")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Sodium-22-radiolabeled silica nanoparticles as new radiotracer for biomedical applications: in vivo positron emission tomography imaging, biodistribution, and biocompatibility
Authors Al Faraj A , Alotaibi B, Pasha Shaik A, Shamma K, Al Jammaz I, Gerl J
Received 1 August 2015
Accepted for publication 12 September 2015
Published 8 October 2015 Volume 2015:10(1) Pages 6293—6302
DOI https://doi.org/10.2147/IJN.S93523
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Achraf Al Faraj,1 Basem Alotaibi,2 Abjal Pasha Shaik,3 Khaled Z Shamma,1 Ibrahim Al Jammaz,2 Jürgen Gerl4
1Molecular and Cellular Imaging Lab, Department of Radiological Sciences, College of Applied Medical Sciences, King Saud University, 2Cyclotron and Radiopharmaceutical Department, King Faisal Specialist Hospital and Research Centre, 3Department of Clinical Lab Sciences, College of Applied Medical Sciences, King Saud University, Riyadh, Saudi Arabia; 4GSI Helmholtzzentrum für Schwerionenforschung GmbH, Darmstadt, Germany
Abstract: Despite their advantageous chemical properties for nuclear imaging, radioactive sodium-22 (22Na) tracers have been excluded for biomedical applications because of their extremely long lifetime. In the current study, we proposed, for the first time, the use of 22Na radiotracers for pre-clinical applications by efficiently loading with silica nanoparticles (SiNPs) and thus offering a new life for this radiotracer. Crown-ether-conjugated SiNPs (300 nm; -0.18±0.1 mV) were successfully loaded with 22Na with a loading efficacy of 98.1%±1.4%. Noninvasive positron emission tomography imaging revealed a transient accumulation of 22Na-loaded SiNPs in the liver and to a lower extent in the spleen, kidneys, and lung. However, the signal gradually decreased in a time-dependent manner to become not detectable starting from 2 weeks postinjection. These observations were confirmed ex vivo by quantifying 22Na radioactivity using γ-counter and silicon content using inductively coupled plasma-mass spectrometry in the blood and the different organs of interest. Quantification of Si content in the urine and feces revealed that SiNPs accumulated in the organs were cleared from the body within a period of 2 weeks and completely in 1 month. Biocompatibility evaluations performed during the 1-month follow-up study to assess the possibility of synthesized nanocarriers to induce oxidative stress or DNA damage confirmed their safety for pre-clinical applications. 22Na-loaded nanocarriers can thus provide an innovative diagnostic agent allowing ultra-sensitive positron emission tomography imaging. On the other hand, with its long lifetime, onsite generators or cyclotrons will not be required as 22Na can be easily stored in the nuclear medicine department and be used on-demand.
Keywords: silica nanoparticles, sodium-22, radiotracer, biodistribution, noninvasive PET imaging, biocompatibility, nanomedicine
Introduction
Positron emission tomography (PET) is a high-performance molecular imaging technique widely used for clinical and pre-clinical investigations of several diseases due to its excellent sensitivity and limitless depth of penetration.1 PET has demonstrated significant potential utilities in oncology, neurology, and cardiology clinical applications.2 The potentiality of this nuclear imaging modality is due to the high sensitivity of the radiotracers, which are labeled by radioactive isotopes, such as radioactive halogens (F-18; Br-76, 77; I-124), carbon-11, and radioactive metals (Ga-68, Cu-64).3 Micro- or even picomoles of radiolabeled materials are usually sufficient for PET-image generation and thus the radiotracers will occupy only a minor part of the radio-pharmaceutics and have no pharmacologically relevant activity.4
Following in vivo administration of the radiotracer, PET imaging is based on the coincident detection of two 511 keV γ-rays traveling at 180° to each other from their point of emission. These γ-ray pairs originate when the positrons from the nuclear decay of the radioisotope interact (annihilate) with electrons in the surrounding tissue.5 Since the positrons rapidly lose their kinetic energy, the point of γ-ray emission stays typically within a fraction of a millimeter around the location of the tracer molecules. To avoid radiation damage to the patient, common PET tracers have short half-lives (ie, approximately 20 minutes for 11C and 110 minutes for 18F).Consequently, the radioisotopes must be generated onsite and quickly administered to the subject before they decay. This requires at each hospital offering PET, huge and costly cyclotrons to produce most of the isotopes or specific generators (for 68Ga and 82Rb production).
On the other hand, despite the existence of a wide choice of PET radio-pharmaceutics, they suffer from many limitations including non-specificity and necessity of administration of relatively high radiation doses to the patients in order to produce sufficient contrast in the site of interest. Thus, the field of molecular imaging and PET urgently required the introduction of new PET imaging probes with high affinity and specificity for routine clinical use.6 While attaching highly specific antibodies to the tracer compounds would obviously be an ideal solution to optimize the image contrast while reducing the applied dose, this procedure is unfortunately not chemically possible for most interesting radiotracers.
Nanotechnology has gained significant interest in the biomedical field and the use of nanomedicine for both therapeutic and diagnostic applications has increased exponentially.7 Nanomaterials have unique physical properties that allow them to be used as imaging probes to specifically target and sensitively identify diseases areas,8 and potentially improve the biodistribution and pharmacokinetic of the nanocarriers.9 Over the last decade, several nanoparticle formulations have been developed for radionuclide imaging with the aim of enhancing the sensitivity and identification of specific targets.10,11 Therefore, nanocarriers loaded with radionuclides may offer an elegant and novel possibility for harnessing this promising technology.
Furthermore, additional radioactive isotopes, excluded until now because of their chemical properties or long lifetime, such as isotopes with advantageous γ-decay properties, may become usable. One such isotope is sodium-22 (22Na), which decays by positron emission and emits a third γ-ray with 1,275 keV energy in addition to the two 511 keV γ-rays. The physical principal of triple γ-ray PET imaging has been recently discussed.12 The three quanta can uniquely allow determining the location of the emitting nucleus in 3D with reduced background and increased sensitivity. Thus, the measurement time and/or the applied activity can be reduced drastically.
