")
Back to Journals » Nature and Science of Sleep » Volume 14
Sleep, Respiration and Nocturnal Paroxysmal Events in Joubert Syndrome: A Case Report
Authors Peraita-Adrados R
Received 2 April 2022
Accepted for publication 22 August 2022
Published 26 August 2022 Volume 2022:14 Pages 1485—1492
DOI https://doi.org/10.2147/NSS.S369097
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sarah L Appleton
Rosa Peraita-Adrados
Sleep and Epilepsy Unit- Clinical Neurophysiology Service, University General Hospital and Research Institute Gregorio Marañón, University Complutense of Madrid (UCM), Madrid, Spain
Correspondence: Rosa Peraita-Adrados, Email [email protected]
Introduction: Joubert syndrome is a rare disorder, characterized by a complex midbrain malformation caused by defects in the structure and/or function of the primary cilium.
Case Report: A 15-year-old boy with mild intellectual disability, hypotonia, mild ataxia, and abnormal eye movements diagnosed as having Joubert Syndrome since childhood, was referred to the Sleep Unit because spells of apnea while sleeping. He did not complain of snoring or daytime somnolence. The macro and microstructure of sleep and the comorbidities such respiratory abnormalities, periodic legs movements (PLM) and paroxysmal motor arousals (PA) and minimal motor events (MME) are described for the first time in Joubert syndrome.
Results: EEG was normal. Video-polysomnography revealed a nocturnal disturbed sleep and periods of hyperpnea accompanied by body movements and followed by a periodic breathing lasting several minutes with no oxygen desaturation. The arousals provoked by apneas triggered paroxysmal motor events with dystonic movements in the hand and right foot accompanied by a spontaneous Babinski. Brain MRI showed the typical “molar tooth sign”.
Conclusion: Joubert syndrome is a heterogeneous disease. Epileptic seizures have been reported in some cases. Video-PSG is mandatory for the identification of nocturnal breathing abnormalities and sleep-related motor paroxysmal episodes.
Keywords: Joubert syndrome, molar tooth sign, hyperpnea and central periodic breathing, nocturnal paroxysmal motor events, sleep fragmentation and arousals
Plain Language Summary
Joubert syndrome is a clinically heterogeneous disease. Significantly, variability in phenotype and genetics has been described.
Joubert syndrome is under-diagnosed and should always be considered in children with non-progressive ataxia, intellectual disability and episodes of hyperpnea and apnea.
The presence of molar tooth sign its present in 85% of the cases of Joubert syndrome.
The epileptic seizures have been reported with lack of details regarding the electro-clinical semiology and without a clear-cut circadian presentation schedule.
The possible physiological mechanism triggering the sleep paroxysmal motor events has been described such as respiratory arousals and motor arousals (PLM) as the usual triggers in nocturnal frontal epileptic seizures and in parasomnias.
A Video-PSG is mandatory for the identification of breathing abnormalities during sleep as well as comorbidities with PLM, parasomnias and related motor seizures during sleep.
Introduction
Joubert Syndrome (JS) first described in 1969,1 is a rare, autosomal recessive disorder, clinically heterogeneous that combine neurological signs: generally poorly controlled movements and mild to moderate intellectual disability, with variable multiorgan involvement, mainly of retina, kidneys, liver and skeleton. Brain MRI is characterized by a complex midbrain-hindbrain malformation with the absence or underdevelopment of the cerebellar vermis and a broad spectrum of other phenotypic findings caused by defects in the structure and/or function of the primary cilium.2 There is a hallmark neuroradiology feature in the brain MRI called “molar tooth sign” (MTS) present in 85% of the cases.3 Abnormal migration defects, mainly periventricular nodular heterotopia, and cortical organization defects such as polymicrogyria have also been reported.4
The incidence has not been precise, it might be between 1–9/100.000.
