")
Back to Journals » Cancer Management and Research » Volume 11
SLC5A1 promotes growth and proliferation of pancreatic carcinoma via glucose-dependent AMPK/mTOR signaling
Authors Gao HF, Chen LY, Cheng CS , Chen H, Meng ZQ, Chen Z
Received 21 November 2018
Accepted for publication 11 February 2019
Published 12 April 2019 Volume 2019:11 Pages 3171—3185
DOI https://doi.org/10.2147/CMAR.S195424
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Hui-Feng Gao,1,2,* Lian-Yu Chen,1,2,* Chien-Shan Cheng,1,2 Hao Chen,1,2 Zhi-Qiang Meng,1,2 Zhen Chen1,2
1Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, People’s Republic of China; 2Department of Integrative Oncology, Fudan University Shanghai Cancer Center, Shanghai 200032, People’s Republic of China
*These authors contributed equally to this work
Background: Accumulating studies have reported that aberrant expression of SLC5A1 is a negative prognostic factor to various cancer patients.
Purpose: Pancreatic cancer tissue has also shown to harbor higher expression of SLC5A1, however how SLC5A1 mediates pancreatic cancer cells growth remains unclear.
Methods: In this study, we examined the mRNA and protein expressions of SLC5A1 in human pancreatic tissue and various cell lines. The in vitro and in vivo roles of SLC5A1 in pancreatic cancer were investigated through stably transfected pancreatic cells with shRNA plasmid targeting SLC5A1.
Results: Our results observed SLC5A1 was over-expressed in human pancreatic cancer tissues as well as most pancreatic cancer cell lines. Both in vitro and in vivo inhibition of SLC5A1 retarded pancreatic cancer cell growth and progression. The SLC5A1 knockdown mediated growth suppression is mainly regulated by reduced cellular glucose uptake by pancreatic cancer cells. Our further mechanistic observation showed that inhibition of SLC5A1 induced AMPK-dependent mTOR suppression and pharmacological inhibition of AMPK rescued the effect of SLC5A1 blockade. Further protein-protein interaction analysis showed association of SLC5A1 with EGFR and knockdown of EGFR also showed decreased cellular survival and glucose uptake by pancreatic cancer cells.
Conclusion: Our findings postulated SLC5A1/EGFR as the potential therapeutic target of pancreatic cancer patients.
Keywords: pancreatic cancer, SLC5A1, EGFR, cancer cell survival
Introduction
Pancreatic cancer is one of the biggest causes of death-leading cancers worldwide. The 5-year survival rate of pancreatic cancer patients is less than 5% and most of the patients are diagnosed at a very advanced stage owing to its asymptomatic characteristic.1 According to the worldwide statistic in 2012, it is estimated 338,000 new cases are diagnosed every year and the annual mortality rate is close to the incidence rate with 331,000 death cases reported.2 To date, the best potential treatment for non-metastasized patients is pancreatectomy, yet only 15–20% of patients are suitable for surgical resection and more than 65% of patients undergoing surgery show disease recurrence.3 As for advanced patients, the chemotherapy gemcitabine, a type of nucleoside analog is commonly used, however this therapy merely prolongs patients’ survival for 0.9 to 4.2 months. Most of the patients rapidly acquire resistance towards gemcitabine, resulting in the poor response and prognosis of patients to gemcitabine.4 Therefore, identifying the tumor promoting driver is urgently needed in order to facilitate the development of effective therapies towards pancreatic cancer.
Glucose is the primary energy source for tumor growth and progression, therefore increased demand for glucose and altered glucose metabolism are common scenarios in tumor cells. The up-regulation of GLUT-1 has been well reported in supporting the increased glucose uptake by tumor cells.5 However, there is another class of glucose transporter, which is easily negligible, namely sodium-dependent glucose transporter (SGLT) that belongs to the SLC5A gene family. Regardless of the extracellular glucose content, SGLTs carry glucose into cells against the sodium ion concentration gradient.6 There are in total six isoforms of SGLTs in SLC5A gene family, ranging from SGLT1 to SGLT6. However, among all these six SGLTs, only SGLT1 and SGLT2 functions as glucose transporter across cell membrane. SGLT1 and SGLT2 are encoded by SLC5A1 and SLC5A2 respectively. In particular, aberrant expression of SLC5A1 was recently observed in different types of human cancers including colorectal cancer,7,8 hepatocellular carcinoma,9 prostate cancer,10 cervical cancer,11 ovarian cancer12 as well as oral squamous cell carcinoma.13 Recent study has postulated the importance of SLC5A genes in mediating pancreatic cancer survival.14,15 However, how SLC5A1 mediates pancreatic cancer cell survival is still not well understood.