However, the extremely long lifetime of 22Na (half-life [T1/2]= 2.6 years) has excluded the administration of this isotope to living species. Loading 22Na with biocompatible nanocarriers, and optimizing their biodistribution, fate, and clearance by controlling their size, surface coating, and charge, will offer a new life for this radioisotope by successfully accomplishing the proposed approach. Grafting antibodies or selective targeting agents to 22Na-loaded nanocarriers can additionally provide a unique and innovative way to mark selectively specific cells with these nano-radiotracers, and ultra-sensitive and fast 3D imaging will become possible for biomedical investigations. On the other hand, with its long lifetime, onsite generators or cyclotrons will not be required as 22Na can be easily stored in the nuclear medicine department and be used on-demand.
Among the wide range of nanoscale materials being investigated for biomedical applications, silica nanoparticles (SiNPs) possess amazing advantages, including straightforward synthesis, relatively low cost, extremely high stability in organic solvents, and easy surface modification. In addition, SiNPs are usually considered to have low toxicity. They have been used for many applications, including in cosmetics, foods, medical diagnosis, cancer therapy, and drug delivery.13
The purpose of this study was to investigate, for the first time, the possibility of using radioactive 22Na for pre-clinical applications by efficiently loading with silica-based nanocarriers. Noninvasive PET imaging, in vivo biodistribution, and biocompatibility of the intravenously injected nanocarriers to the mice model were longitudinally assessed in a 1-month follow-up study.
Materials and methods
Sodium-loaded SiNPs design
SiNPs (ie, silicon dioxide: SiO2) with a diameter of 300 nm were obtained from Micromod Partikeltechnologie GmbH, Rostock, Germany. Unless otherwise specified, all the chemical materials were purchased from Sigma Aldrich, MO, USA.
In order to prepare the crown-ether-conjugated SiNPs, an amino arm was first introduced using 3-aminopropyltriethoxysilane. The resulting SiO2-NH2 construct was then reacted with succinic anhydride in pyridine to introduce a carboxylic acid functional group. After activation of the surface of carboxylated silica particles with N, N′-dicyclohexyl carbodiimide in chloroform, crown ethers (ie, 4-amino-benzo-15-crown-5) were conjugated to the nanoparticles surface via amide bonds (Figure 1A).
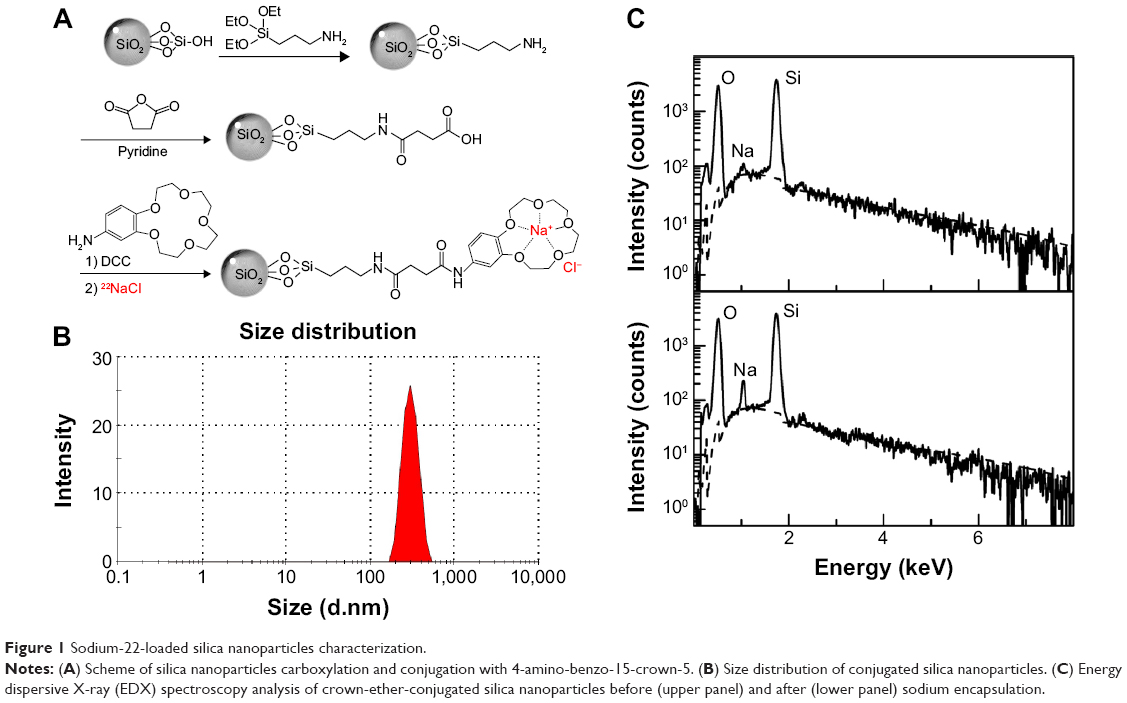 | Figure 1 Sodium-22-loaded silica nanoparticles characterization. |
22Na radionuclide with an initial radioactivity of 2 mCi (74 MBq) was purchased from PerkinElmer, MA, USA, as sodium chloride (22NaCl) solution in water. 22Na encapsulation was performed by incubating 1 mg of the crown-ether-conjugated SiNPs, at room temperature for 30 minutes, with 22NaCl suspension diluted at various activity levels (25, 50, 100, or 200 μCi). Triethylamine base (1 μL) was added to the mixture to prevent protonation of the oxygen atoms in the crown ether. Finally, the nanoconjugates were washed two times with deionized water following centrifugation for 15 minutes and the loading efficacy was quantified using the G4-W γ-counter (Gamma Products Inc., Palos Hills, IL, USA).