Despite years of JS gene discovery, the genetic cause cannot be identified in up to 30% of individuals, depending on the cohort, sequencing method, and criteria for pathogenic variants. All these genes encode for proteins of the primary cilium, “ciliopathies”. Primary cilia are known to play key roles in the development and functioning of several cell types: neurons, retinal photoreceptors, kidney tubules and bilis duct. In the developing cerebellum and brainstem, these organelles regulate major signal transduction pathways, and have been implicated in both in neuronal cell proliferation and axonal migration.5 The prognosis depends mostly on renal and hepatic complications that, if not timely diagnosed and managed, represent the major causes of death in JS patients.
From the first publications, respiratory abnormalities were characterized by attacks of tachypnea alternating with respiratory pauses; hyperpnea-apnea episodes or episodic tachypnea, apnea, snoring and Pediatric Sleep Questionnaire scores suggestive of sleep-related breathing disorders.6–8 Centrally mediated brainstem dysregulation triggers episodic hyperpnea and apnea. At birth, some infants have severe manifestations of breathing control requiring mechanical ventilation and/or tracheostomy in rare cases. Early detection and improved understanding of sleep-related breathing abnormalities may contribute to improve the prognosis for patients with Joubert syndrome.6
We report the clinical, cranial MRI and video-polysomnography (v-PSG) features of an adolescent diagnosed as having Joubert Syndrome in childhood who presented paroxysms of hyperpnea followed by a central periodic breathing and sleep-related paroxysmal motor events during sleep. The macro and microstructure of sleep and the comorbidities, respiratory abnormalities, periodic legs movements (PLM), paroxysmal motor arousals (PA) and minimal motor events (MME) are described for the first time in Joubert syndrome.
The children’s parents gave their informed consent to participate in the study as well as the consent to publish the data, and the institution’s research Ethics Committee approved the study.
Case Report
A 15-year-old boy with mild intellectual disability, hypotonia, mild ataxia, and abnormal eye movements, diagnosed as having Joubert Syndrome since childhood, was referred to the Sleep Unit because his parents noticed that he experienced spells of apnea while sleeping since childhood and more frequent in the last two years. He did not complain of excessive daytime somnolence or fatigue.
He was born after normal pregnancy and cesarean section, newborn weight 3530 gr, normal male karyotype 46 XY. He had no family history of consanguinity, neurological or sleep disorders. A mild hypotonia was observed during breastfeeding. He presented a delayed gait at 2.5 years, a global maturational delay including language, attentional and learning difficulties. The patient goes to a special education center due to his intellectual disability. No seizures have been reported and the patient was untreated.
Results
Clinical examination discard renal or liver involvement. His anthropometric measurements were as follows: weight, 63.4 kg; height, 172 cm; BMI, 21.5 kg/m2.
The neuro-ophthalmology examination showed a complex non-paralytic strabismus and oculomotor apraxia with saccades and associated cephalic jerks that have improved over time. Absence of retinal dystrophy.
The Pediatric Daytime Sleepiness Scale (PDSS) score was 12 and the waking standard EEG recording was normal. Two v-PSG recordings (EEG, EOG, EKG, submental and tibialis anterior EMG, respiration and oxygen saturation) were performed. The parameters analyzed were sleep efficiency index, sleep latency, Rem latency, wake after sleep onset (WASO), percentages of stages N1, N2, N3, and REM in relation to Total Sleep Time; total number of awakenings (from any sleep stages to wakefulness); apnea-hypopnea (AHI) and periodic leg movements (PLM) indexes. The data were manually scored according to the American Academy of Sleep Medicine (AASM - Version 2.6). The first v-PSG revealed a disturbed macro and microstructure of nocturnal sleep, long sleep latency, long REM latency, increased WASO, high sleep fragmentation index (number of awakenings), decreased efficiency index, only 2 sleep cycles and low percentage of stage REM, as shown in the hypnogram. These abnormalities were interpreted as the consequence of “the first night effect”. A second v-PSG performed two months later showed a normal sleep macrostructure (five sleep cycles) with the persistence of a high fragmentation index and with unchanged respiratory and motor patterns (Figure 1) (Table 1).
