")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
Slowly Progressive Type 1 Diabetes Mellitus: Current Knowledge And Future Perspectives
Authors Nishimura A , Matsumura K, Kikuno S, Nagasawa K, Okubo M, Mori Y, Kobayashi T
Received 30 July 2019
Accepted for publication 18 October 2019
Published 28 November 2019 Volume 2019:12 Pages 2461—2477
DOI https://doi.org/10.2147/DMSO.S191007
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Akihiro Nishimura,1 Kimio Matsumura,1 Shota Kikuno,1 Kaoru Nagasawa,1 Minoru Okubo,1 Yasumichi Mori,1 Tetsuro Kobayashi2
1Department of Endocrinology and Metabolism, Toranomon Hospital, Tokyo, Japan; 2Division of Immunology and Molecular Medicine, Okinaka Memorial Institute for Medical Research, Tokyo, Japan
Correspondence: Tetsuro Kobayashi
Okinaka Memorial Institute for Medical Research, 2-2-2 Toranomon, Minato-Ku, Tokyo, Japan
Tel +81-3-3588-1111
Fax +81-3-3582-7068
Email [email protected]
Abstract: Slowly progressive type 1 insulin-dependent diabetes mellitus (SPIDDM), sometimes referred to as latent autoimmune diabetes in adults (LADA), is a heterogeneous disease that is often confused with type 1 and type 2 diabetes. As a result, there were few diagnostic criteria for this disorder until 2012, when the Japan Diabetes Society established criteria that could be used in clinical practice. A primary question is whether pathologic markers for type 1 or type 2 diabetes are present in the pancreas of patients with SPIDDM, because the phenotype of SPIDDM is similar to both type 1 and type 2 diabetes. Recent studies clarified pathologic findings in the pancreas of patients with SPIDDM, which included T-cell-mediated insulitis, a marker of type 1 diabetes; pseudoatrophic islets (islets specifically devoid of beta cells), another hallmark of type 1 diabetes; and a lack of amylin (ie, islet amyloid polypeptide) deposition to the islet cells, a pathologic marker of type 2 diabetes. In terms of preventing the loss of beta-cell function in patients with SPIDDM, several studies have shown that some drugs, including dipeptidyl peptidase-4 inhibitors, are effective. There is an increased need for early diagnosis of SPIDDM to preserve beta-cell function. This review presents updated findings on the pathogenesis and immunologic findings of the affected pancreas, diagnostic markers, risk factors for progression of beta-cell dysfunction, epidemiology, clinical features, diagnostic strategies, prevention strategies, and clinical options for patients with SPIDDM.
Keywords: slowly progressive type 1 diabetes mellitus; SPIDDM, latent autoimmune diabetes in adults; LADA, insulitis, PanIN, GAD antibody
Introduction
Slowly progressive type 1 insulin-dependent diabetes mellitus (SPIDDM),1–4 sometimes referred to as latent autoimmune diabetes in adults (LADA),5,6 is the most prevalent clinical subtype of type 1 diabetes mellitus in Japan.3 In terms of nomenclature, SPIDDM is not always synonymous with LADA. This is because SPIDDM includes patients with any age of onset of diabetes (AOD), whereas LADA restricts the AOD to adults. Additionally, genetic differences potentially influence the different clinical phenotypes.7,8 The term SPIDDM is used in this review when the reported cases meet the diagnostic criteria for SPIDDM set by the Japan Diabetes Society9 and/or when the authors of the manuscripts cited used SPIDDM in their reports.1–4
Both SPIDDM and LADA are characterized by late age at onset; progressive beta-cell failure, which is associated with an initial non-insulin-requiring state and an ultimate insulin-dependent state over several years, and persistent islet cell autoantibodies, including glutamic acid decarboxylase autoantibodies (GADAb) and islet cell antibodies (ICAs).1–4,9,10 SPIDDM and LADA are conceptual phenotypes between classic acute-onset type 1 diabetes (AT1D) and type 2 diabetes (T2D), and the term “Type 1.5” diabetes was also proposed to describe these disorders.6 Because of the wide range of clinical phenotypes and the overlap between features of type 1 diabetes (T1D) and T2D, diagnostic criteria that could be used in routine clinical practice were not established until recently. In 2012, the Japan Diabetes Society organized a nationwide Research Committee on Type 1 Diabetes and established diagnostic criteria for SPIDDM.9 These criteria are helpful in recruiting subjects for intervention trials aimed at preventing progressive beta-cell failure using various pharmacologic options, such as insulin, metformin, dipeptidyl peptidase-4 (DPP4) inhibitors, and/or glucagon-like peptide 1 analogs. The most challenging clinical issue is how to intervene before beta-cell failure in patients with SPIDDM.11–14
This review presents updated findings on the pathogenesis and immunologic findings of the affected pancreas, diagnostic markers, risk factors for progression of beta-cell dysfunction, epidemiology, clinical features, diagnostic strategies, prevention strategies, and clinical options for patients with SPIDDM.
Pathogenesis And Immunologic Findings Of The Pancreas In Patients With SPIDDM
It is unclear whether beta-cell-specific inflammation of islet cells (insulitis), a pathologic marker of T1D, or amylin (islet amyloid polypeptide [IAPP]) deposition, a specific marker of T2D, are present in the pancreas of patients with SPIDDM. This lack of clarity occurs because the phenotype of SPIDDM is similar in part with both T1D and T2D. Similarly, it is unclear whether pancreatic exocrine inflammation occurs in the pancreas of patients with SPIDDM. Radiologic changes seen on a ductogram of the pancreas showed the presence of chronic pancreatitis-like dilatation, narrowing of pancreatic ducts, and atrophy of exocrine pancreas in patients with SPIDDM.15,16 Shimada et al17 reported the presence of CD4+ T-cell dominant islet inflammation in a patient with GADAb-positive, non-insulin dependent diabetes in the early stage of SPIDDM. In a study using pancreatic scintigraphy with interleukin-2 radiolabeled with 99mTc (99mTc-IL-2) and contrast-enhanced magnetic resonance imaging, pancreatic accumulation of 99mTc-IL-2 was detected in patients with LADA.18 Furthermore, 99mTc-IL-2 uptake was inversely correlated with duration of disease (r = 0.7; P = 0.03). Systematic pathologic examination of the pancreas of patients with SPIDDM is warranted to resolve the many questions regarding the role of pancreatic inflammation and amylin-positive amyloid deposition in this disorder.4 Between 1982 and 2014, we obtained pancreatic tissue during autopsies of patients with various types of diabetes including SPIDDM, fulminant T1D (FT1D), acute-onset type 1 diabetes (AT1D), autoimmune pancreatitis, and T2D.15,19 The pancreatic ducts of cases with SPIDDM showed marked pancreatic intraepithelial neoplasia (PanIN lesions) (Figure 1A).20,21 PanIN lesions are precancerous pancreatic ductal changes with Kras mutations, which are characterized by tall columnar cells and significant mucinous secretions. Secreted mucin-rich content sometimes builds up around pancreatic ducts and blocks the secretion of pancreatic digestive enzymes. As a result, leaked/digestive enzymes cause cell necrosis and acute inflammation and ultimately lead to obstructive chronic pancreatitis. Pancreatic ducts that are obstructed by mucous plaque become dilated and cystic.20,21 Our previous report16 showed that the dilated and narrowed portions of pancreatic ducts in patients with SPIDDM are linked to three different pathological changes. First, distinct pathologic changes of the pancreas in patients with SPIDDM include persistent insulitis with preserved beta cells (Figure 1B), major histocompatibility complex (MHC) class I hyper-expression of the islet cells, and exocrine pancreatic inflammation related to PanIN lesions.20,21 Chronic inflammatory milieu due to obstructive chronic pancreatitis induces the release of self-antigens from islet cells and the exocrine pancreas and enhance MHC class I expression of beta cells. These inflammatory conditions attract autoreactive T cells to islet beta cells with resulting insulitis, a definitive finding in patients with T1D.22 Second, the presence of pseudoatrophic islet cells (islet cells devoid of beta cells), another hallmark of T1D,22 has been noted in all cases of SPIDDM. This pathologic finding indicates that beta cells are specifically destroyed through an immunologic mechanism, despite extensive exocrine pancreatic inflammation. Third, IAPP amyloid deposition to the islet cells, a pathologic marker of T2D,23 was not observed in the pancreas of patients with SPIDDM, suggesting that the mechanism(s) of beta-cell failure in SPIDDM is/are not the same as in T2D, despite transient clinical phenotypes similar to T2D in patients with SPIDDM. As outlined in Figure 1A, “PanIN lesions are associated with an atrophied, fibrous pancreatic lobe”. Fibrosis in the exocrine pancreas may alternatively be referred to as desmoplastic reaction and it is a result of the activation of the pancreatic stellate cells (PSC) with subsequent extracellular matrix (ECM) production and deposition (Figure 1A). Interestingly it was reported that chronic hyperglycaemia itself may activate PSCs, induce ECM production and therefore may potentially contribute to the development of this pathology.24 Furthermore, Carstensen et al reported that patients with T1DM are at higher risk for pancreatic cancer development, independently from the exogenous insulin dose applied, which potentially also raises a role for chronic hyperglycaemia in the neoplasia risk development.25
Diagnostic Markers, Strategies And Criteria Of SPIDDM
Diagnostic Markers: Islet Cell Autoantibodies
Five islet autoantibodies such as GAD antibody, insulin-associated antigen 2 (IA-2), insulin autoantibodies (IAA), zinc transporter 8 antibodies (ZnT8) and ICA have been detected in patients with LADA. Among these autoantibodies, GADAb is the most common and sensitive marker in patients with SPIDDM and adult-onset AT1D.26–30 In addition, GADAb autoantibodies persist for a long time after disease onset and are not influenced by age at onset. In contrast, the four other islet cell autoantibodies are negatively associated with age at onset. In a Japanese nationwide survey conducted between April 2004 and December 2009, the frequency of patients with SPIDDM among patients with T2D was 10%.31 Patients with SPIDDM showed a significantly higher frequency of GADAb than IA-2Ab or IAA and showed the highest frequency of single positive autoantibodies among GADAb, IA-2Ab, and IAA than patients with AT1D.31 In the Action LADA 7 study, 598 of 6156 patients (9.8%) had any islet cell autoantibody present, and GAD, IA-2, and ZnT8 autoantibodies were identified in 8.8%, 2.3%, and 1.8%, of patients, respectively.32 In that study, 144 of 598 (24.8%) patients had two or more islet cell autoantibodies present.
The association between GADAb titers and further progression of beta-cell dysfunction was studied with a large number of patients with diabetes without insulin treatment for up to 15 years (from 1995 to 2005) using a well-standardized GADAb assay from the Diabetes Antibody Standardization Program 2005.33 Subjects did not require insulin and had a disease duration between 6 months and 5 years. Subjects were divided into those with a high titer of GADAb (10 ≥U/mL), a low titer of GADAb (<10 U/mL), and GADAb-negative T2D. GADAb-positive subjects progressed to the insulin-dependent stage during the follow-up period (Figure 2A). The progression rate was significantly higher in GADAb-positive groups and IA-2Ab or ICA-positive groups than in those with T2D (Figure 2B). GADAb antibodies were a risk factor for insulin-dependency irrespective of titer (hazard ratio [HR]: 4.69–11.21), age of onset <45 years (HR: 2.18), BMI <22 kg/m2 (HR: 2.40), fasting C-peptide <1.0 ng/mL at entry (HR: 4.61), and independent autoantibodies of ICA and IA-2 from GADAb (HR: 3.05–3.90). In contrast, Murao et al34 reported that thyroid autoantibodies, including anti-thyroglobulin antibody (TgAb) or anti-thyroid peroxidase antibody (TPOAb), were independent risk factors for beta cell failure. The association between islet cell autoantibodies and the deterioration of beta-cell function is described later.