Nanocarrier characterizations
UV–Vis analysis
UV-Vis spectrophotometer analysis was performed using a BioTek Epoch 2 multiplate reader (BioTek Instruments, Inc., Winooski, VT, USA) to measure the loading of 4-amino-benzo-15-crown-5 on the SiNPs. The absorbance spectra (optical density) were measured at 298 nm to quantify the difference in the crown ether concentration in the supernatant before and after reaction with the carbodiimide-activated nanoparticles.
Size distribution and surface charge
The mean diameter of crown-ether-conjugated SiNPs and the zeta potential based on dynamic light scattering to determine the electrical surface charge of the nanocarriers before and after their conjugation were measured using a Zetasizer Nano ZS90 (Malvern Instruments, UK) with a particle concentration of 1 mg/mL in ultrapure water at 25°C. Results were expressed as zeta potential (mV) or mean diameter (nm) ± standard deviation on an average of three measurements.
Energy dispersive X-ray analysis
Energy dispersive X-ray spectroscopy analysis of crown-ether-conjugated SiNPs was performed before and after encapsulation of non-radioactive sodium (NaCl solution) with a DSM960 scanning electron microscope (Carl Zeiss, Oberkochen, Germany).
Experimental protocol
All animal procedures were performed in accordance with the National guidelines for the care of laboratory animals and the study was approved by the Ethical Committee of the College of Applied Medical Sciences (agreement number: CAMS06/3334). Female Balb/c mice (6 weeks old) used in this study were housed in a protected environment in a temperature-controlled ventilated cabinet and given free access to sterilized food and water on a 12-hour light/dark cycle.
Mice were intravenously injected with 100 μL suspension of either free 22Na (22NaCl solution) or 22Na-loaded SiNPs (22Na-SiNPs) at a concentration of 100 μg per mice with a corresponding radioactivity loading of 10 μCi. Noninvasive PET imaging, biodistribution, and biocompatibility were then assessed in a longitudinal follow-up study for up to 1 month following intravenous injection (n=6/group/time point). A group of mice (n=3/group/time point) was kept in isolated metabolic cages (Tecniplast, Buguggiate, Italy) and the urine and feces were collected separately.
Noninvasive PET imaging
Mice were imaged using a small-animal PET R4 rodent model scanner (Siemens Healthcare, Erlangen, Germany) at 5 minutes, 4 hours, 24 hours, 48 hours, 72 hours, 1 week, 2 weeks, and 1 month following 22Na or 22Na-SiNPs injection under 1.5%–2% isoflurane anesthesia. Images were reconstructed by a two-dimensional ordered-subsets expectation maximization algorithm. No background correction was performed. Regions of interest were drawn over the liver, spleen, kidneys, and lung. The radioactivity concentration within regions of interest were obtained from the mean value within the multiple organs of interest, averaged on three consecutive slices, and then converted to the percentage of injected dose per gram (%ID/g).
Biodistribution evaluations
Following PET imaging at 24 hours, 72 hours, 1 week, and 2 weeks, animals were sacrificed by overdose anesthesia and the blood, liver, spleen, kidneys, lung, and heart were harvested. Tissue samples were accurately weighed, and 22Na radioactivity was quantified using the γ-counter and expressed as %ID/g.
In another set of animals, blood, tissue samples, and collected urine and feces were mineralized in 2 mL ultrapure nitric acid (Panreac, Barcelona, Spain) and digested in a Mars6 Xpress microwave digestion system (CEM, NC, USA). Silicon content was then sensitively quantified using an inductively coupled plasma-mass spectrometry (ICP-MS) (Aurora M90, Bruker Daltonics, MA, USA) and reported as microgram of Silicon per gram of tissue (μg Si/g).
Data obtained from both radioactivity measurements and silica quantifications using the ICP-MS were linearly fitted and statistically correlated.
In vivo biocompatibility assessments
Oxidative stress evaluation using TBARS assay
The ability of the developed sodium-loaded SiNPs to cause an increase in oxidative stress with an estimation of lipid peroxidation using the thiobarbituric acid reactive substances (TBARS) assay (R&D Systems, Inc., Minneapolis, MN, USA) was assessed as per the manufacturer’s instructions on plasma and liver tissue cell lysate samples at different investigation time points following injection. All experiments per condition and time point were conducted in triplicates. Blood samples were stored in heparin vacutainer tubes. Samples were centrifuged for 15 minutes at 1,000× g and plasma samples were collected and immediately analyzed or stored at -20°C until further analyses. For obtaining cell lysates from tissues, liver biopsies were minced, the filtrates were collected and treated with cell lysis buffer for 30 minutes. All cell lysate samples were centrifuged before processing for oxidative stress assessments. Plasma and cell lysate samples were incubated with acid reagent for 15 minutes. The samples were centrifuged and supernatants were treated with 2-thiobarbituric acid (TBA) reagent and incubated for a further 2–3 hours at 45°C–50°C. The absorbance of the samples was measured through optical density measurements at 532 nm using the BioTek Epoch 2 multiplate reader.
DNA damage assessment (comet assay)
Comet assay was used as a simple and effective method to assess the possibility of the developed nanocarriers to cause DNA damage in blood and liver tissues. Peripheral blood mononuclear cells from blood and cells isolated following mincing of liver tissues were prepared for comet assay as per the manufacturer’s protocol (R&D Systems, Inc.). For liver tissue, small pieces were minced finely and cell suspensions were recovered in phosphate buffered saline. Cells from blood and tissue samples were counted, pelleted, and re-suspended at 1×105 cells/mL in ice-cold phosphate buffered saline. Cells were immobilized in low melting agarose, lysed with lysing solution for 1 hour, immersed in alkaline unwinding buffer for 20 minutes, and electrophoresed at 21 V for 30 minutes. Slides were fixed before sliver staining. The cells were observed under an Olympus BX53 microscope (Olympus Corporation, Tokyo, Japan). Data for DNA damage were presented as mean ± SD (μm) for an average of 100 cells. In the case of DNA damage, the denatured and cleaved DNA fragments migrate out of the cells in the presence of electric potential as a “comet tail”, while undamaged DNA remains within the cell membrane as the “comet head”.