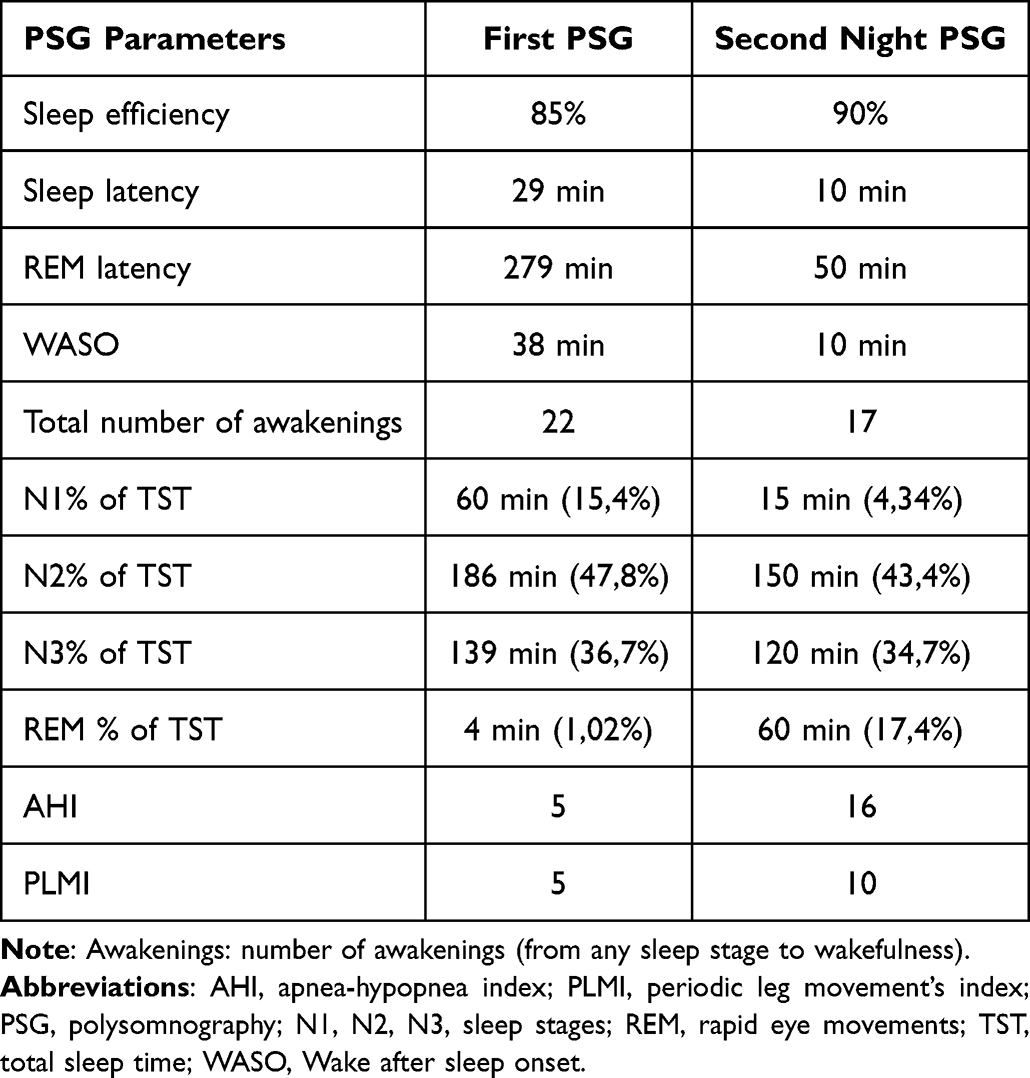 |
Table 1 Sleep, Respiratory and PLM Parameters of the First and Second PSG Recordings |
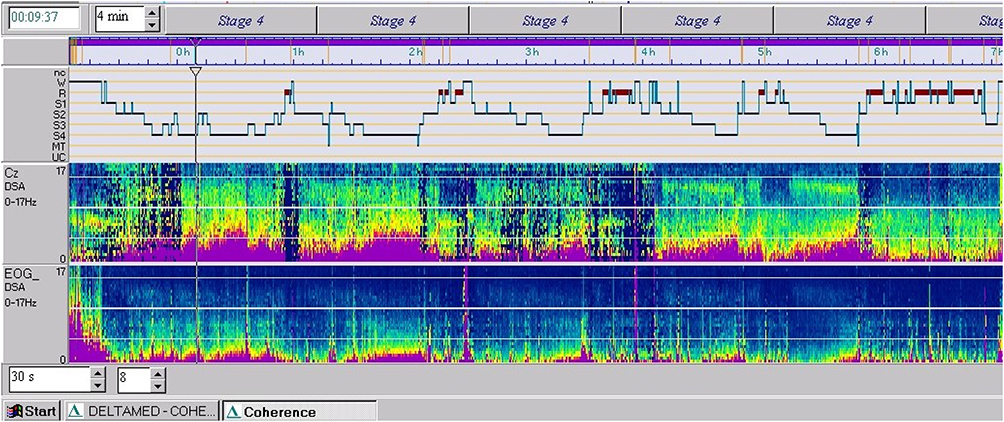 |
Figure 1 Hypnogram and power spectrum analysis (PSA of Cz and EOG). |
Respiration (airflow, thoracic and abdominal effort and SaO2) was normal with periods of hyperpnea preceded by a body movement and followed by a central periodic breathing lasting for several minutes with no significant oxygen desaturation (SaO2) (Figure 2).
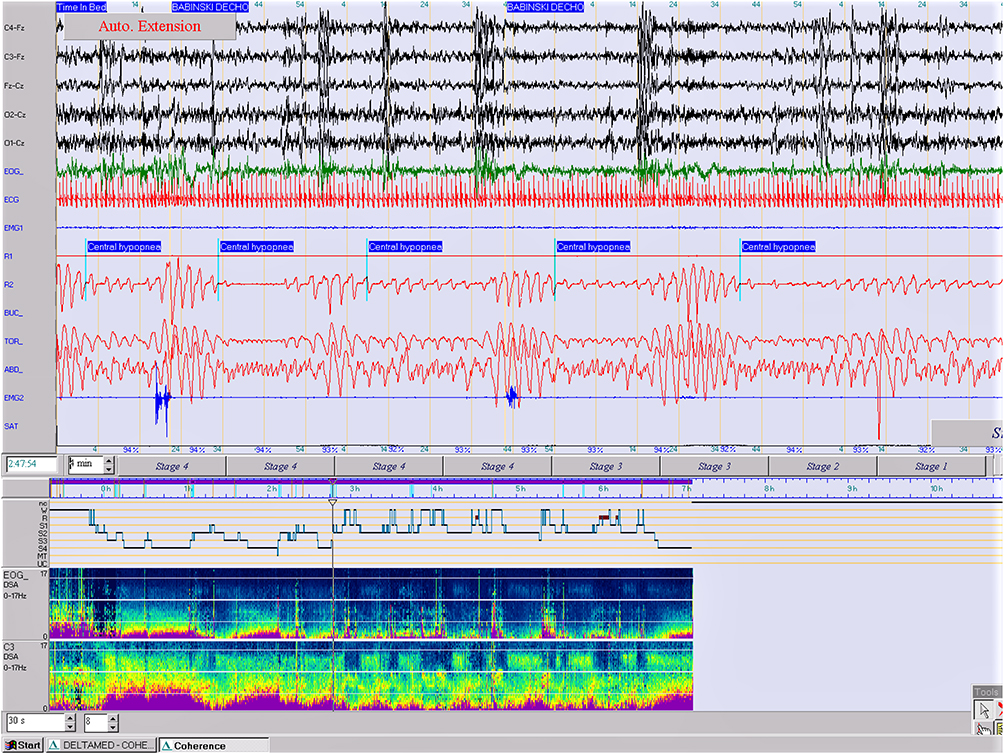 |
Figure 2 Central periodic breathing lasting for 4 minutes with no significant oxygen desaturation (epoch of 4 min., 10 μV). (PSA of EOG and C3). |
The most outstanding finding was the appearance of highly stereotyped paroxysmal motor events accompanied in the EEG by a theta-delta rhythmic activity and followed by a stage shift and/or a postural change and a short awakening. The arousals provoked by episodes of hyperpnea triggered these minimal motor events with dystonic movements in the right hand and foot and a spontaneous Babinski (isolated or repeated), which lasted 5 to 10 seconds. On other occasions, a hyperextension of arms and dystonic movements of the right hand and a right Babinski followed the hyperpnoea were observed (Figure 3). These paroxysmal motor events appeared throughout the night in the absence of clear-cut epileptiform interictal epileptic abnormalities in wakefulness and/or sleep. In addition, periodic leg movements (PLM) - that systematically provoked an arousal - were recorded.