In this study, we aimed to investigate the role of SLC5A1 in mediating the in vitro and in vivo survival of pancreatic cancer. The expressions of SLC5A1 in human pancreatic cancer tissue and normal pancreas tissues were extracted from three (Gene Expression Omnibus) GEO datasets and compared. We also investigated the expressions of SLC5A1 in a panel of pancreatic cancer cell lines. SLC5A1 was further knocked down in pancreatic cancer cells and the cellular survival was determined in in vitro and in vivo models. The downstream signaling of SLC5A1 was validated by quantitative poly-chain reaction (qPCR) and western blotting analysis. Our further mechanistic observation showed that the association of SLC5A1 with EGFR mediated pancreatic cancer cell growth via AMPK/mTOR signaling.
Materials and methods
Cell culture
The human pancreatic cancer cell lines Mia PaCa-2, BxPC-3, Panc-1, human pancreatic cells UACC-462 and hTERT-HPNE were purchased from ATCC (Manassas, VA, USA) and cultured based on the ATCC established guidelines. Human KP3 was obtained from JCRB cell bank (Tokyo, Japan) while murine pancreatic adenocarcinoma cell line Panc-2 was obtained from Frederick National Laboratory for Cancer Research (Frederick, MD, USA). All pancreatic cancer cells were either cultured in DMEM or RPMI1640 medium supplemented with 10% of fetal bovine serum (FBS) and 1% of penicillin/streptomycin. UACC-462 was cultured in Leibovitz’s L-15 medium supplemented with 5% of FBS. hTERT-HPNE cells were cultured with a mixture of 75% DMEM (no glucose; Thermo Fisher Scientific, Waltham, MA, USA) and 25% Medium M3 Base (InCell, Frisco, TX, USA) supplemented with 5% FBS, 10 ng/mL human recombinant epidermal growth factor, 1 g/L glucose, and 750 ng/mL puromycin. All cells were maintained in 37°C humidified incubator supplemented with 5% CO2.
Orthotopic pancreatic cancer model
The orthotopic pancreatic cancer mouse model was established based on previous protocol16 and was approved by the ethics committee of Fudan University. The protocol of animal study has followed the international guidelines of animal experiments, especially the UKCCCR guidelines for the welfare of animals in experimental Neoplasia, United Kingdom Co-ordinating Committee on Cancer Research, UKCCCR (1997), NIH Guidelines for Survival Rodent Surgery, Guide for the Care and Use of Laboratory Animals, NRC (2011), the assessment and alleviation of pain and distress in research animals, NHMRC (2007), and NIH Guidelines for Endpoints in Animal Study Proposals. In brief, the 5-week old female C57/BL/6N mice were anesthetized using ketamine/xylazine solution. The 1×106 shSLC5A1 and shControl Panc-2 cells in phosphate buffered saline (PBS) and Matrigel matrix (1:1) were prepared. The left flank of abdominal skin of anesthetized mice was exposed and the cells were injected to the pancreas tail of the mice. After tumor implantation, the tumor growth was monitored weekly by luciferin in vivo imaging. By the end of experiment, the mice were sacrificed by overdose of pentobarbital and tumor-bearing pancreas were removed and weighed.
Cell viability and colony formation assay
The cells were seeded onto 24-well plate at a cell density of 1×104. The cells were harvested every 2 days for viable cell count using a hemocytometer. As for colony formation assay, the cells were seeded at a cell density of 500 and allowed to grow for 14 days. Fresh medium was supplemented to the cell every other day. By the end of the experiment, the cells were fixed with 4% paraformaldehyde for 30 minutes followed by 2 hours of crystal violet staining.
Cell cycle assay
For cell cycle assay, the cells were seeded on 6-well plate at cell density of 1×105 and allowed to grow for 72 hours. The cells were then detached from the plate and fixed with cold ethanol overnight. After that, the cells were counterstained with propidium iodide with RNase A before subjected to flow cytometry analysis.