Statistical analysis
Data presented as the mean standard deviation were analyzed by t-test using SPSS v12.0 (SPSS Inc., Chicago, IL, USA) software. P-values <0.05 were considered significant for all tests.
Results and discussion
SiNPs characterization and sodium loading efficacy
SiNPs were successfully conjugated with crown ether. The density of 4-amino-benzo-15-crown-5 on the SiNP surface, quantified by measuring the disappearance of crown ether in the supernatant before and after conjugation with the activated nanoparticles using a UV-vis spectrophotometer at 298 nm, was found to be 50 nmol per mg of nanoparticles. Crown-ether-conjugated SiNPs were found to have a hydrodynamic diameter equal to 289.9 nm (Figure 1B). The surface charge (zeta potential) increased from -42.1±0.6 mV before to -0.18±0.1 mV after crown ether conjugation, confirming the successful binding of the crown ether with a close to neutral surface charge.
SiNPs were preferred in the current study to validate our approach of sodium complexation due to their relatively good biocompatibility from one side13 and their high solubility in organic solvents allowing stable conjugation of the amino-benzo-crown ether from the other side. Gokel et al have reviewed the importance of the crown ethers as molecular sensors for ions, molecular scaffolds, and complexing agents.14 Apart from their high affinities for alkali ions such as sodium, crown ethers were reported to help forming stable complexes and inhibiting materials aggregation.15 4-Amino-benzo-15-crown-5 was preferred for selective and efficient sodium complexation.
Energy dispersive X-ray analysis confirmed the encapsulation of sodium with the presence of characteristic peak observed at 1.04 keV (Figure 1C). Quantification of radioactive 22Na encapsulation by the crown-ether-conjugated SiNPs revealed a labeling efficiency of 86.7%±1.0%, 94.7%±1.2%, 98.1%±1.4%, and 58.3%±0.9% following incubation with 22Na diluted to 25, 50, 100, and 200 μCi, respectively. Addition of triethylamine base was crucial to obtain high loading efficacy by preventing protonation of the oxygen atoms in the crown ether.
Noninvasive imaging and in vivo biodistribution
After characterizing the 22Na-loaded crown-ether-conjugated SiNPs, a single dose of 100 μg suspended in 100 μL and having a corresponding 10 μCi radioactivity dose, was intravenously injected to each mouse. PET imaging was performed to monitor their biodistribution in mice and compared to free 22Na (22NaCl solution) diluted to the same radioactivity level. We intentionally preferred a relatively low dose of nanoparticles equivalent to 5 mg/kg and low radioactivity dose as one of the advantages of our approach was that the radioactivity level of loaded 22Na remains stable during a 1-month investigation study. Intravenous injection was preferred as it gives direct access to the blood circulation and thus a rapid distribution all over the entire body.16
Following intravenous injection, 22Na-SiNPs were successfully detected using a noninvasive PET scanner (Figure 2A). Quantitatively, a transient accumulation in the liver and to a lower extent in the kidneys, spleen, and lung were detected 5 minutes postinjection with %ID/g corresponding to 22.35%±3.21%, 8.43%±1.92%, 11.44%±2.81%, and 4.12%±1.58% in the liver, spleen, kidneys, and lung, respectively. Interestingly, signal in the liver progressively decreased in a time-dependent manner to become not visibly detectable in PET images starting from 2 weeks postinjection. The transient accumulation of 22Na-SiNPs in the kidneys reached its maximum at 24 hours time point and then gradually decreased to become not detectable starting from 2 weeks. However, no signal in the spleen and lung was detected starting from 1 week postinjection of SiNPs loaded with 22Na.
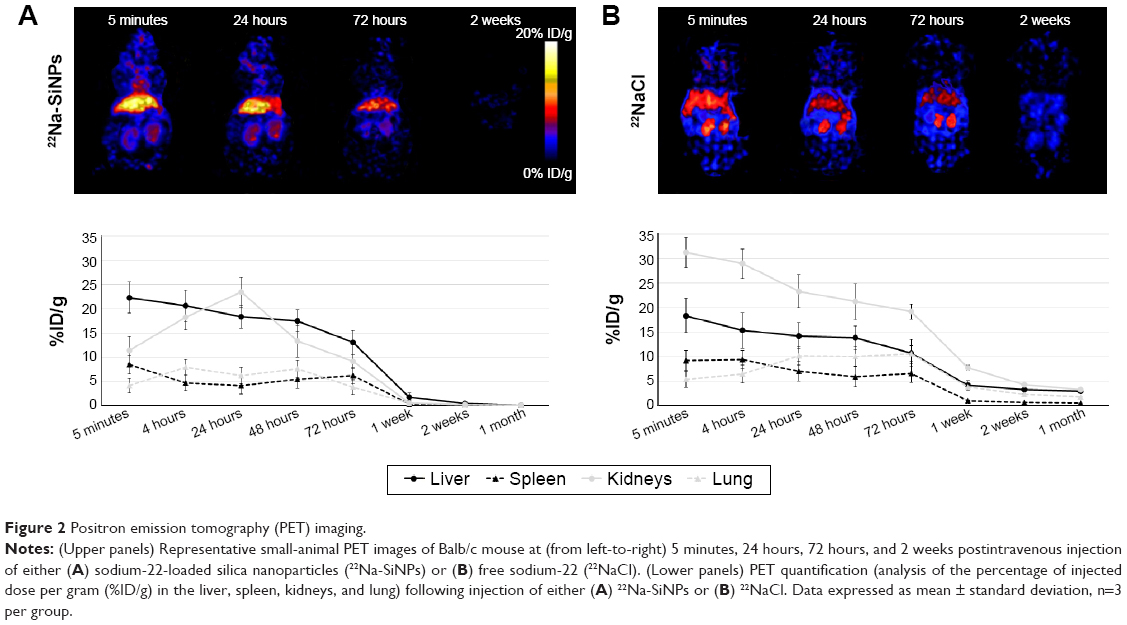 | Figure 2 Positron emission tomography (PET) imaging. |
On the other hand, free 22Na injected as 22NaCl solution (Figure 2B) revealed a higher transient accumulation in the kidneys with 31.22%±3.08% and to a lower extent in the liver with 18.33%±3.52% at 5 minutes time point. While the signal arising from free 22Na gradually decreased in a time-dependent manner in all groups, it remains clearly detectable in PET images in all organs of interest even after 1 month postinjection.