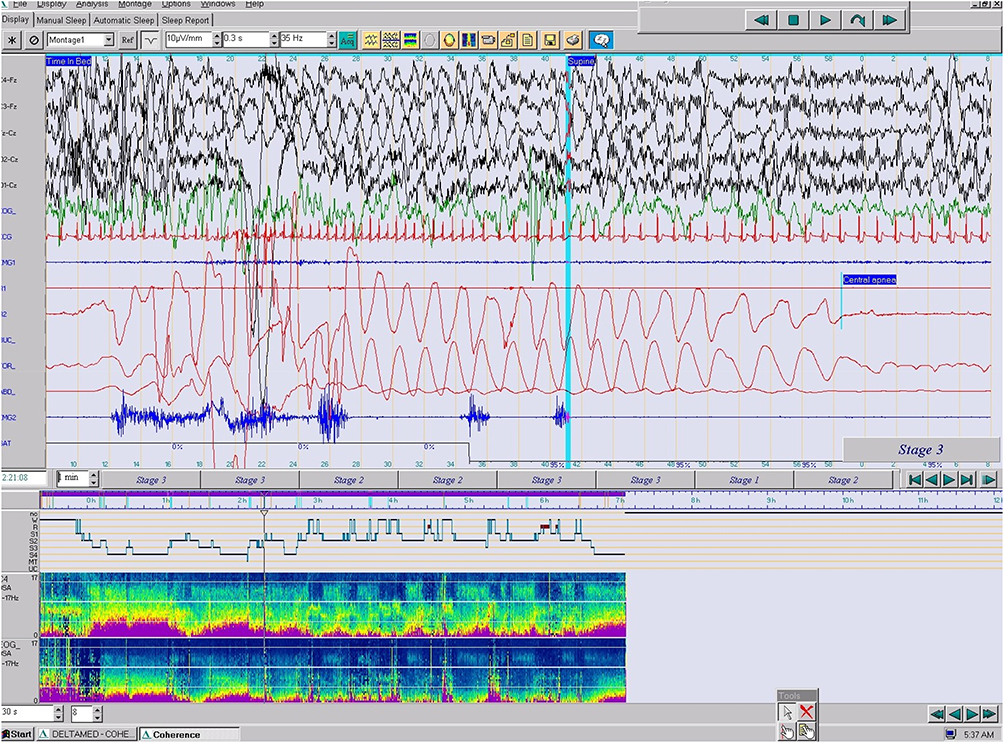 |
Figure 3 The v-PSG showed highly stereotyped arousals provoked by a central apnea and paroxysmal motor events accompanied in the EEG by a theta-delta rhythmic activity and followed by stage shifts and short awakenings with dystonic movements in the right hand and foot, a spontaneous Babinski and a postural change in stage N3. Tachycardia and taquipnea are present (epoch of 1 min., 10 μV). |
Brain MRI showed the “molar tooth sign” (MTS) on axial image with these three components: midline cerebellar vermis hypoplasia, deepened interpeduncular fossa, and thick, elongated superior cerebellar peduncles (Figure 4).