Patients with LADA, especially with high GADAb titers, have been shown to have other organ-specific autoantibodies such as thyroid peroxidase (TPO), antiparietal cells, or tissue transglutaminase antibodies.35,36 In particular, more than 20% of the patients with LADA had positive TPO antibodies35,36 suggesting the need for general screening for TPO antibodies in all patients with LADA.
Several, but not all, studies showed bimodal distribution of GADAb in patients with SPIDDM or LADA, suggesting the presence of two distinct forms of the disease in the ~1/10th of patients who are antibody positive at all.32,35,37–39 Compared with the low GADAb titer group, the high GADAb titer group was younger, had a higher HbA1c level, lower BMI, lower prevalence of metabolic syndrome and its components, higher prevalence of other autoantibodies including IA-2 and TPO antibodies, and higher prevalence of high/moderate human leukocyte antigen (HLA) risk genotypes; these findings suggest a greater similarity to AT1D.35,36,38,39 However, these findings cannot rule out the possibility that patients with early-phase AT1D were erroneously included in the study population.35,36,38,39
In addition to the GADAb titer, antibody-binding epitopes of GADAb are associated with the clinical phenotype of diabetes.40–43 For example, the binding of GAD65Ab with N-terminal 83 residues was shown to be inversely correlated with the period in which insulin was not required.42 The associations between GADAb titer or antibody-binding epitopes and beta-cell function are described later.
Positive findings for IA-2 antibodies have different clinical meanings in patients with LADA. Patients with positive findings for IA-2 alone have a clinical phenotype more similar to T2D, whereas patients with positive findings for both IA-2 and GADAb have a clinical phenotype more similar to AT1D.44 The NIRAD study analyzed IA-2 epitope immunoreactivity and showed that the IA-2(256–760) antibodies represent a new sensitive marker for the study of the humoral IA-2 immunoreactivity in patients with LADA.45 In addition, the frequency of IA-2(256–760) antibodies increased with increasing BMI, whereas the frequency of GAD and intracytoplasmic (IC) IA-2IC(605–979) antibodies decreased with increasing BMI.46 However, the clinical utility of IA-2 antibodies is questionable because the prevalence of these antibodies differs by country.47,48
Diagnostic Strategies
Theoretically, all patients with newly diagnosed diabetes, and in particular T1D, should be tested for GADAb autoantibodies because: 1) epidemiologic studies indicate that 2% to 10% of patients with newly diagnosed diabetes show positive findings, which may indicate the presence of SPIDDM, 2) the prevalence of SPIDDM is increasing, and 3) some intervention strategies to slow or stop the decline in beta-cell function that occurs with SPIDDM are available (described below). There is, however, no recommendation to test islet cell autoantibodies, including GADAb, in all patients with newly diagnosed diabetes; this lack of recommendation is likely due to the high costs of testing. The American Diabetes Association’s “Standards of Medical Care in Diabetes 2019” recommends screening for a panel of autoantibodies only in children and adolescents with overweight/obesity in whom the diagnosis of T2D is being considered, in the setting of a research trial, and in first-degree family members of a proband with T1D.49,50
In most clinical settings, islet cell autoantibodies are generally tested only when SPIDDM is suspected. In Japan, the costs for measuring GADAb, IA2Ab, and ICA autoantibodies are covered by the life insurance system.
In addition to patients with newly diagnosed diabetes, there may be a need to test for islet cell autoantibodies in patients diagnosed with T2D who have poor glycemic control. This is because some case reports showed that islet cell autoantibodies became positive in patients with T2D in whom findings were previously negative.51
Diagnostic Criteria Of SPIDDM