Following PET imaging at 24 hours, 72 hours, 1 week, and 2 weeks, 22Na radioactivity was ex vivo quantified in the blood, liver, spleen, kidneys, lung, and heart using the γ-counter (Figure 3). Overall, quantification of %ID/g in the liver, spleen, kidneys, and lung confirmed noninvasive PET readouts with a transient accumulation in the liver, spleen, and kidneys and to a lower extent in the lung and heart. 22Na-SiNPs gradually decreased in a time-dependent manner to be fully eliminated starting from 2 weeks postinjection, while free 22Na were still detectable even after 1 month postinjection. Radioactivity quantification in the blood revealed 36.67%±2.68% and 38.78%±1.32% of 22Na-SiNPs injected dose detected at 24 hours and 72 hours, but then significantly decreased to 1.49%±0.98% and 0.13%±0.03% at 1 week and 2 weeks investigation time point, respectively. In mice group injected with free 22NaCl, 17.18%±0.97% of %ID/g was still present in the blood at 1 week postinjection.
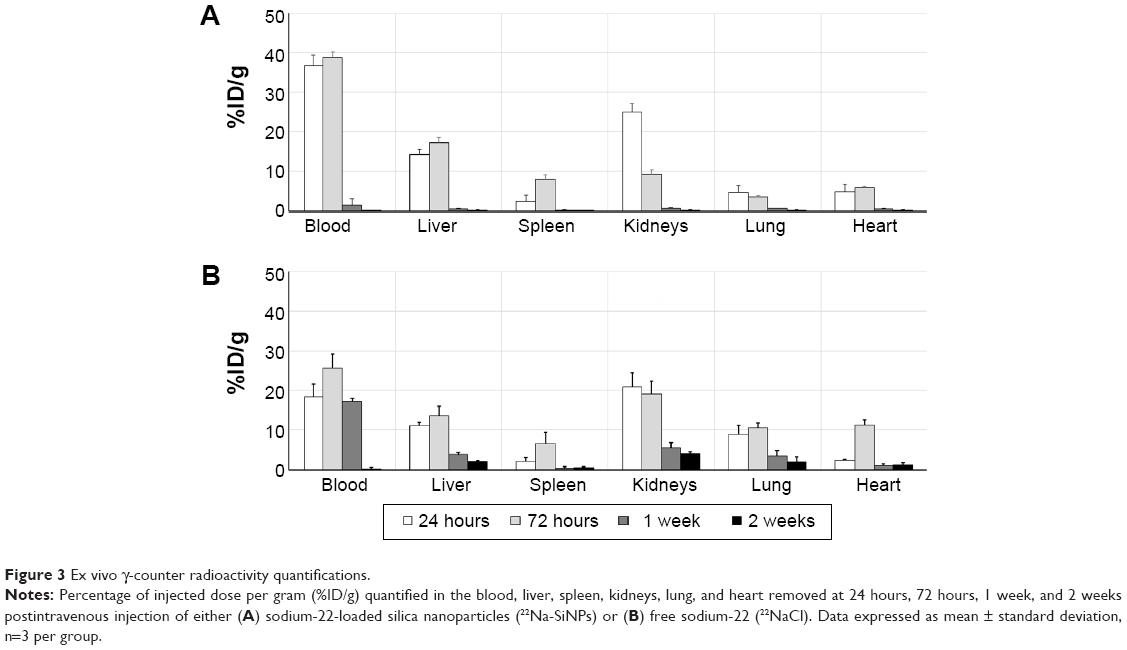 | Figure 3 Ex vivo γ-counter radioactivity quantifications.