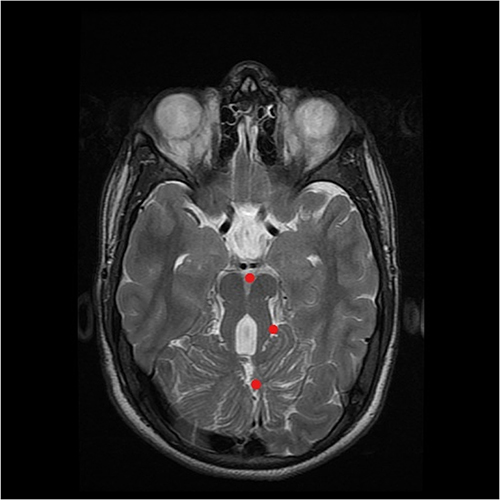 |
Figure 4 T2-weighted MRI in axial view at the level of the midbrain displaying the molar tooth sign, which is created by a combination of deep interpeduncular fossa, slender superior cerebellar peduncles and enlarged IV ventricle. The midline cerebellar vermis is severely hypoplastic. |
A CPAP therapy was discarded because the absence of daytime somnolence and acceptable school level and behaviour. Antiepileptic treatment was not administered.
Discussion
In Joubert syndrome, neurological symptoms consist of intellectual disability and delayed language and motor skills. Intellectual deficit is not a mandatory feature and exceptional cases may have borderline - as in this case - or even normal. Early hypotonia is observed in nearly all cases, and can be recognized in the neonatal period or infancy, evolving into truncal ataxia and development delay. Oculomotor apraxia and inability to follow objects visually with compensatory head movements, is also common. One of the organs most frequently involved is retinal dystrophy, due to progressive degeneration of photoreceptor cells.
The presence of a neuroradiological hallmark, designated as the “molar tooth sign” (MTS) on brain MRI was clear in our case.
Centrally mediated brainstem dysregulation triggers episodic hyperpnea and apnea. At birth, some infants have severe manifestations of breathing control requiring mechanical ventilation and/or tracheostomy in rare cases. Early diagnostic and understanding of sleep-related breathing abnormalities may contribute to improve the prognosis for patients with JS.6
We found in our case the typical respiratory pattern with alternate episodes of hyperpnea and apnea as well as episodic hyperpnea during sleep without SaO2 desaturation (Figure 5). The arousals provoked by apneas triggered paroxysmal motor events that have not been previously described in this syndrome. Epilepsy and EEG abnormalities are rare features in the series of JS seen in approximately 3% of patients although there are no consistent predictive radiologic findings 9–12. Two previously reported patients with dentate nuclei abnormality had epilepsy. It is not proven whether cerebellar abnormalities could contribute to seizure generation.13 In exceptional cases, seizures resistant to treatment have compromised long-term survival; these are more likely in patients with structural brain malformations in addition to the MTS. In our patient the arousals provoked by central apneas triggered paroxysmal motor events with dystonic movements in the hand and right foot accompanied by a spontaneous Babinski reflex which lasted 1 to 5 seconds. These motor stereotyped episodes appeared in clusters throughout the night.
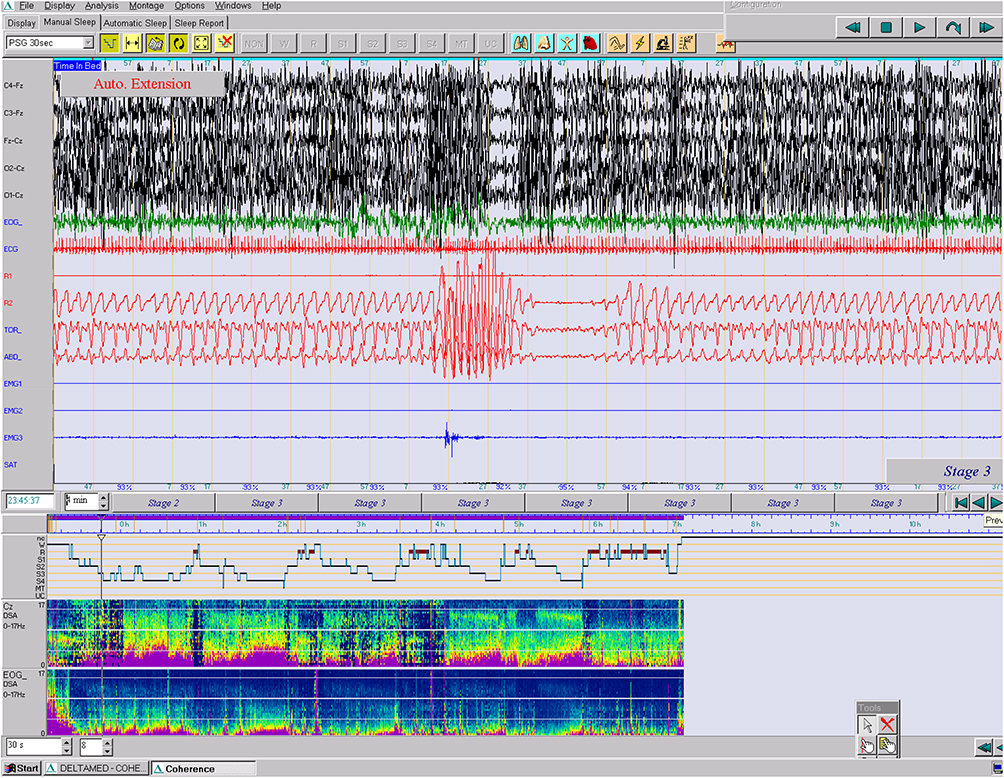 |