Due to the great heterogeneity of SPIDDM or LADA among different ethnic groups and geographic areas, common diagnostic criteria are not specific and may lead to false-positive findings. Recently, the Japan Diabetes Society established diagnostic criteria for SPIDDM9 (Table 4). On the other hand, the Immunology of Diabetes Society (IDS) established diagnostic criteria for LADA in 2005.52 The IDS criteria require; 1) Adult age at onset of diabetes, 2) the presence of circulating islet autoantibodies and 3) lack of a requirement for insulin for at least 6 months after diagnosis. These two criteria are similar in the requirements for positive islet autoantibodies and insulin independency at onset. However, there are at least three differences between these criteria. First, unlike LADA criteria, age of onset of SPIDDM is not restricted to populations older than 35 years.4,6 Second, the Japan Diabetes Society cutoff criteria for duration from the onset of diabetes to insulin-dependency was established based on comparisons with patients with AT1D:53 For patients with AT1D, the duration from onset of diabetes to the insulin-requiring state was <3 months. A diagnosis of SPIDDM can only be made after a 3-month period in which no insulin is required. Third, only GADAb and/or ICA are applied for islet autoantibodies in the Japan Diabetes Society criteria whereas all kinds of islet cell autoantibodies are applied in the Immunology of Diabetes Society criteria.
 |
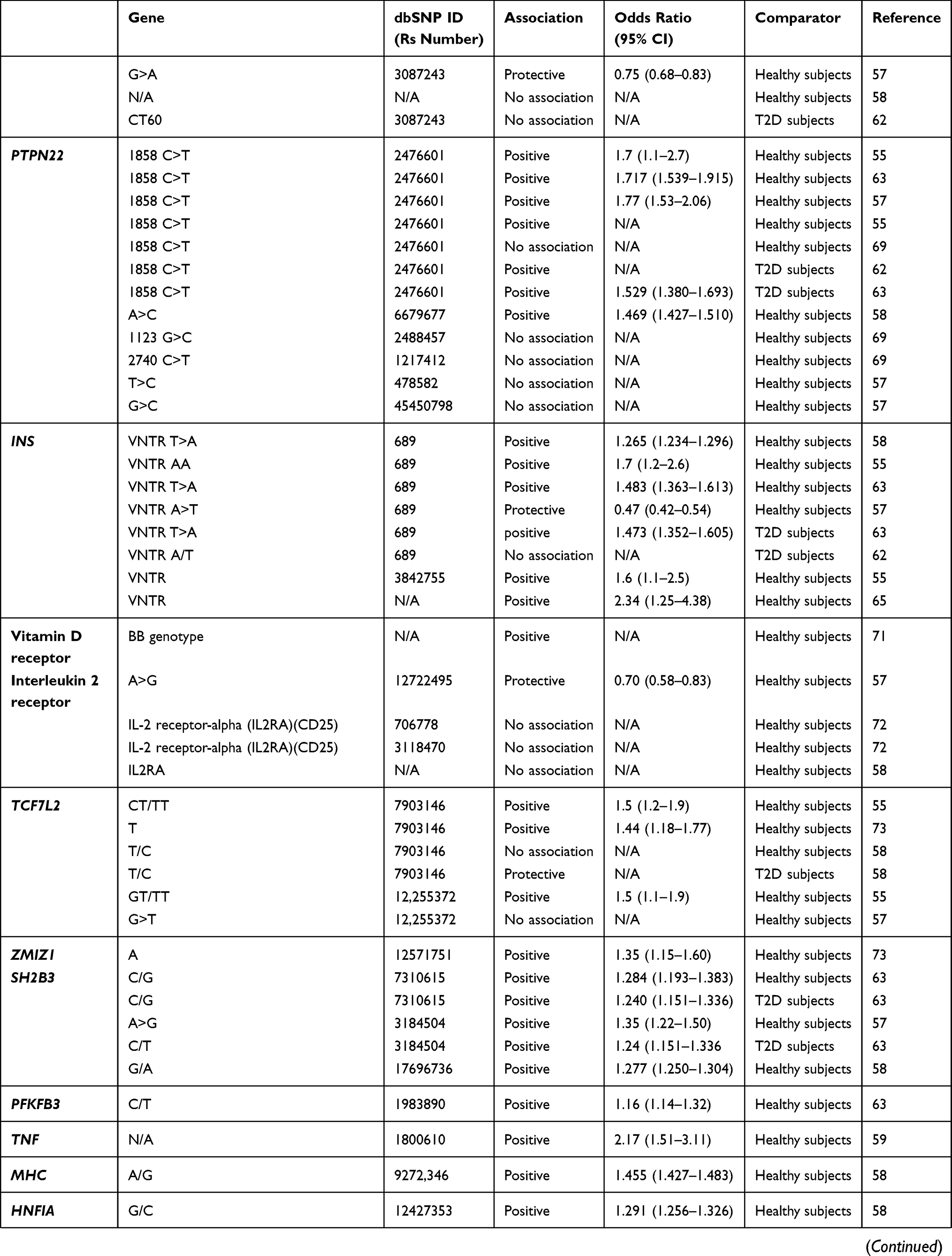 | 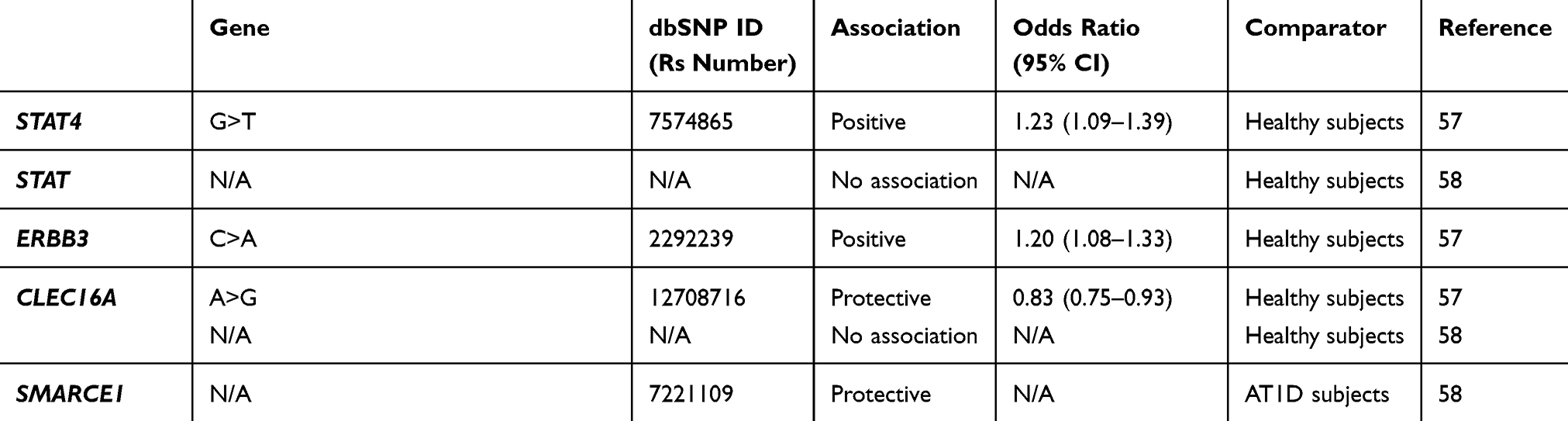
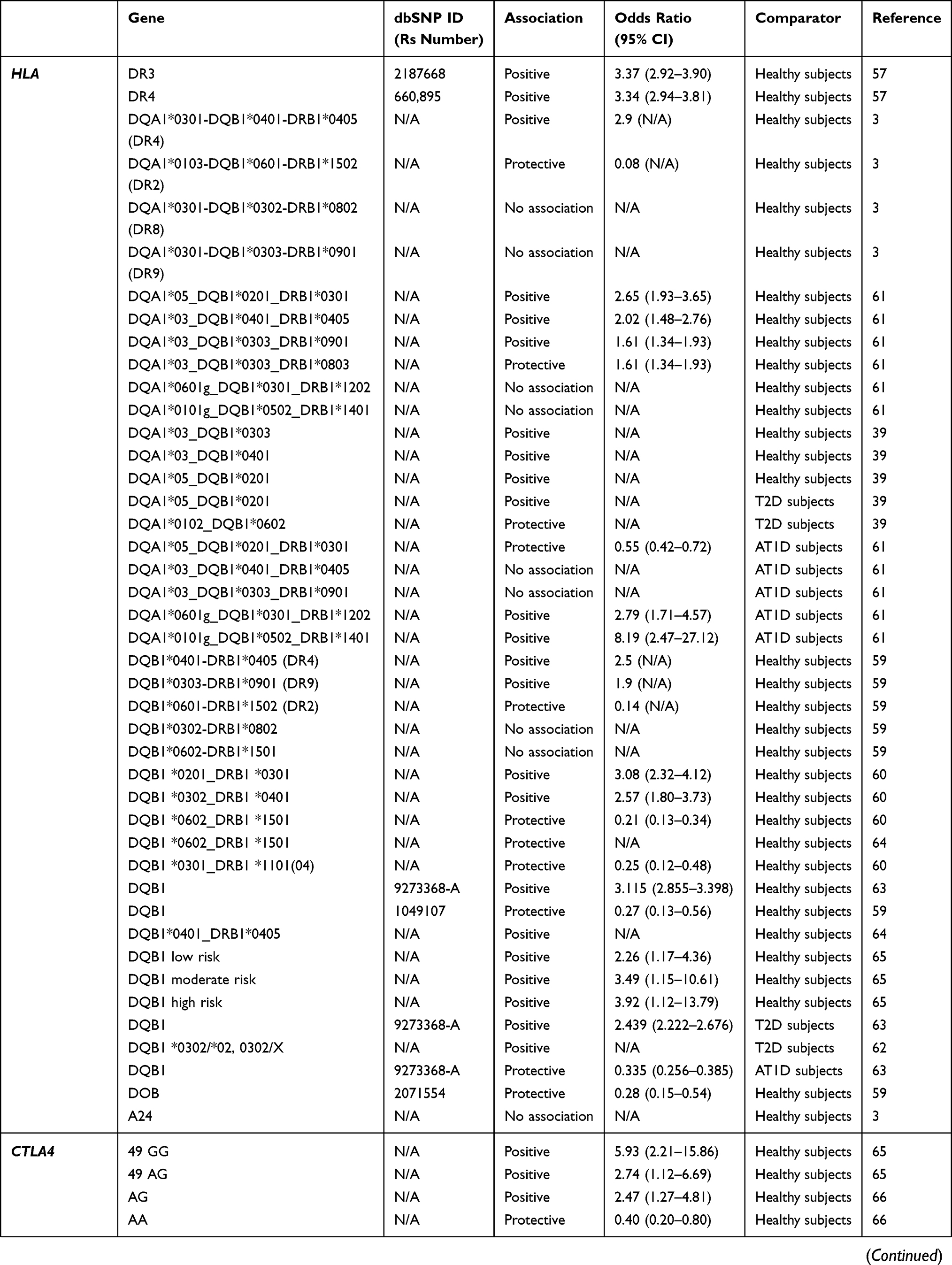
Table 1 Genes Associated Or Not Associated With SPIDDM Or LADA |
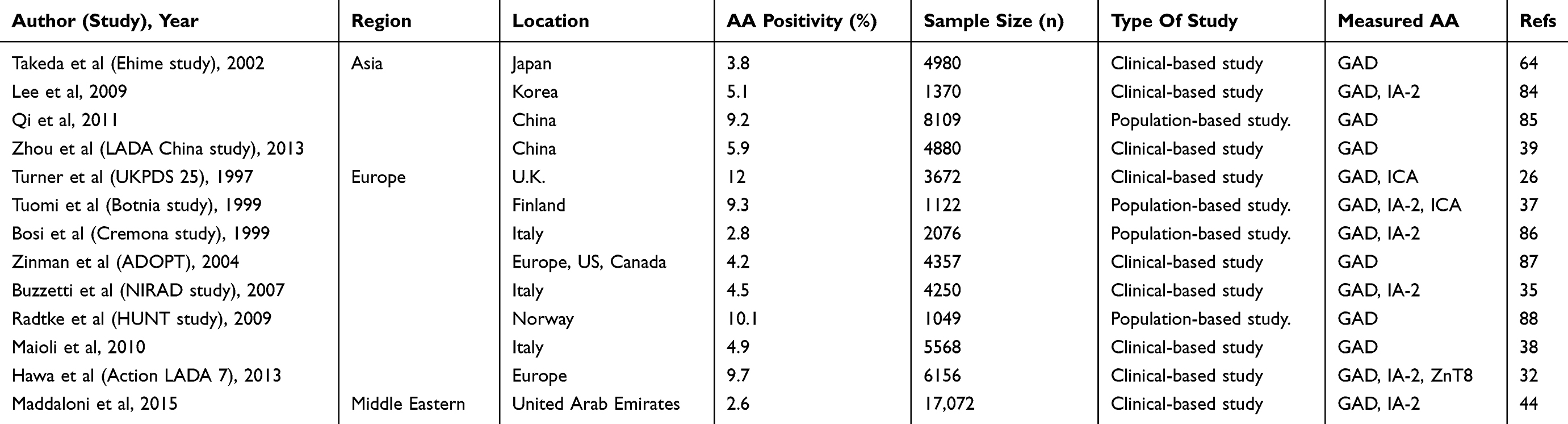 |
Table 2 Prevalence Of Islet Cell Autoantibody Positivity Among Patients With Diabetes |
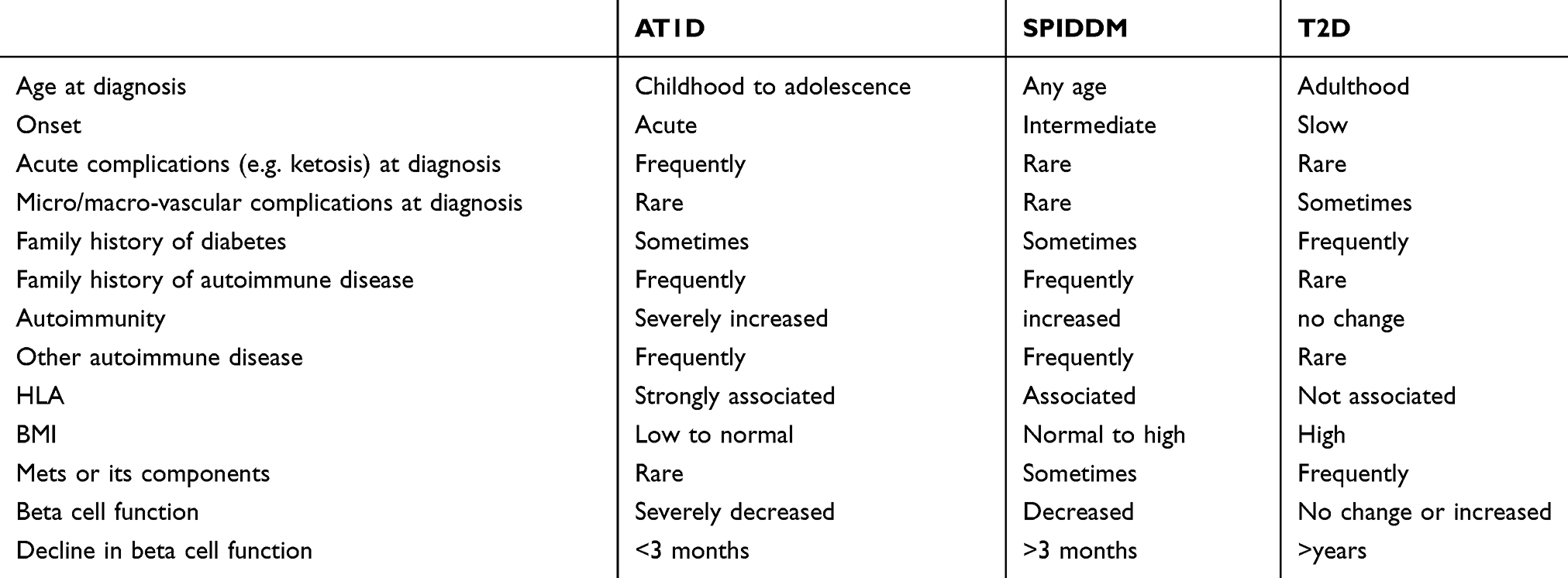 |
Table 3 Clinical Characteristics Of SPIDDM Compared With AT1D And T2D |
 |
Table 4 Diagnostic Criteria For Slowly Progressive Insulin-Dependent Type 1 Diabetes Mellitus (2012) |
Genetics
LADA is reported to share genetic features with both AT1D and T2D, although it is more similar to AT1D54–58 (Table 1). In a population-based study assessing the association between family history of T1D or T2D and LADA, both family history of T1D and T2D were associated with an increased risk for LADA. However, compared with a family history of T2D, a family history of T1D was more strongly associated with higher GADAb antibody levels, lower C-peptide levels, and a lower prevalence of low-risk HLA genotypes.54 In another study that compared the frequencies of several genes associated with AT1D or T2D among patients with LADA, AT1D, T2D, and healthy controls, the frequencies of AT1D-associated genes were higher in patients with LADA compared with patients with T2D or controls, and the frequencies of T2D-associated genes increased to the same extent in patients with LADA or T2D compared with controls.55
Human Leukocyte Antigen (HLA)
In a Japanese population, HLA-DQ haplotypes were examined in patients with AT1D and SPIDDM.3 In patients with AT1D, three haplotypes, DQA1*03:01-DQB1*04:01-DRB1*04:05 (DR4), DQA1*03:01-DQB1*03:02-DRB1*08:02: (DR8), and DQA1*03:01-DQB1*03:03-DRB1*09:01 (DR9) showed an increased prevalence, whereas only DQA1*03:01-DQB1*04:01-DRB1*04:05 (DR4) was significantly increased in patients with SPIDDM. Additionally, DQA1*01:03-DQB1*06:01-DRB1*15:02 (DR2) was decreased in both AT1D and SPIDDM. These results indicate that SPIDDM is related to specific susceptible and protective HLA genotypes, but fewer susceptible HLA genotypes, including increased HLA-DR4 and decreased DR2, are associated with SPIDDM compared with AT1D. We also studied nationwide data from the Japan Diabetes Society involving the increased number of subjects with SPIDDM, AT1D, and FT1D and confirmed previous results.59 That is, a high proportion of patients with AT1D had four types of susceptible haplotype combinations, HLA-DR4/4, -DR9/9, -DR4/9, and -DR4/8, whereas patients with SPIDDM had only two susceptible haplotype combinations, HLA-DR4/4 and DR4/8. These results suggest that the differences in number of susceptible genes influence DR-beta, DQ-alpha, and DQ-beta heterodimer expression encoded by DRB1, DQA1, and DQB1 genes and determine the rate of beta-cell damage in patients with AT1D and SPIDDM. In other words, the number of T1D-related HLA haplotypes determines the clinical subtype of T1D (AT1D or SPIDDM). Another characteristic feature of SPIDDM is a lack of association with class I HLA: HLA-A24.3 HLA-A24 is associated with complete destruction of beta-cell function in patients with AT1D.15 The lack of association of HLA-A24 with complete destruction of beta cells in patients with SPIDDM may be related to a gradually progressive clinical course and preserved beta-cell function in this disorder.