|
After assessing the biodistribution of 22Na-SiNPs using noninvasive PET and γ-counter quantifications by measuring the radioactivity arising from the radioactive 22Na tracers, the biodistribution of SiNPs was further assessed by sensitively measuring Si content in the different samples using ICP-MS and compared to control mice injected with physiological saline (0.9% NaCl) (Figure 4A). The advantage of ICP-MS is that the organs are normally digested for analysis, which allows direct measurement of the atomic composition of organs.17
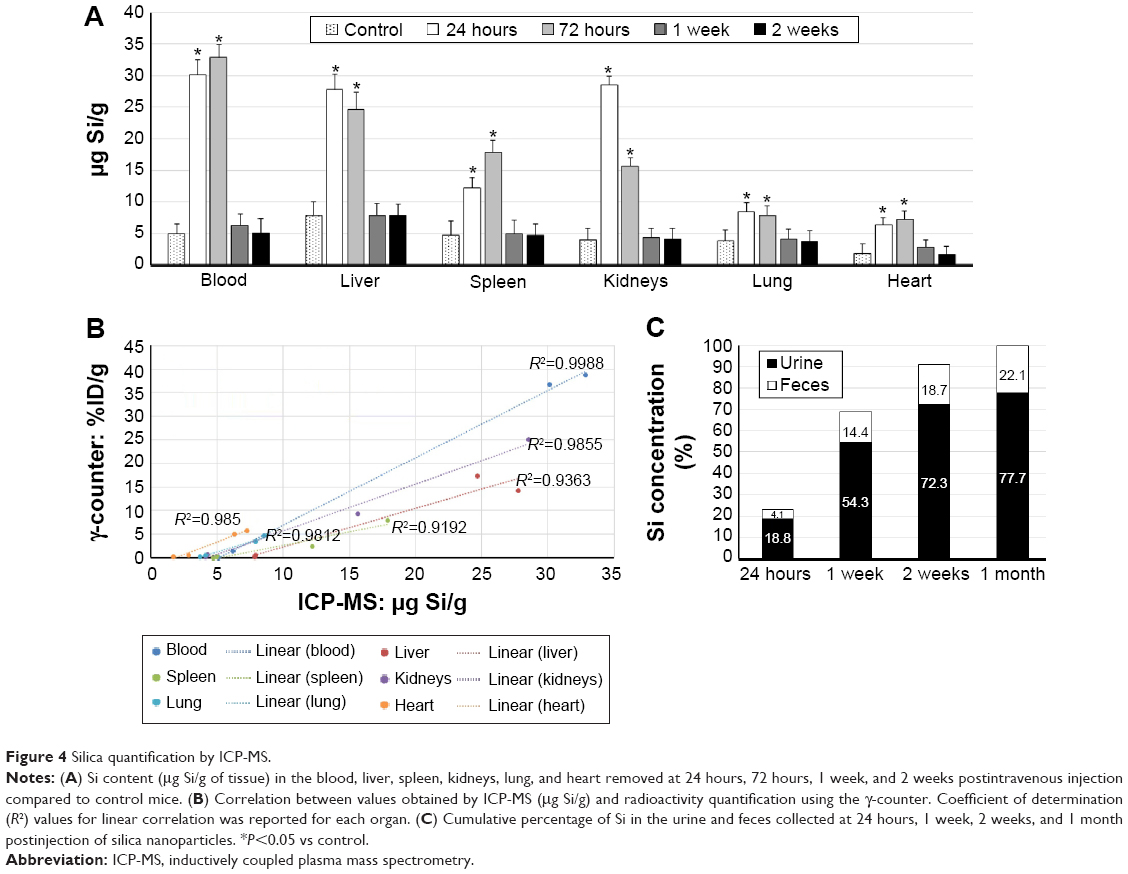 | Figure 4 Silica quantification by ICP-MS. |
Compared to intrinsic Si content in control mice, ICP-MS revealed a statistically significant increase in SiNPs present in the blood, liver, spleen, and kidneys, and to a much lower extent in the lung and heart, at 24 hours and 72 hours postinjection. However, Si content regained control values and become non-statistically significant starting from 1 week postinjection (P<0.05).
Correlating by linearly fitting data obtained using the γ-counter and ICP-MS revealed an excellent coefficient of determination R2 ranging from 0.92 to 0.99 for the different investigated organs of interest (Figure 4B). The high R2 values suggested that encapsulation of 22Na within the crown-ether-conjugated SiNPs remained stable, at least until the nanocarriers reached the different organs of interest during the first hours following intravenous injection.
Finally, the cumulative percentage of SiNPs eliminated from the body was assessed in urine and feces collected from mice at different investigations time points by quantifying Si content using ICP-MS (Figure 4C). Results showed that 22.9%, 68.7%, 91%, and 99.8% were eliminated in 24 hours, 1 week, 2 weeks, and 1 month postinjection, respectively. Thus, SiNPs accumulated in the organs were cleared from the body within a period of 2 weeks and completely in 1 month.
Several parameters such as size and surface characteristics (ie, coating, charge, shape, etc) were reported to affect the biodistribution, pharmacokinetics, and bioavailability of the nanoparticles. Nanoparticles for therapy need to have a long retention time for targeting and therapy.18 By understanding these parameters, nanoparticles can be designed for improved circulation and targeted delivery, thus creating more efficient and safe therapeutic and diagnostic probes.19
For example, Decuzzi et al have evaluated the effect of particle size and shape on the biodistribution of SiNPs having 0.7–3 μm diameters. The particles were primarily retained by reticuloendothelial system organs such as the liver, spleen, and lungs. As the nanoparticles size decreased, a greater percentage was more retained in the liver.20 Cho et al have also assessed the impact of size on tissue distribution and elimination of intravenously injected SiNPs having 50, 100, and 200 nm diameter. As size increased, more nanoparticles were trapped by macrophages before being cleared via urine and bile and fully eliminated in 4 weeks.21 Besides, Yu et al examining the effect of surface charge showed that amine surface modification reduced lung accumulation but resulted in increased liver and spleen accumulation.22
On the other hand, the same parameters that affect the biodistribution of nanoparticles are known to play an essential role in the toxicity of nanoparticles. However, the effects of these parameters were not yet fully understood in vivo. For prospective pre-clinical applications, the biocompatibility of the developed nanocarriers was evaluated.
In vivo biocompatibility assessments
Oxidative stress evaluation (TBARS assay)
The possibility of SiNPs to induce oxidative stress in blood and liver were assessed by estimating lipid peroxidation via the release of malondialdehyde and measured using the TBARS assay. Assessments were performed over a range of seven investigation time points from 4 hours until 1 month. A significant increase in oxidative stress in plasma samples was evident at 24 hours and 48 hours following nanoparticles injection. However, the levels reached normalcy from 72 hours and remained comparable with no statistically significant differences at 1 week, 2 weeks, and 1 month. Since liver is the major organ for clearance of nanoparticles, a significant and expected increase in oxidative stress was observed from 4 hours and at 24 hours and 48 hours postinjection of SiNPs (Figure 5A). However, similar to plasma samples, the increases in lipid peroxidation at early time points (ie, up to 48 hours) gradually decreased to become statistically comparable to control values after 72 hours. The transient increase in lipid peroxidation at early time points in both plasma and liver samples indicated that the synthesized nanoparticles might cause initial increases in oxidative stress. However, this level of increase can easily revert to normal levels over a short time period and remain stable throughout the remaining investigation time points up to 1 month.
