(5):567–569. | ||
Anderson G, Lane PJ, Jenkinson EJ. Generating intrathymic microenvironments to establish T-cell tolerance. Nat Rev Immunol. 2007;7(12):954–963. | ||
Ouyang W, Beckett O, Flavell RA, Li MO. An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity. 2009;30(3):358–371. | ||
Takahashi T, Kuniyasu Y, Toda M, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10(12):1969–1980. | ||
Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6(11):1142–1151. | ||
D’Armiento JM, Scharf SM, Roth MD, et al. Eosinophil and T cell markers predict functional decline in COPD patients. Respir Res. 2009;10:113. | ||
Barceló B, Pons J, Ferrer JM, Sauleda J, Fuster A, Agustí AG. Phenotypic characterisation of T-lymphocytes in COPD: abnormal CD4+CD25+ regulatory T-lymphocyte response to tobacco smoking. Eur Respir J. 2008;31(3):555–562. | ||
Weaver CT, Hatton RD. Interplay between the TH17 and TReg cell lineages: a (co-)evolutionary perspective. Nat Rev Immunol. 2009;9(12):883–889. | ||
Lee YK, Mukasa R, Hatton RD, Weaver CT. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol. 2009;21(3):274–280. | ||
Vargas-Rojas MI, Ramírez-Venegas A, Limón-Camacho L, Ochoa L, Hernández-Zenteno R, Sansores RH. Increase of Th17 cells in peripheral blood of patients with chronic obstructive pulmonary disease. Respir Med. 2011;105(11):1648–1654. | ||
Di Stefano A, Caramori G, Gnemmi I, et al. T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol. 2009;157(2):316–324. | ||
Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. | ||
Lanckacker EA, Robays LJ, Joos GF, Vermaelen KY. A new danger in the air: how pulmonary innate immunity copes with man-made airborne xenobiotics. J Innate Immun. 2010;2(2):96–106. | ||
Ruddy MJ, Shen F, Smith JB, Sharma A, Gaffen SL. Interleukin-17 regulates expression of the CXC chemokine LIX/CXCL5 in osteoblasts: implications for inflammation and neutrophil recruitment. J Leukoc Biol. 2004;76(1):135–144. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.