There is an independent and racial association between LADA and HLA haplotypes and susceptible genes associated with AT1D in the Caucasian population. The primary susceptible haplotypes in Caucasian patients with LADA were DRB1 0301_DQB1 0201, and DRB1 0401_DQB1 0302, whereas DRB1 1501_DQB1 0602 had a protective effect.60 The frequencies of these HLA haplotypes, however, were lower than those seen in patients with AT1D37,61 and the frequency of HLA-DQB1 risk genotypes and alleles was significantly lower compared with patients with AT1D diagnosed after the age of 35.62 In the genome-wide association study (GWAS) of LADA, the authors reported the attenuation of the key type 1-associated HLA haplotype (HLA-DQB1, rs9273368-A) frequencies in LADA.63 In addition, the frequency for AT1D protective haplotypes was significantly higher in Caucasian patients with LADA compared with patients with AT1D.61
The association between HLA haplotype and SPIDDM or LADA is also seen in Asian patients. In a study of LADA in a Chinese population, HLA diabetes-susceptible haplotypes were more frequent in patients with LADA (63.9%) compared to patients with T2D (47.1%) and controls (43.2%), whereas HLA diabetes-protective haplotypes was less frequent in patients with LADA (22.8%) compared to patients with T2D (33.3%) and controls (32.7%).39 The contribution of HLA-DRB1-DQA1-DQB1 loci to LADA susceptibility, however, differs from that in Caucasian patients with LADA.61
The presence of AT1D susceptibility or protective HLA haplotypes is associated with clinical characteristics in patients with LADA.35,37,64 The prevalence of high or moderate HLA-risk genotypes was associated with high GADAb titers35,39 and younger age at disease onset,64 whereas protective HLA haplotypes are associated with older age at onset of diabetes, high BMI, resistance to insulin-deficient state, low titer of GADAb, and low frequency of other organ-specific autoantibodies.64 In another study, DR3 was associated with positive findings for GADAb, but absence of IA-2A, and DR4 was associated with positive findings for IA-2A.57
Cytotoxic T Lymphocyte-Associated Antigen 4 (CTLA4)
Several studies showed an association between CTLA4 and LADA,57,65,66 whereas Andersen et al showed that CTLA4 was associated only with AT1D.62 In the 1990s, Van der Auwera et al showed that the presence of a G-containing CTLA4 genotype confers a moderate but significant increased relative risk for insulin-dependent diabetes mellitus that is independent of age.67 That study suggested a weak association between CTLA4 and LADA. In a Japanese population with SPIDDM and other subtypes of T1D, a G-containing genotype was poorly associated with T1D but was associated with autoimmune thyroid disease complicated with T1D.68 In a study of a Caucasian population, Cosentino et al showed that the heterozygous A/G genotype was significantly more frequent among 80 patients with LADA (69%) than among 85 healthy subjects of similar age and geographic location (47%).66
Protein Tyrosine Phosphatase, Non-Receptor 22 Gene (PTPN22)
A systematic and trans-rational association and linkage study including 1690 Japanese samples, 180 Korean samples, and 472 Caucasian samples demonstrated that the minor allele of a single-nucleotide polymorphism (SNP) at nucleotide position 1858 (rs2476601, +1858C>T) was associated with AT1D in Japanese and Caucasian populations.69 In the Japanese population, we can identify five novel SNPs, but not the +1858C>T SNP. Of these two frequent SNPs, −1123G>C, and +2740C>T were in strong linkage disequilibrium (LD) with a combined group consisting of subjects with AT1D and SPIDDM. The −1123G>C promoter SNP was associated with AT1D but not SPIDDM in the Japanese population (odds ratio [OR] = 1.42, 95% confidence interval [CI] =1.07–1.89, P=0.015). This association was also observed in Korean patients with T1D (Mantel–Haenszel χ2 = 6.543, P = 0.0105, combined OR = 1.41, 95% CI = 1.09–1.82). Furthermore, the affected family-based control (AFBAC) association test and the transmission disequilibrium analysis of multiplex families of European descent from the British Diabetes Association (BDA) Warren Repository indicated that the association was stronger for −1123G>C compared to +1858C>T,69 suggesting a weak predisposition for SPIDDM associated with the PTPN22 polymorphism.
In contrast, a Caucasian population of patients with LADA showed an increased frequency of PTPN22 risk genotypes and alleles compared with patients with T2D, whereas the frequency was significantly lower compared with patients with AT1D diagnosed after age 35.62
Insulin Gene (INS)
In a Caucasian population, LADA was independently associated with a variable number of tandem repeats in the insulin gene (INS VNTR).55,57,65 The AA genotype of rs689, referring to the class I allele in INS VNTR, was increased in both patients with classic AT1D and patients with LADA compared with control subjects55 and in Japanese patients with T1D including SPIDDM.70 Similar association with INS rs689 and LADA in case subjects of European ancestry versus population control subjects was found in GWAS.63
Vitamin D Receptor
Vitamin D receptors are expressed on T cells and affect cytokine responses. The BB genotype was higher in patients with SPIDDM and AT1D than in controls.71
Interleukin-2 Receptor-Alpha (IL-2R alpha) Gene
IL-2R alpha genes are expressed on regulatory T cells. We found a strong association with IL-2R alpha genes in patients with AT1D, but not in those with SPIDDM or FT1D.72
Transcription Factor 7 Like 2 (TCF7L2)
An association between LADA and TCF7L2, a known risk allele for T2D, has been reported,55,57,73,74 but findings remain inconclusive.57
Zinc Finger MIZ-Type Containing 1 (ZMIZ1)
One previous report showed that variants in the ZMIZ1 (rs12571751, p = 4.1 × 10−5) loci was strongly associated with LADA.73
SH2B Adaptor Protein 3 (SH2B3)
Two GWAS results showed independent association between SH2B3 rs7310615 and LADA.58,63 In addition, Mishra et al showed that SH2B3 signal remained strongly associated and had stronger OR in the restricted subset of GADA+ IA2A+ LADA cases (OR = 1.48; P = 5.93 x 10−6) compared with whole LADA cases (OR = 1.277; P = 1.10 x 10−5).58
6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3 (PFKFB3)
In the recently published GWAS of LADA, the authors reported a novel signal at the known type 1 diabetes locus harboring PFKFB3, encoding a regulator of glycolysis and insulin signaling in T2D and inflammation and autophagy in autoimmune disease.63
Risk Factors For Progression Of Beta-Cell Dysfunction In SPIDDM
There are five risk factors for loss of beta-cell function in patients with SPIDDM or LADA: 1) numbers of autoantibody for pancreatic beta-cell (multiple antibodies), 2) high GADAb titer, 3) lower BMI, 4) HLA and 5) sulfonylurea treatment after diagnosis.
Number Of Autoantibodies
Several longitudinal studies have shown that patients with SPIDDM or LADA with multiple islet autoantibodies are likely to show a more rapid progression to insulin-dependency, suggesting that the number of positive islet cell autoantibodies reflects the intensity of the autoimmune response to islet cell damage.26,28,75–77 In Japanese patients with SPIDDM, GADAb-positive and ICA-positive findings lead to insulin-dependency in 100% of patients during 5 years, whereas only 47% of the population with GADAb-positive findings alone progressed to insulin-dependency 5 years after diagnosis.10
In the UKPDS 25, GADAb-positive findings alone or both GADAb- and ICA-positive findings lead to the need for insulin therapy for 52% and 68% of patients older than 45, respectively.26
High GADAb Titer
Patients with SPIDDM or LADA with a high GADAb titer have an increased risk for insulin dependency compared with patients with a low GADAb titer (<10 AU/mL: <180 U/mL according to the World Health Organization [WHO] standard samples).26,35,78 In our long-term, follow-up Japanese study (mean follow up, 107 months), 65% of patients with a high GADAb titer (≥10 AU/mL: ≥180 WHO U/mL according to WHO standard samples) progressed to insulin dependency, whereas 24% of patients with a low GADAb titer (<180 WHO U/mL) patients progressed to insulin dependency (Figure 2A).33 Notably, the reduction rate of serum C-peptide response to oral glucose was significantly faster in the high and low GADAb titer groups than in patients with T2D (Figure 2B). In the NIRAD 7 study, 71.1% of patients with a high GADAb titer progressed to insulin dependency, compared with 41.6% of patients with a low GADAb titer and 20.9% of patients with T2D (P<0.0001 for both).76 However, these studies used different GADAb cut-off values. The use of a standardized GADAb titer cut-off value is needed to determine the accuracy of GADAb titers for predicting the progression to beta-cell dysfunction.79
Lower BMI
Lower BMI (<22 kg/m2) is an independent risk factor for progression to insulin-dependency in patients with SPIDDM who do not require insulin.33 The NIRAD study showed that a BMI <25 kg/m2 can identify patients with LADA who will require insulin therapy within 4 years of diagnosis.38 A low BMI is related to decreased pancreatic digestive function, as evidenced by a small atrophic exocrine pancreas80 and low serum levels of pancreatic enzymes in patients with SPIDDM.81 In addition, Japanese individuals with SPIDDM and the T1D-risk HLA (DRB1*04:05-DQB1*04:01) haplotype had a lower BMI.82 Caucasian patients with LADA with a T1D-risk HLA (HLA DR3/4) had a lower BMI than those without the risk HLA phenotype.83 Taken together, the findings of exocrine pancreatic failure in patients with SPIDDM and HLA genes associated with SPIDDM may indicate that HLA genes are related to extensive exocrine advanced pancreatic failure in patients with reduced BMI and SPIDDM.
HLA
In a multivariate analysis, Maioli et al showed that HLA DRB1-DQB1 genotypes in patients at high risk for AT1D had a significant effect on disease progression within 4 years of diagnosis.38
Sulfonylurea Treatment After Diagnosis
In the NIRAD study, treatment with a sulfonylurea in the first year after diagnosis of diabetes was associated with faster progression to insulin dependency.76 In a Tokyo study, patients randomly assigned to a sulfonylurea treatment group had a faster progression to insulin dependency compared with patients treated with insulin.11
Epidemiology
AT1D is the most common type of diabetes in children and adolescents. However, LADA is more common than AT1D in patients older than 30 years who develop diabetes.32,44 A large body of evidence shows that the prevalence of SPIDDM or LADA varies from 2% to 10% (Table 2).26,32,35,37–39,44,64,84–88 This variation is due to several factors such as ethnicity, country, types of autoantibodies assessed, assay methods, sample age, or study type (clinical, registry, or population based). In Japan, the prevalence of SPIDDM ranges from 4% to 10% among patients tentatively diagnosed with non-insulin requiring diabetes in a referral hospital-based study (Table 2).31 In general, the prevalence of SPIDDM or LADA is similar in Europe, northern America, and Asia. It is noteworthy that the incidence of SPIDDM or LADA in Asian countries, including Japan, is similar to that in northern Europe. In contrast, the annual incidence of AT1D in Japan is extremely low (2/104). The comparable incidence of LADA with a Caucasian population may suggest that environmental factors rather than a genetic predisposition have a larger influence on the occurrence of SPIDDM or LADA.