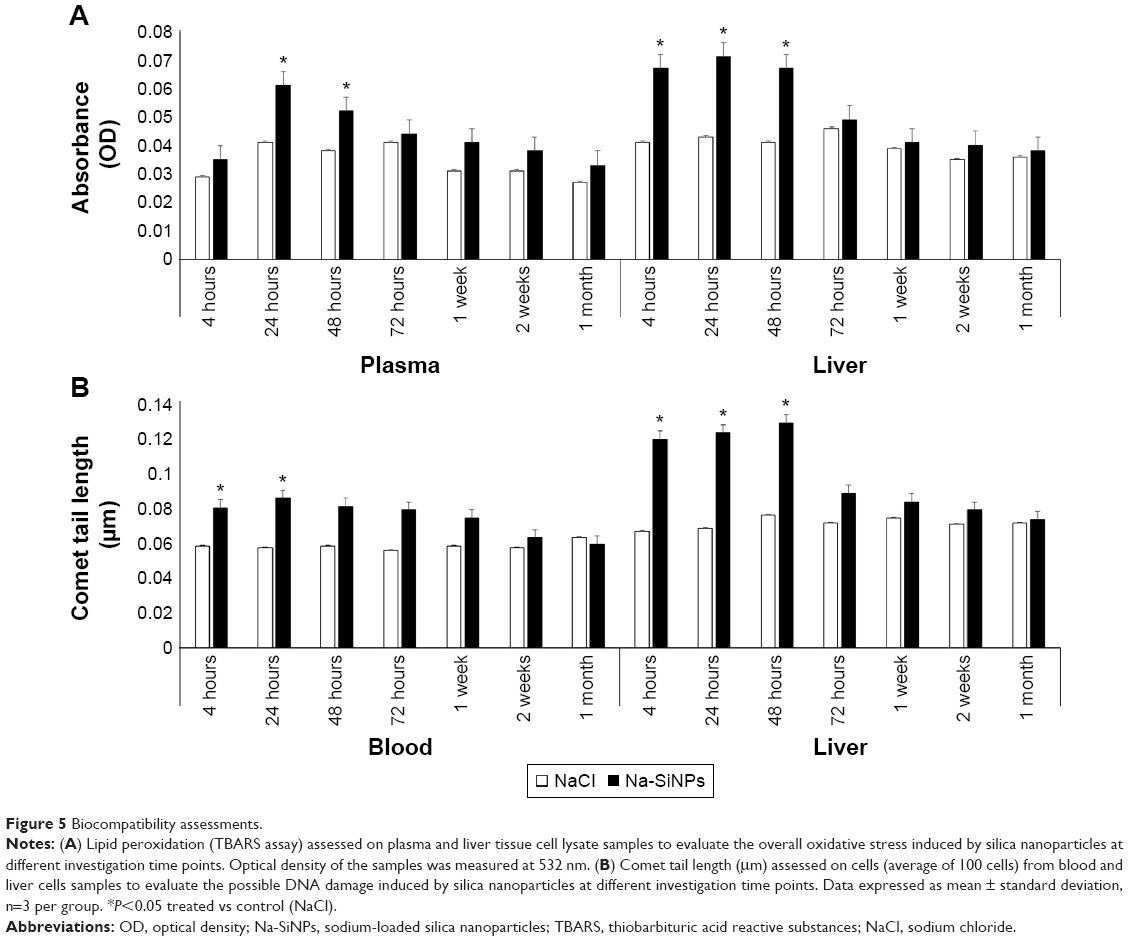 | Figure 5 Biocompatibility assessments. |
DNA damage assessment (comet assay)
To investigate the effect of SiNPs on DNA damage, a comet assay analysis was performed on cells from blood and liver. In blood cells, DNA damage was identified at early exposures of 4 hours (0.085 μm, P<0.05) and 24 hours (0.091 μm, P<0.05). However, starting from 48 hours postexposure, no statistically significant increase in DNA damage was tabulated across all the remaining time points (48 hours: 0.086 μm; 72 hours: 0.084 μm; 1 week: 0.079 μm; 2 weeks: 0.067 μm; 1 month: 0.063 μm).
The DNA damage in liver cells corresponds to the results of oxidative stress. A significant increase in comet tail lengths was observed at 4 hours (0.127 μm, P<0.05), 24 hours (0.131 μm, P<0.05), and 48 hours (0.137 μm, P<0.05) while no significant increase was detected after 72 hours (72 hours: 0.094 μm; 1 week: 0.089 μm; 2 weeks: 0.084 μm; 1 month: 0.078 μm) compared to control group receiving NaCl (Figure 5B). A significant decrease in the level of DNA damage was observed at later time points starting from 48 hours in blood and 72 hours in liver cell samples.
Corroborating oxidative stress and DNA damage evaluations have thus confirmed the safety of the SiNPs used in the current study.
Conclusion
The extremely long half-life of 2.6 years have limited the use of 22Na for biomedical applications even with its outstanding chemical properties ideal for noninvasive nuclear imaging. We have proposed in the current study to load 22Na with crown-ether-conjugated SiNPs and therefore the biodistribution and clearance of 22Na radiotracers will be driven by the nanocarriers.