Figure 5 Paroxysm of hyperpnea followed by a central apnea lasting 20 sec and preceded by a PLM with no significant oxygen desaturation (epoch of 4 min., 10 μV). (PSA of C and EOG). |
We have previously published a cohort of Spanish children - none diagnosed as a Joubert syndrome - who underwent v-PSG for suspected sleep-related breathing disorders (SBD) to ascertain the eventual presence of epileptic seizures. In total, 25 children were diagnosed with SBD, and 4 out of 25 (16%) children met the criteria for OSA and epilepsy, with epileptiform discharges and/or seizures during sleep. We diagnosed benign epilepsy with centro-temporal spikes in two cases, partial symptomatic epilepsy in one, and Nocturnal Frontal Lobe Epilepsy (NFLE) in another, while we found paroxysmal arousals in two patients.14 In a later study in children with NFLE, we found a high comorbidity with other sleep disorders, such as SDB, and we found a disturbed nocturnal sleep as indicated by an increased number of stage shifts and arousals, a higher percentage of light sleep stages, decreased percentages of slow-wave sleep and REM sleep, and diminished sleep efficiency. Among children diagnosed with NFLE, seven were referred for sleep disordered breathing (SDB) as in this Joubert case. Standard EEG was normal in 21 out of 24 cases.15
Arousal fluctuations play an important role in triggering minimal motor events (MME) during sleep in NFLE; the epileptic discharge acts as a trigger for the appearance of behaviors that are the expression of inborn motor patterns, related to central pattern generators (CPG), and mainly located outside the cerebral cortex. According to Tassinari et al, the concept may be extended to parasomnias, whose motor expressions are the same as in epileptic seizures since they result from the activity of the same CPG.16
Recent findings indicate that, in a single epileptic patient, highly stereotyped MME can occur in either the presence or absence of an epileptiform discharge as in this case.17 According to our previous series, a high percentage of children with NFLE are often misdiagnosed. The nocturnal disturbed sleep could be associated with other sleep disorders and especially with OSA.
Recently, a new epileptic syndrome characterized by the occurrence of sleep-related hypermotor seizures of variable complexity and duration had been described. Seizures usually arise in the frontal lobe, but extrafrontal seizure onset zones as well have been described.18
Finally, we would like to emphasize that sleep-related hypermotor epilepsy provoke nocturnal sleep disruption with high fragmentation, and the presence of other sleep disorders (SDB, PLM, parasomnias) may be a trigger for nocturnal paroxysmal events as in the case of the present study.