Clinical Features Of SPIDDM Or LADA Compared With AT1D And T2D
Table 3 shows clinical characteristics of SPIDDM or LADA compared with AT1D and T2D. These three subtypes of diabetes, however, form a continuum of varying severity in terms of immune and metabolic dysfunction,89 and the clinical characteristics of SPIDDM or LADA are extremely broad.
Asian patients with LADA have a lower BMI compared with Caucasian patients with LADA. In a joint collaborative project between Italy and Korea, Caucasian patients with LADA had a significantly higher BMI than Asian patients with LADA (27 ± 5.1 kg/m2 versus 25.3 ± 3.3 kg/m2, respectively, p <0.01) although the mean age and HbA1c values were nearly identical.90 Similar lower BMI values were observed in the study from Japan and China.39,64
Among patients with SPIDDM or LADA, there are some differences in clinical characteristics between high-GADAb titer patients and low-GADAb titer patients.26,39,87 Compared with patients with low GADAb titers, patients with high GADAb titers had more prominent traits of insulin deficiency and more severe autoimmunity, resulting in higher A1C levels, lower BMI, and a lower prevalence of metabolic syndrome and its components.
Mirco/Macro Complications
To date, the risk of micro and macrovascular complications in patients with SPIDDM or LADA compared with patients with AT1D, patients with T2D, or healthy controls are not well known, as almost all studies are cross-sectional or statistically underpowered.
In terms of microvascular complications, a few studies showed a lower prevalence of diabetic kidney disease in patients with LADA compared with patients with T2D. For example, the Fremantle Diabetes Study showed that microalbuminuria was less frequent both at baseline and during follow-up in the LADA subgroup with relatively shorter duration of disease compared with the GAD antibody-negative patients after adjustment for other risk factors.91 A Chinese study showed that the prevalence of diabetic retinopathy and diabetic nephropathy was significantly lower in patients with LADA compared with patients with T2D.92 These findings could be explained by the fact that patients with T2D may go undiagnosed for a longer period than patients with SPIDDM or LADA and thus be more likely to experience complications of chronic hyperglycemia.93,94 No study has shown statistically significant differences in the prevalence of neuropathy between patients with LADA and T2D after adjusting for several risk factors, although some studies showed a numerically higher prevalence of neuropathy in patients with LADA.91,95–97 A study assessing nerve conduction parameters showed more frequent peripheral nerve dysfunction in patients with LADA compared with patients with T2D in limited subgroups.92
The prevalence of macrovascular disease in patients with SPIDDM or LADA is similar to that of patients with T2D but higher than in healthy individuals.91,98 Theoretically, patients with SPIDDM may have lower macrovascular complications compared with patients with T2D because patients with SPIDDM have a better lipid profile, lower blood pressure, and lower BMI. On the other hand, patients with SPIDDM have worse glycemic control compared with patients with T2D due to reduced beta-cell function and a higher risk for hypoglycemia due to the greater number of patients treated with insulin. In a follow up to the HUNT study, increased risks for all-cause mortality (HR, 1.55), cardiovascular disease (1.87), and ischemic heart disease (2.39) were observed in patients with LADA compared with individuals without diabetes. Patients with T2D had similarly high risks compared with those without diabetes.98 The Fremantle Diabetes Study showed that GADAb-positive patients with LADA had a similar prevalence and incidence of coronary heart disease and cerebrovascular disease and cardiac and all-cause mortality compared with the GADAb-negative patients.91
Prevention Strategies And Clinical Options For SPIDDM
Preservation of beta-cell function is the principal treatment strategy for SPIDDM or LADA. In SPIDDM, residual beta-cell function is particularly important for the prevention of diabetic complications99,100 because beta cells have been shown to be preserved in patients with SPIDDM even after 10 years.15 Our prospective study, published in 1996,101 demonstrated that small doses of insulin before the pre-insulin requiring stage of SPIDDM were associated with seroconversion of ICA from positive to negative findings, suggesting that some kind of immunologic modulation is associated with low insulin doses. An extended multicenter, randomized, non-blinded clinical study (Tokyo study) of 60 GADAb-positive non-insulin requiring patients with diabetes for ≤5 years reported that patients treated with insulin early showed greater preservation of the C-peptide response than patients treated with a sulfonylurea.11 In addition, a low-GADAb titer and preserved C-peptide response at entry were independent markers for the effective prevention of insulin dependency. In our case report, changes in treatment from insulin to an alpha-glucosidase inhibitor resulted in increases in the GADAb titer, suggesting that activation of an immunologic assault to beta cells may contribute to the development of SPIDDM.102 The Nationwide study demonstrated that seroconversion of GADAb from negative to positive and an increase in HbA1c level in patients with T2D occurred more frequently in non-insulin-treated patients.51 A meta-analysis recommended early insulin treatment instead of sulfonylureas as being preferable to preserve residual beta-cell function in patients with LADA.103
DPP-4 inhibitors are effective in preventing loss of beta-cell function for long-term periods in patients with SPIDDM or LADA.14 It was reported that the fasting serum DPP-4 activity independently from the ICA or GAD status was increased in patients with T1DM in 2011.104 Subsequently others found that DPP-4 inhibitors are effective in preventing loss of beta-cell function for long-term periods in patients with SPIDDM or LADA. Our 4-year, open-label, randomized control trial (SPAN-S study) demonstrated that sitagliptin treatment was more potent than insulin treatment at preventing beta-cell deterioration in patients with SPIDDM (Figure 3).14 In this study, we compared our present data with those in the Tokyo Study as a historical control. This study indicated that treatment by sitagliptin significantly influenced the longitudinal changes of the C-peptide responses to the oral glucose tolerance test with a more increased direction than insulin and sulfonylurea. Additional studies have also reported preservation of beta cell function in patients with SPIDDM or LADA treated with DPP-4 inhibitors, including sitagliptin for 1 year,13 linagliptin,105 and saxagliptin.106 The possible mechanisms for the effect of DPP-4 inhibitors may be related to findings in NOD mice models in which DPP4-inhibitors decrease CD4+ T cell migration107 and increase CD4(+)CD25(+)FoxP3(+) regulatory T cells.108
Candidates for combination treatment for preserving beta cell function in SPIDDM include metformin,12 which changes gut microbiota profiles, and pioglitazone, although results are inconclusive.109,110
Conclusion
Prevention of slowly progressive beta-cell failure in patients with SPIDDM is a promising target for intervention. Addressing the unique inflammatory changes associated with PanIN lesions of the pancreatic duct in patients with SPIDDM may also be an attractive target to slow or stop the progression of beta-cell dysfunction in these patients.
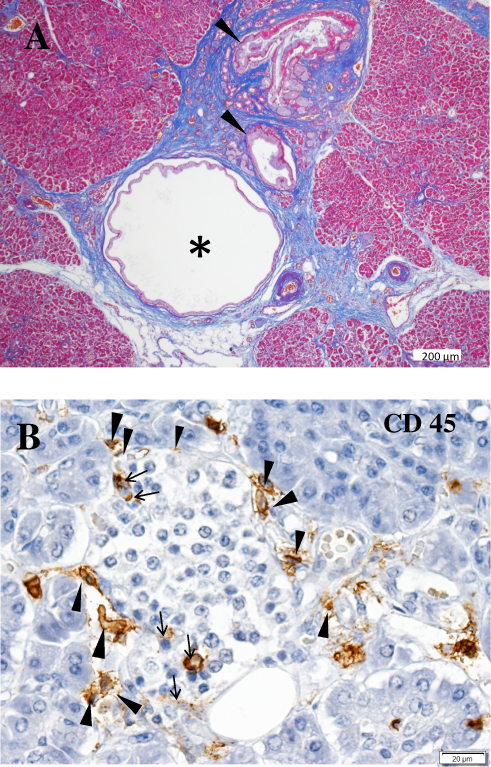 |
Figure 1 Characteristic features of the pancreas in patients with SPIDDM. (A) Pancreatic intraductal papillous neoplasia (PanIN) lesion frequently observed in the patients with SPIDDM. Azan staining. PanIN lesion of pancreatic ducts (arrowheads) is associated with an atrophied, fibrous pancreatic lobe (stained blue), a dilated pancreatic duct (asterisk) and extensive mononuclear cell infiltration. (B) Insulitis in a patient with SPIDDM. CD45 (leukocyte common antigen) + mononuclear cells (MNCs) are infiltrated around (arrowheads) and into the islet cells (arrows). Most of the CD45+ cells are CD8+ T cells and CD68+ macrophages. Aida K, Fukui T, Jimbo E, et al. Distinct inflammatory changes of the pancreas of slowly progressive insulin-dependent (Type 1) Diabetes. Pancreas. 2018;47(9):1101–1109.21 |
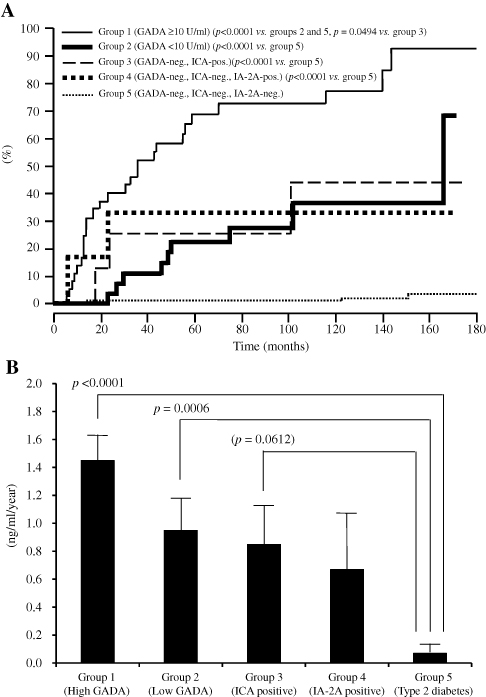 |