SiNPs with 300 nm and neutral surface charge were successfully loaded with 22Na with a loading efficacy of up to 98.1%±1.4%. Following their intravenous injection, noninvasive PET imaging was performed to monitor their biodistribution in mice and compared to free 22Na in a 1-month longitudinal study. A transient accumulation of 22Na-SiNPs was detected in the liver and to a lower extent in the spleen, kidneys, and lung. However, the signal gradually decreased in a time-dependent manner to become not detectable starting from 2 weeks postintravenous injection. These observations were confirmed ex vivo by quantifying 22Na radioactivity using the γ-counter and Silicon content using the ICP-MS in the blood and the different organs of interest. The high correlation between those results suggested that encapsulation of 22Na within the crown-ether-conjugated SiNPs remained stable, at least until the nanocarriers reached the different organs of interest during the first hours following injection. Quantification of Si content in the urine and feces revealed that SiNPs accumulated in the organs were cleared from the body within a period of 2 weeks and completely in 1 month.
Biocompatibility evaluations performed during the 1-month follow-up study to assess the possibility of synthesized silica nanocarriers to induce oxidative stress or DNA damage. Results revealed a transient effect during the first 48 hours but at a level that can easily revert and be stable over a short time period confirming the safety of SiNPs for pre-clinical investigations.
This study showed for the first time the possibility of safe administration of 22Na radiotracers by loading within crown-ether-conjugated SiNPs, which were eliminated from the mice body in 2 weeks based on the physicochemical characteristics of the synthesized nanocarriers. 22Na-loaded nanocarriers can thus provide an innovative diagnostic agent allowing ultra-sensitive PET imaging, on the other hand, with its long lifetime, onsite generators or cyclotrons will not be required as 22Na can be easily stored in the nuclear medicine department and be used on-demand. As future perspectives, these 22Na nanocarriers could be further conjugated with a specific targeting agent for a better diagnosis and therapy of cancerous or inflammatory diseases.
Acknowledgments
This project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, Kingdom of Saudi Arabia (award number 12-MED2535). The funder has no role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Disclosure
The authors report no conflicts of interest in this work.
References
Phelps ME. Positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci U S A.2000;97(16):9226–9233. | ||
Vallabhajosula S, Solnes L, Vallabhajosula B. A broad overview of positron emission tomography radiopharmaceuticals and clinical applications: what is new? Semin Nucl Med.2011;41(4):246–264. | ||
Zhu A, Shim H. Current molecular imaging positron emitting radiotracers in oncology. Nucl Med Mol Imaging.2011;45(1):1–14. | ||
Matthews PM, Rabiner EA, Passchier J, Gunn RN. Positron emission tomography molecular imaging for drug development. Br J Clin Pharmacol.2012;73(2):175–186. | ||
Garcia EV. Physical attributes, limitations, and future potential for PET and SPECT. J Nucl Cardiol.2012;19(Suppl 1):S19–S29. | ||
Jiang L, Tu Y, Shi H, Cheng Z. PET probes beyond (18)F-FDG. J Biomed Res.2014;28(6):435–446. | ||
Rizzo LY, Theek B, Storm G, Kiessling F, Lammers T. Recent progress in nanomedicine: therapeutic, diagnostic and theranostic applications. Curr Opin Biotechnol.2013;24(6):1159–1166. | ||
Sheng Y, Liao LD, Thakor NV, Tan MC. Nanoparticles for molecular imaging. J Biomed Nanotechnol.2014;10(10):2641–2676. | ||
Moros M, Mitchell SG, Grazu V, de la Fuente JM. The fate of nanocarriers as nanomedicines in vivo: important considerations and biological barriers to overcome. Curr Med Chem.2013;20(22):2759–2778. | ||
Ting G, Chang CH, Wang HE, Lee TW. Nanotargeted radionuclides for cancer nuclear imaging and internal radiotherapy. J Biomed Biotechnol.2010;2010. doi:10.1155/2010/953537. | ||
Srivatsan A, Chen X. Recent advances in nanoparticle-based nuclear imaging of cancers. Adv Cancer Res.2014;124:83–129. | ||
Cussonneau J-P; Inventor. Process for locating a position radionuclide, applications and device for implenting same; 2010. | ||
Jaganathan H, Godin B. Biocompatibility assessment of Si-based nano- and micro-particles. Adv Drug Deliv Rev.2012;64(15):1800–1819. | ||
Gokel GW, Leevy WM, Weber ME. Crown ethers: sensors for ions and molecular scaffolds for materials and biological models. Chem Rev.2004;104(5):2723–2750. | ||
Tian Y, Zhang X, Li Y, et al. Crown ethers attenuate aggregation of amyloid beta of Alzheimer’s disease. Chem Commun.2014;50(99):15792–15795. | ||
Hirn S, Semmler-Behnke M, Schleh C, et al. Particle size-dependent and surface charge-dependent biodistribution of gold nanoparticles after intravenous administration. Eur J Pharm Biopharm.2011;77(3):407–416. | ||
Liberman A, Mendez N, Trogler WC, Kummel AC. Synthesis and surface functionalization of silica nanoparticles for nanomedicine. Surf Sci Rep.2014;69(2–3):132–158. | ||
Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res.2008;14(5):1310–1316. | ||
Ernsting MJ, Murakami M, Roy A, Li SD. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J Control Release.2013;172(3):782–794. | ||
Decuzzi P, Godin B, Tanaka T, et al. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release.2010;141(3):320–327. | ||
Cho M, Cho WS, Choi M, et al. The impact of size on tissue distribution and elimination by single intravenous injection of silica nanoparticles. Toxicol Lett.2009;189(3):177–183. | ||
Yu T, Hubbard D, Ray A, Ghandehari H. In vivo biodistribution and pharmacokinetics of silica nanoparticles as a function of geometry, porosity and surface characteristics. J Control Release.2012;163(1):46–54. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.