Acknowledgments
We deeply thank Dr. Rogelio Simón, Dr. Ana Camacho and Dr. Fernando Mateo from Child Neurology Division, 12 de Octubre University Hospital, Madrid, Spain, for referring the case to the Sleep and Epilepsy Unit; and to Dr. Ana Martínez de Aragón, Department of Radiology from the same Hospital, for providing the Cranial MRI images.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19(9):813–825. doi:10.1212/wnl.19.9.813
2. Yachnis AT, Rorke LB. Cerebellar and brainstem development: an overview in relation to Joubert syndrome. J Child Neurol. 1999;14(9):570–573. doi:10.1177/088307389901400904
3. Maria BL, Quisling RG, Rosainz LC, et al. Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance. J Child Neurol. 1999;14(6):368–376. doi:10.1177/088307389901400605
4. Dixon-Salazar T, Silhavy JL, Marsh SE, et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004;75(6):979–987. doi:10.1086/425985
5. Van De Weghe JC, Giordano JL, Mathijssen IB, et al. TMEM218 dysfunction causes ciliopathies, including Joubert and Meckel syndromes. HGG Adv. 2021;2(1):100016. doi:10.1016/j.xhgg.2020.100016
6. Kamdar BB, Nandkumar P, Krishnan V, Gamaldo CE, Collop NA. Self-reported sleep and breathing disturbances in Joubert syndrome. Pediatr Neurol. 2011;45(6):395–399. doi:10.1016/j.pediatrneurol.2011.09.005
7. De Jonghe M, Schroell M, Degremont-Weitzel C. [Joubert syndrome]. Bull Soc Sci Med Grand Duche Luxemb. 1994;131(2):19–23. French. PMID: 7820905
8. Andermann F, Andermann E, Ptito A, Fontaine S, Joubert M. History of Joubert syndrome and a 30-year follow-up of the original proband. Child Neurol. 1999;14(9):565–569. PMID: 10488900. doi:10.1177/088307389901400903
9. Calleja-Pérez B, Fernández-Jaén A, Martínez-Bermejo A, Pascual-Castroviejo I. Joubert syndrome: a report of 5 cases. Rev Neurol. 1998;26(152):548–550.
10. Amin OS, Shwani SS. Ataxia, hyperpnoea and mental retardation: was it the molar tooth? BMJ Case Rep. 2010;2010:bcr1020092331–bcr1020092331. doi:10.1136/bcr.10.2009.2331
11. Barreirinho J, Teixeira N, Moreira C, Bastos S, Gonçalves C, Barbot MC. Joubert’s syndrome: report of 12 cases. Rev Neurol. 2001;32(9):812–817.
12. Ahmetgjekaj I, Rahman M, Hyseni F, et al. A case report of Joubert syndrome with renal involvement and seizures in a neonate. Radiol Case Rep. 2021;16(5):1075–1079. doi:10.1016/j.radcr.2021.02.031
13. Kuchukhidze G, Rauchenzauner M, Gotwald T, Janecke A, Trinka E. Hypoplasia of deep cerebellar nuclei in Joubert syndrome. Pediatr Neurol. 2009;40(6):474–476. PMID: 19433286. doi:10.1016/j.pediatrneurol.2008.12.007
14. Miano S, Bachiller C, Gutiérrez M, Salcedo A, Villa MP, Peraita-Adrados R. Paroxysmal activity and seizures associated with sleep breathing disorder in children: a possible overlap between diurnal and nocturnal symptoms. Seizure. 2010;19(9):547–552. doi:10.1016/j.seizure.2010.07.015
15. Miano S, Peraita-Adrados R. Nocturnal frontal lobe epilepsy is often misdiagnosed as sleep disorders in children: a case series. Rev Neurol. 2013;56(5):257–267.
16. Tassinari CA, Cantalupo G, Högl B, et al. Neuroethological approach to frontolimbic epileptic seizures and parasomnias: the same central pattern generators for the same behaviours. Rev Neurol (Paris). 2009;165(10):762–768. doi:10.1016/j.neurol.2009.08.002
17. Terzaghi M, Sartori I, Mai R, et al. Sleep-related minor motor events in nocturnal frontal lobe epilepsy. Epilepsia. 2007;48:335–341. doi:10.1111/j.1528-1167.2006.00929.x
18. Gibbs SA, Proserpio P, Francione S, et al. Seizure duration and latency of hypermotor manifestations distinguish frontal from extrafrontal onset in sleep-related hypermotor epilepsy. Epilepsia. 2018;59(9):e130–e134. doi:10.1111/epi.14517
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.