Figure 2 (A) Longitudinal changes of patients who progressed from the non-insulin-requiring stage to an insulin-dependent stage (IDS) in five study groups. IDS is defined as the state when integrated values of serum C-peptide levels at 0, 30, 60, 90, and 120 min during a 100-g oral glucose tolerance test (sigma C-peptide) reach <5 ng/mL. (B) Reduction rate of sigma C-peptide in group 1 (patients with GADAb titer ≥10 AU/mL [≥180 WHO U/mL]), group 2 (patients with GADAb titer <10 AU/mL [<180 WHO U/mL]), group 3 (patients without GADAb and with islet cell antibodies [ICA], group 4 (patients without GADAb or ICA antibodies and with insulin-associated antigen 2 antibodies [IA-2A]), and group 5 (patients with T2D without GADAb, ICA, or IA-2A antibodies).Note: Copyright © 2015. Japan Diabetes Society. Reproduced from Tanaka S, Okubo M, Nagasawa K, et al. Predictive value of titer of GAD antibodies for further progression of beta cell dysfunction in slowly progressive insulin-dependent (type 1) diabetes (SPIDDM). Diabetol Int. 2016;7(1):42–52.33 |
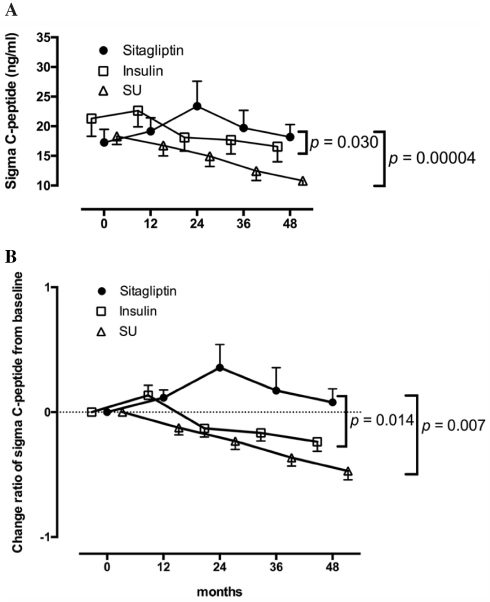 |
Figure 3 Longitudinal changes in the C-peptide response to the oral glucose tolerance test for 48 months in patients treated with sitagliptin in the Tokyo study. Patients with 48 months of follow-up are shown. Data are expressed as the mean ± SEM. In both the ∑C-peptide values (A) and change ratios from baseline (B), a repeated-measures analysis of variance revealed a significant interaction between time and treatment assignment (sitagliptin or insulin; p = 0.030 and p = 0.014, respectively) as well as between time and treatment assignment (sitagliptin or sulfonylurea; p = 0.00004 and p = 0.007, respectively) in the longitudinal changes. Note: Reproduced from Awata T, Shimada A, Maruyama T, et al. Possible Long-Term Efficacy of Sitagliptin, a Dipeptidyl Peptidase-4 Inhibitor, for Slowly Progressive Type 1 Diabetes (SPIDDM) in the Stage of Non-Insulin Dependency: An Open-Label Randomized Controlled Pilot Trial (SPAN-S). Diabetes Ther. 2017;8(5):1123-1134. Creative commons license and disclaimer available from: http://creativecommons.org/licenses/by/4.0/legalcode.14 |
Acknowledgments
The authors would like to thank the late Taro Maruyama, Saitama Social Insurance Hospital, for his generous assistance in obtaining the study samples. This work was supported by a research grant from Japan Society for the Promotion of Science KAKENHI (grant 2459319).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kobayashi T, Itoh T, Kosaka K, Sato K, Tsuji K. Time course of islet cell antibodies and beta-cell function in non-insulin-dependent stage of type I diabetes. Diabetes. 1987;36(4):510–517. doi:10.2337/diab.36.4.510
2. Kobayashi T, Nakanishi K, Sugimoto T, et al. Maleness as risk factor for slowly progressive IDDM. Diabetes Care. 1989;12(1):7–11. doi:10.2337/diacare.12.1.7
3. Kobayashi T, Tamemoto K, Nakanishi K, et al. Immunogenetic and clinical characterization of slowly progressive IDDM. Diabetes Care. 1993;16(5):780–788. doi:10.2337/diacare.16.5.780
4. Kobayashi T. Subtype of insulin-dependent diabetes mellitus (IDDM) in Japan: slowly progressive IDDM–the clinical characteristics and pathogenesis of the syndrome. Diabetes Res Clin Pract. 1994;24(Suppl):S95–S99. doi:10.1016/0168-8227(94)90234-8
5. Stenstrom G, Gottsater A, Bakhtadze E, Berger B, Sundkvist G. Latent autoimmune diabetes in adults: definition, prevalence, beta-cell function, and treatment. Diabetes. 2005;54(Suppl 2):S68–S72. doi:10.2337/diabetes.54.suppl_2.S68
6. Leslie RD, Palmer J, Schloot NC, Lernmark A. Diabetes at the crossroads: relevance of disease classification to pathophysiology and treatment. Diabetologia. 2016;59(1):13–20. doi:10.1007/s00125-015-3789-z
7. Kobayashi T, Tanaka S, Harii N, et al. Immunopathological and genetic features in slowly progressive insulin-dependent diabetes mellitus and latent autoimmune diabetes in adults. Ann N Y Acad Sci. 2006;1079:60–66. doi:10.1196/annals.1375.009
8. Pozzilli P, Guglielmi C. Immunomodulation for the prevention of SPIDDM and LADA. Ann N Y Acad Sci. 2006;1079:90–98. doi:10.1196/annals.1375.012
9. Tanaka S, Ohmori M, Awata T, et al. Diagnostic criteria for slowly progressive insulin-dependent (type 1) diabetes mellitus (SPIDDM) (2012): report by the Committee on Slowly Progressive Insulin-Dependent (Type 1) Diabetes Mellitus of the Japan Diabetes Society. Diabetol Int. 2015;6(1):1–7.
10. Kobayashi T, Nakanishi K, Okubo M, Murase T, Kosaka K. GAD antibodies seldom disappear in slowly progressive IDDM. Diabetes Care. 1996;19(9):1031. doi:10.2337/diacare.19.9.1031a
11. Maruyama T, Tanaka S, Shimada A, et al. Insulin intervention in slowly progressive insulin-dependent (type 1) diabetes mellitus. J Clin Endocrinol Metab. 2008;93(6):2115–2121. doi:10.1210/jc.2007-2267
12. Hirata T, Shimada A, Morimoto J, Maruyama T. Slowly progressive type 1 diabetes treated with metformin for five years after onset. Intern Med. 2013;52(23):2635–2637. doi:10.2169/internalmedicine.52.9522
13. Zhao Y, Yang L, Xiang Y, et al. Dipeptidyl peptidase 4 inhibitor sitagliptin maintains beta-cell function in patients with recent-onset latent autoimmune diabetes in adults: one year prospective study. J Clin Endocrinol Metab. 2014;99(5):E876–E880. doi:10.1210/jc.2013-3633
14. Awata T, Shimada A, Maruyama T, et al. Possible long-term efficacy of sitagliptin, a dipeptidyl peptidase-4 inhibitor, for Slowly Progressive Type 1 Diabetes (SPIDDM) in the stage of non-insulin-dependency: an open-label randomized controlled pilot trial (SPAN-S). Diabetes Ther. 2017;8(5):1123–1134. doi:10.1007/s13300-017-0299-7
15. Nakanishi K, Kobayashi T, Miyashita H, et al. Relationships among residual beta cells, exocrine pancreas, and islet cell antibodies in insulin-dependent diabetes mellitus. Metabolism. 1993;42(2):196–203. doi:10.1016/0026-0495(93)90035-M
16. Nakanishi K, Kobayashi T, Miyashita H, et al. Exocrine pancreatic ductograms in insulin-dependent diabetes mellitus. Am J Gastroenterol. 1994;89(5):762–766.
17. Shimada A, Imazu Y, Morinaga S, et al. T-cell insulitis found in anti-GAD65+ diabetes with residual beta-cell function. A case report. Diabetes Care. 1999;22(4):615–617. doi:10.2337/diacare.22.4.615
18. Signore A, Capriotti G, Chianelli M, et al. Detection of insulitis by pancreatic scintigraphy with 99mTc-labeled IL-2 and MRI in patients with LADA (Action LADA 10). Diabetes Care. 2015;38(4):652–658. doi:10.2337/dc14-0580
19. Kobayashi TNK, Sugimoto T, Murase T, Kosaka K. Histopathological changes of the pancreas in islet cell antibodies (ICA)-positive subjects before and the clinical onset of insulin-dependent diabetes melitus (IDDM). Diabetes. 1988;37:24A.
20. Kobayashi T, Aida K, Fukui T, et al. Pancreatic ductal hyperplasia/dysplasia with obstructive chronic pancreatitis: an association with reduced pancreatic weight in type 1 diabetes. Diabetologia. 2016;59(4):865–867. doi:10.1007/s00125-016-3867-x
21. Aida K, Fukui T, Jimbo E, et al. Distinct inflammatory changes of the pancreas of slowly progressive insulin-dependent (Type 1) Diabetes. Pancreas. 2018;47(9):1101–1109. doi:10.1097/MPA.0000000000001144
22. Campbell-Thompson ML, Atkinson MA, Butler AE, et al. The diagnosis of insulitis in human type 1 diabetes. Diabetologia. 2013;56(11):2541–2543. doi:10.1007/s00125-013-3043-5
23. Sugawara K, Kobayashi T, Nakanishi K, et al. Marked islet amyloid polypeptide-positive amyloid deposition: a possible cause of severely insulin-deficient diabetes mellitus with atrophied exocrine pancreas. Pancreas. 1993;8(3):312–315. doi:10.1097/00006676-199305000-00005
24. Kiss K, Baghy K, Spisak S, et al. Chronic hyperglycemia induces trans-differentiation of human pancreatic stellate cells and enhances the malignant molecular communication with human pancreatic cancer cells. PloS One. 2015;10(5):e0128059. doi:10.1371/journal.pone.0128059
25. Carstensen B, Read SH, Friis S, et al. Cancer incidence in persons with type 1 diabetes: a five-country study of 9000 cancers in type 1 diabetic individuals. Diabetologia. 2016;59(5):980–988. doi:10.1007/s00125-016-3884-9
26. Turner R, Stratton I, Horton V, et al. UKPDS 25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction of insulin requirement in type 2 diabetes. UK Prospective Diabetes Study Group. Lancet. 1997;350(9087):1288–1293. doi:10.1016/S0140-6736(97)03062-6
27. Graham J, Hagopian WA, Kockum I, et al. Genetic effects on age-dependent onset and islet cell autoantibody markers in type 1 diabetes. Diabetes. 2002;51(5):1346–1355. doi:10.2337/diabetes.51.5.1346
28. Lampasona V, Petrone A, Tiberti C, et al. Zinc transporter 8 antibodies complement GAD and IA-2 antibodies in the identification and characterization of adult-onset autoimmune diabetes: Non Insulin Requiring Autoimmune Diabetes (NIRAD) 4. Diabetes Care. 2010;33(1):104–108. doi:10.2337/dc08-2305
29. Kawasaki E. Type 1 diabetes and autoimmunity. Clin Pediatr Endocrinol. 2014;23(4):99–105. doi:10.1297/cpe.23.99
30. Genovese S, Bazzigaluppi E, Goncalves D, et al. Clinical phenotype and beta-cell autoimmunity in Italian patients with adult-onset diabetes. Eur J Endocrinol. 2006;154(3):441–447. doi:10.1530/eje.1.02115
31. Tanaka S, Awata T, Shimada A. Clinical characteristics of slowly progressive insulin-dependent (type 1) diabetes mellitus (SPIDDM): 1st subcommittee report on SPIDDM, committee on type 1 diabetes, Japan Diabetes Society. J Japan Diab Soc. 2011;54(1):65–75.
32. Hawa MI, Kolb H, Schloot N, et al. Adult-onset autoimmune diabetes in Europe is prevalent with a broad clinical phenotype: action LADA 7. Diabetes Care. 2013;36(4):908–913. doi:10.2337/dc12-0931
33. Tanaka S, Okubo M, Nagasawa K, et al. Predictive value of titer of GAD antibodies for further progression of beta cell dysfunction in slowly progressive insulin-dependent (type 1) diabetes (SPIDDM). Diabetol Int. 2016;7(1):42–52. doi:10.1007/s13340-015-0211-5
34. Murao S, Kondo S, Ohashi J, et al. Anti-thyroid peroxidase antibody, IA-2 antibody, and fasting C-peptide levels predict beta cell failure in patients with latent autoimmune diabetes in adults (LADA) – a 5-year follow-up of the Ehime study. Diabetes Res Clin Pract. 2008;80(1):114–121. doi:10.1016/j.diabres.2008.01.024
35. Buzzetti R, Di Pietro S, Giaccari A, et al. High titer of autoantibodies to GAD identifies a specific phenotype of adult-onset autoimmune diabetes. Diabetes Care. 2007;30(4):932–938. doi:10.2337/dc06-1696
36. Zampetti S, Capizzi M, Spoletini M, et al. GADA titer-related risk for organ-specific autoimmunity in LADA subjects subdivided according to gender (NIRAD study 6). J Clin Endocrinol Metab. 2012;97(10):3759–3765. doi:10.1210/jc.2012-2037
37. Tuomi T, Carlsson A, Li H, et al. Clinical and genetic characteristics of type 2 diabetes with and without GAD antibodies. Diabetes. 1999;48(1):150–157. doi:10.2337/diabetes.48.1.150
38. Maioli M, Pes GM, Delitala G, et al. Number of autoantibodies and HLA genotype, more than high titers of glutamic acid decarboxylase autoantibodies, predict insulin dependence in latent autoimmune diabetes of adults. Eur J Endocrinol. 2010;163(4):541–549. doi:10.1530/EJE-10-0427
39. Zhou Z, Xiang Y, Ji L, et al. Frequency, immunogenetics, and clinical characteristics of latent autoimmune diabetes in China (LADA China study): a nationwide, multicenter, clinic-based cross-sectional study. Diabetes. 2013;62(2):543–550. doi:10.2337/db12-0207
40. Falorni A, Gambelunghe G, Forini F, et al. Autoantibody recognition of COOH-terminal epitopes of GAD65 marks the risk for insulin requirement in adult-onset diabetes mellitus. J Clin Endocrinol Metab. 2000;85(1):309–316. doi:10.1210/jcem.85.1.6301
41. Achenbach P, Hawa MI, Krause S, et al. Autoantibodies to N-terminally truncated GAD improve clinical phenotyping of individuals with adult-onset diabetes: action LADA 12. Diabetologia. 2018;61(7):1644–1649. doi:10.1007/s00125-018-4605-3
42. Kobayashi T, Tanaka S, Okubo M, Nakanishi K, Murase T, Lernmark A. Unique epitopes of glutamic acid decarboxylase autoantibodies in slowly progressive type 1 diabetes. J Clin Endocrinol Metab. 2003;88(10):4768–4775. doi:10.1210/jc.2002-021529
43. Jin P, Huang G, Lin J, Luo S, Zhou Z. Epitope analysis of GAD65 autoantibodies in adult-onset type 1 diabetes and latent autoimmune diabetes in adults with thyroid autoimmunity. Acta Diabetologica. 2011;48(2):149–155. doi:10.1007/s00592-010-0250-0
44. Maddaloni E, Lessan N, Al Tikriti A, Buzzetti R, Pozzilli P, Barakat MT. Latent autoimmune diabetes in adults in the United Arab Emirates: clinical features and factors related to insulin-requirement. PloS One. 2015;10(8):e0131837. doi:10.1371/journal.pone.0131837
45. Tiberti C, Giordano C, Locatelli M, et al. Identification of tyrosine phosphatase 2(256-760) construct as a new, sensitive marker for the detection of islet autoimmunity in type 2 diabetic patients: the non-insulin requiring autoimmune diabetes (NIRAD) study 2. Diabetes. 2008;57(5):1276–1283. doi:10.2337/db07-0874
46. Buzzetti R, Spoletini M, Zampetti S, et al. Tyrosine phosphatase-related islet antigen 2(256-760) autoantibodies, the only marker of islet autoimmunity that increases by increasing the degree of BMI in obese subjects with type 2 diabetes. Diabetes Care. 2015;38(3):513–520. doi:10.2337/dc14-1638
47. Mishra R, Hodge KM, Cousminer DL, Leslie RD, Grant SFA. A global perspective of latent autoimmune diabetes in adults. Trends Endocrinol Metab. 2018;29(9):638–650. doi:10.1016/j.tem.2018.07.001
48. Park Y, Wintergerst KA, Zhou Z. Clinical heterogeneity of type 1 diabetes (T1D) found in Asia. Diabetes Metab Res Rev. 2017;33:7. doi:10.1002/dmrr.v33.7
49. American Diabetes Association. Classification and diagnosis of diabetes: standards of medical care in diabetes-2019. Diabetes Care. 2019;42(Suppl 1):S13–S28. doi:10.2337/dc19-S002
50. American Diabetes Association. Children and adolescents: standards of medical care in diabetes-2019. Diabetes Care. 2019;42(Suppl 1):S148–S164. doi:10.2337/dc19-S013
51. Oikawa Y, Shimada A, Awata T, et al. Clinical features of cases of seroconversion of anti-glutamic acid decarboxylase antibody during the clinical course of type 2 diabetes: a nationwide survey in Japan. Diabetol Int. 2017;8(3):306–315. doi:10.1007/s13340-017-0312-4
52. Fourlanos S, Dotta F, Greenbaum CJ, et al. Latent autoimmune diabetes in adults (LADA) should be less latent. Diabetologia. 2005;48(11):2206–2212. doi:10.1007/s00125-005-1960-7
53. Kawasaki E, Maruyama T, Imagawa A, et al. Diagnostic criteria for acute-onset type 1 diabetes mellitus (2012): report of the Committee of Japan Diabetes Society on the research of fulminant and acute-onset type 1 diabetes mellitus. J Diabetes Invest. 2014;5(1):115–118. doi:10.1111/jdi.12119
54. Hjort R, Alfredsson L, Andersson T, et al. Family history of type 1 and type 2 diabetes and risk of latent autoimmune diabetes in adults (LADA). Diabetes Metab. 2017;43(6):536–542. doi:10.1016/j.diabet.2017.05.010
55. Cervin C, Lyssenko V, Bakhtadze E, et al. Genetic similarities between latent autoimmune diabetes in adults, type 1 diabetes, and type 2 diabetes. Diabetes. 2008;57(5):1433–1437. doi:10.2337/db07-0299
56. Pettersen E, Skorpen F, Kvaloy K, Midthjell K, Grill V. Genetic heterogeneity in latent autoimmune diabetes is linked to various degrees of autoimmune activity: results from the Nord-Trondelag Health Study. Diabetes. 2010;59(1):302–310. doi:10.2337/db09-0923
57. Howson JM, Rosinger S, Smyth DJ, Boehm BO, Todd JA. Genetic analysis of adult-onset autoimmune diabetes. Diabetes. 2011;60(10):2645–2653. doi:10.2337/db11-0364
58. Mishra R, Chesi A, Cousminer DL, et al. Relative contribution of type 1 and type 2 diabetes loci to the genetic etiology of adult-onset, non-insulin-requiring autoimmune diabetes. BMC Medicine. 2017;15(1):88. doi:10.1186/s12916-017-0846-0
59. Kawabata Y, Ikegami H, Awata T, et al. Differential association of HLA with three subtypes of type 1 diabetes: fulminant, slowly progressive and acute-onset. Diabetologia. 2009;52(12):2513–2521. doi:10.1007/s00125-009-1539-9
60. Desai M, Zeggini E, Horton VA, et al. An association analysis of the HLA gene region in latent autoimmune diabetes in adults. Diabetologia. 2007;50(1):68–73. doi:10.1007/s00125-006-0513-z
61. Luo S, Lin J, Xie Z, et al. HLA genetic discrepancy between latent autoimmune diabetes in adults and type 1 diabetes: LADA China study no. 6. J Clin Endocrinol Metab. 2016;101(4):1693–1700. doi:10.1210/jc.2015-3771
62. Andersen MK, Lundgren V, Turunen JA, et al. Latent autoimmune diabetes in adults differs genetically from classical type 1 diabetes diagnosed after the age of 35 years. Diabetes Care. 2010;33(9):2062–2064. doi:10.2337/dc09-2188
63. Cousminer DL, Ahlqvist E, Mishra R, et al. First genome-wide association study of latent autoimmune diabetes in adults reveals novel insights linking immune and metabolic diabetes. Diabetes Care. 2018;41(11):2396–2403. doi:10.2337/dc18-1032
64. Takeda H, Kawasaki E, Shimizu I, et al. Clinical, autoimmune, and genetic characteristics of adult-onset diabetic patients with GAD autoantibodies in Japan (Ehime Study). Diabetes Care. 2002;25(6):995–1001. doi:10.2337/diacare.25.6.995
65. Haller K, Kisand K, Pisarev H, et al. Insulin gene VNTR, CTLA-4 +49A/G and HLA-DQB1 alleles distinguish latent autoimmune diabetes in adults from type 1 diabetes and from type 2 diabetes group. Tissue Antigens. 2007;69(2):121–127. doi:10.1111/tan.2007.69.issue-2
66. Cosentino A, Gambelunghe G, Tortoioli C, Falorni A. CTLA-4 gene polymorphism contributes to the genetic risk for latent autoimmune diabetes in adults. Ann N Y Acad Sci. 2002;958:337–340. doi:10.1111/j.1749-6632.2002.tb03000.x
67. Van der Auwera BJ, Vandewalle CL, Schuit FC, et al. CTLA-4 gene polymorphism confers susceptibility to insulin-dependent diabetes mellitus (IDDM) independently from age and from other genetic or immune disease markers. The Belgian diabetes registry. Clin Experiment Immunol. 1997;110(1):98–103. doi:10.1111/cei.1997.110.issue-1
68. Ikegami H, Awata T, Kawasaki E, et al. The association of CTLA4 polymorphism with type 1 diabetes is concentrated in patients complicated with autoimmune thyroid disease: a multicenter collaborative study in Japan. J Clin Endocrinol Metab. 2006;91(3):1087–1092. doi:10.1210/jc.2005-1407
69. Kawasaki E, Awata T, Ikegami H, et al. Systematic search for single nucleotide polymorphisms in a lymphoid tyrosine phosphatase gene (PTPN22): association between a promoter polymorphism and type 1 diabetes in Asian populations. Am J Med Genet A. 2006;140(6):586–593. doi:10.1002/ajmg.a.31124
70. Awata T, Kawasaki E, Ikegami H, et al. Insulin gene/IDDM2 locus in Japanese type 1 diabetes: contribution of class I alleles and influence of class I subdivision in susceptibility to type 1 diabetes. J Clin Endocrinol Metab. 2007;92(5):1791–1795. doi:10.1210/jc.2006-2242
71. Shimada A, Kanazawa Y, Motohashi Y, et al. Evidence for association between vitamin D receptor BsmI polymorphism and type 1 diabetes in Japanese. J Autoimmunity. 2008;30(4):207–211. doi:10.1016/j.jaut.2007.09.002
72. Kawasaki E, Awata T, Ikegami H, et al. Genetic association between the interleukin-2 receptor-alpha gene and mode of onset of type 1 diabetes in the Japanese population. J Clin Endocrinol Metab. 2009;94(3):947–952. doi:10.1210/jc.2008-1596
73. Andersen MK, Sterner M, Forsen T, et al. Type 2 diabetes susceptibility gene variants predispose to adult-onset autoimmune diabetes. Diabetologia. 2014;57(9):1859–1868. doi:10.1007/s00125-014-3287-8
74. Lukacs K, Hosszufalusi N, Dinya E, Bakacs M, Madacsy L, Panczel P. The type 2 diabetes-associated variant in TCF7L2 is associated with latent autoimmune diabetes in adult Europeans and the gene effect is modified by obesity: a meta-analysis and an individual study. Diabetologia. 2012;55(3):689–693. doi:10.1007/s00125-011-2378-z
75. Bottazzo GF, Bosi E, Cull CA, et al. IA-2 antibody prevalence and risk assessment of early insulin requirement in subjects presenting with type 2 diabetes (UKPDS 71). Diabetologia. 2005;48(4):703–708. doi:10.1007/s00125-005-1691-9
76. Zampetti S, Campagna G, Tiberti C, et al. High GADA titer increases the risk of insulin requirement in LADA patients: a 7-year follow-up (NIRAD study 7). Eur J Endocrinol. 2014;171(6):697–704.
77. Verge CF, Gianani R, Kawasaki E, et al. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes. 1996;45(7):926–933. doi:10.2337/diab.45.7.926
78. Liu L, Li X, Xiang Y, et al. Latent autoimmune diabetes in adults with low-titer GAD antibodies: similar disease progression with type 2 diabetes: a nationwide, multicenter prospective study (LADA China Study 3). Diabetes Care. 2015;38(1):16–21. doi:10.2337/dc14-1770
79. Kobayashi T, Tanaka S, Shimada A, Maruyama T. High titer of autoantibodies to GAD identifies a specific phenotype of adult-onset autoimmune diabetes: response to Buzzetti et al. Diabetes Care. 2007;30(11):e126. author reply e127. doi:10.2337/dc07-1104
80. Sasamori H, Fukui T, Hayashi T, et al. Analysis of pancreatic volume in acute-onset, slowly-progressive and fulminant type 1 diabetes in a Japanese population. J Diabetes Invest. 2018;9(5):1091–1099. doi:10.1111/jdi.12816
81. Nakanishi K, Matsumura T, Kobayashi T. Slowly progressive IDDM and malnutrition-related diabetes. Diabetes Care. 1996;19(2):178. doi:10.2337/diacare.19.2.178a
82. Hoshina S, Miura J, Uchigata Y. Relationship between HLA haplotype and BMI change in Japanese slowly progressive type 1 diabetes patients. Diabetes Res Clin Pract. 2017;124:81–83. doi:10.1016/j.diabres.2016.12.006
83. Fourlanos S, Elkassaby S, Varney MD, Colman PG, Harrison LC. Higher body mass index in adults at diagnosis of the slowly progressive form of type 1 diabetes mellitus is associated with lower risk HLA genes. Diabetes Res Clin Pract. 2014;104(3):e69–e71. doi:10.1016/j.diabres.2014.03.009
84. Lee SH, Kwon HS, Yoo SJ, et al. Identifying latent autoimmune diabetes in adults in Korea: the role of C-peptide and metabolic syndrome. Diabetes Res Clin Pract. 2009;83(2):e62–e65. doi:10.1016/j.diabres.2008.11.031
85. Qi X, Sun J, Wang J, et al. Prevalence and correlates of latent autoimmune diabetes in adults in Tianjin, China: a population-based cross-sectional study. Diabetes Care. 2011;34(1):66–70. doi:10.2337/dc10-0488
86. Bosi EP, Garancini MP, Poggiali F, Bonifacio E, Gallus G. Low prevalence of islet autoimmunity in adult diabetes and low predictive value of islet autoantibodies in the general adult population of northern Italy. Diabetologia. 1999;42(7):840–844. doi:10.1007/s001250051235
87. Zinman B, Kahn SE, Haffner SM, O’Neill MC, Heise MA, Freed MI. Phenotypic characteristics of GAD antibody-positive recently diagnosed patients with type 2 diabetes in North America and Europe. Diabetes. 2004;53(12):3193–3200. doi:10.2337/diabetes.53.12.3193
88. Radtke MA, Midthjell K, Nilsen TI, Grill V. Heterogeneity of patients with latent autoimmune diabetes in adults: linkage to autoimmunity is apparent only in those with perceived need for insulin treatment: results from the Nord-Trondelag Health (HUNT) study. Diabetes Care. 2009;32(2):245–250. doi:10.2337/dc08-1468
89. Leslie RD, Kolb H, Schloot NC, et al. Diabetes classification: grey zones, sound and smoke: action LADA 1. Diabetes Metab Res Rev. 2008;24(7):511–519. doi:10.1002/dmrr.v24:7
90. Guglielmi C, Palermo A, Pozzilli P. Latent autoimmune diabetes in the adults (LADA) in Asia: from pathogenesis and epidemiology to therapy. Diabetes Metab Res Rev. 2012;28(Suppl 2):40–46. doi:10.1002/dmrr.2345
91. Myhill P, Davis WA, Bruce DG, Mackay IR, Zimmet P, Davis TM. Chronic complications and mortality in community-based patients with latent autoimmune diabetes in adults: the Fremantle Diabetes Study. Diabet Med. 2008;25(10):1245–1250. doi:10.1111/j.1464-5491.2008.02562.x
92. Wang C, Lu J, Lu W, et al. Evaluating peripheral nerve function in asymptomatic patients with type 2 diabetes or latent autoimmune diabetes of adults (LADA): results from nerve conduction studies. J Diabetes Complications. 2015;29(2):265–269. doi:10.1016/j.jdiacomp.2014.11.001
93. Buzzetti R, Zampetti S, Maddaloni E. Adult-onset autoimmune diabetes: current knowledge and implications for management. Nature Rev Endocrinol. 2017;13(11):674–686. doi:10.1038/nrendo.2017.99
94. Pozzilli P, Pieralice S. Latent autoimmune diabetes in adults: current status and new horizons. Endocrinol Metab. 2018;33(2):147–159. doi:10.3803/EnM.2018.33.2.147
95. Isomaa B, Almgren P, Henricsson M, et al. Chronic complications in patients with slowly progressing autoimmune type 1 diabetes (LADA). Diabetes Care. 1999;22(8):1347–1353. doi:10.2337/diacare.22.8.1347
96. Roh MO, Jung CH, Kim BY, Mok JO, Kim CH. The prevalence and characteristics of latent autoimmune diabetes in adults (LADA) and its relation with chronic complications in a clinical department of a university hospital in Korea. Acta Diabetologica. 2013;50(2):129–134. doi:10.1007/s00592-010-0228-y
97. Arikan E, Sabuncu T, Ozer EM, Hatemi H. The clinical characteristics of latent autoimmune diabetes in adults and its relation with chronic complications in metabolically poor controlled Turkish patients with Type 2 diabetes mellitus. J Diabetes Complications. 2005;19(5):254–258. doi:10.1016/j.jdiacomp.2005.02.004
98. Olsson L, Grill V, Midthjell K, Ahlbom A, Andersson T, Carlsson S. Mortality in adult-onset autoimmune diabetes is associated with poor glycemic control: results from the HUNT Study. Diabetes Care. 2013;36(12):3971–3978. doi:10.2337/dc13-0564
99. Nakanishi K, Kobayashi T, Miyashita H, et al. Relationships among islet cell antibodies, residual beta-cell function, and metabolic control in patients with insulin-dependent diabetes mellitus of long duration: use of a sensitive C-peptide radioimmunoassay. Metabolism. 1990;39(9):925–930. doi:10.1016/0026-0495(90)90302-S
100. Nakanishi K, Kobayashi T, Inoko H, Tsuji K, Murase T, Kosaka K. Residual beta-cell function and HLA-A24 in IDDM. Markers of glycemic control and subsequent development of diabetic retinopathy. Diabetes. 1995;44(11):1334–1339. doi:10.2337/diab.44.11.1334
101. Kobayashi T, Nakanishi K, Murase T, Kosaka K. Small doses of subcutaneous insulin as a strategy for preventing slowly progressive beta-cell failure in islet cell antibody-positive patients with clinical features of NIDDM. Diabetes. 1996;45(5):622–626. doi:10.2337/diab.45.5.622
102. Nishimura A, Nagasawa K, Okubo M, Kobayashi T, Mori Y. Exponential increase of glutamic acid decarboxylase (GAD) antibody titer after initiating and stopping insulin in a patient with slowly progressive type 1 diabetes. Endocr J. 2015;62(12):1077–1082. doi:10.1507/endocrj.EJ15-0378
103. Brophy S, Davies H, Mannan S, Brunt H, Williams R. Interventions for latent autoimmune diabetes (LADA) in adults. Cochrane Database Syst Rev. 2011;(9):Cd006165.
104. Varga T, Somogyi A, Barna G, et al. Higher serum DPP-4 enzyme activity and decreased lymphocyte CD26 expression in type 1 diabetes. Pathol Oncol Res. 2011;17(4):925–930. doi:10.1007/s12253-011-9404-9
105. Johansen OE, Boehm BO, Grill V, et al. C-peptide levels in latent autoimmune diabetes in adults treated with linagliptin versus glimepiride: exploratory results from a 2-year double-blind, randomized, controlled study. Diabetes Care. 2014;37(1):e11–e12. doi:10.2337/dc13-1523
106. Buzzetti R, Pozzilli P, Frederich R, Iqbal N, Hirshberg B. Saxagliptin improves glycaemic control and C-peptide secretion in latent autoimmune diabetes in adults (LADA). Diabetes Metab Res Rev. 2016;32(3):289–296. doi:10.1002/dmrr.v32.3
107. Kim SJ, Nian C, McIntosh CH. Sitagliptin (MK0431) inhibition of dipeptidyl peptidase IV decreases nonobese diabetic mouse CD4+ T-cell migration through incretin-dependent and -independent pathways. Diabetes. 2010;59(7):1739–1750. doi:10.2337/db09-1618
108. Tian L, Gao J, Hao J, et al. Reversal of new-onset diabetes through modulating inflammation and stimulating beta-cell replication in nonobese diabetic mice by a dipeptidyl peptidase IV inhibitor. Endocrinology. 2010;151(7):3049–3060. doi:10.1210/en.2010-0068
109. Kawano Y, Irie J, Nakatani H, Yamada S. Pioglitazone might prevent the progression of slowly progressive type 1 diabetes. Intern Med. 2009;48(12):1037–1039. doi:10.2169/internalmedicine.48.1990
110. Shimada A, Shigihara T, Okubo Y, Katsuki T, Yamada Y, Oikawa Y. Pioglitazone may accelerate disease course of slowly progressive type 1 diabetes. Diabetes Metab Res Rev. 2011;27(8):951–953. doi:10.1002/dmrr.v27.8
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.