2-(N-(7(-Nitrobenz02-oxa-1,3-diazol-4-yl)Amino)-2-Deocyglucose (2-NBDG) uptake assay
The 2-NBDG uptake assay was performed according to the manufacturers’ protocol (Biovision, San Francisco, CA, USA). In brief, the cells were seeded to plate for 48 hours and the cells were further incubated with 2-NBDG reagent and glucose uptake enhancer supplemented medium for 30 minutes. The cells were then collected and washed in analysis buffer once before subjected to flow cytometry analysis.
Western blotting
The cell lysates were subjected to Radioimmunopecipitation assay (RIPA) buffer supplemented with protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA) for protein extraction. Extracted protein concentration was then determined using Bradford reagent (Bio-rad) and protein samples were boiled at 95°C for 5 min before separation on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel. The protein was transferred to polyvindylidende fluoride (PVDF) membrane for 2 hours. The membrane was then washed and blocked in 5% bovine serum albumin (BSA) for 2 hours followed by incubation with primary antibody overnight at 4°C. Horse radish peroxidase (HRP)-conjugated antibody was applied and allowed for 2-hours incubation before visualization using Chemidoc imaging system (Bio-rad).
RNA isolation and quantitative real time PCR
The cell lysates were subjected to Trizol reagent (Thermo Fisher Scientific) for RNA extraction, followed by cDNA synthesis using first strand cDNA synthesis kit (Takara, Japan). Quantitative mRNA levels were determined with SYBR master mix (Takara, Japan) using LC480 real time PCR platform (Hoffman-La Roche Ltd, Basel, Switzerland). The relative expression of each gene was calculated by normalizing against GAPDH gene expression. The primers used were as follows, mouse SLC5A1: (forward) 5ʹ TCTGTAGTGGCAAGGGGAAG 3ʹ (reverse) 5ʹ ACAGGGCTTCTGTGTCTTGG 3ʹ; human SLC5A1: (forward) 5′-TCCTGCTTGCTATTTTCTGGA-3′ (reverse) 5′-ATAATCGTGGGACAGTTGCTG-3′; mouse GAPDH: (forward) AGGTCGGTGTGAACGGATTTG (reverse) TGTAGACCATGTAGTTGAGGTCA; human GAPDH (forward) 5′-TCAACGACCACTTTGTCAAGCTCA-3′ (reverse) 5′-GCTGGTGGTCCAGGGGTCTTACT-3′.
Cell transfection
CRISPR gene knock out system was used to prepare the stable and transient silencing of SLC5A1 (SGLT-1) and EGFR. Both CRISPR KO plasmids were purchased from Santa Cruz Biotechnologies (Dallas, Texas, USA). In brief, the cells were incubated with lipofectamine 2000 and plasmids for 48 hours before harvested. For stable selection of SLC5A1, the cell medium was replaced with medium supplemented with puromycin after 24 hours of transduction. Quantitative real time PCR and western blotting analysis were used to validate the expression of SLC5A1.
Statistical analysis
All statistical analysis was performed with Student’s t-test or one-way ANOVA, and P-values <0.05 were statistically significant.
Results
SLC5A1 is over-expressed in human pancreatic cancer and associated with poor prognosis
To examine the clinical significance of SGLTs in pancreatic cancer patients, we firstly analyze the relative expression of SGLTs in pancreatic tumorous tissue and non-tumorous fractions that were extracted from the publicly available GEO dataset (GDS4102 and GDS4336). It was observed that the mRNA expression of SLC5A1 was significantly up-regulated in pancreatic cancerous tissue as compared to the adjacent non-cancerous fraction (Figure 1A). On the other hand, the mRNA expression of another cotransporter, SLC5A2 showed reduced expression in pancreatic tumor tissue as compared to normal tissue. Further analysis on the clinical correlation of SGLTs expression with overall or disease-free survivals of pancreatic cancer patients showed that patients with high expression of SLC5A1 is associated with lower overall survival rate (Figure 1B). However, there is no significant correlation observed in other SGLTs except SMIT1 expression with overall and disease-free survival rate of pancreatic cancer patients (Figure 1C). Then we analyzed the mRNA expression of SGLT1-6 and SMIT1 in pancreatic cancer cell line Panc-1. We found that expression of SGLT1 and SMIT1 is significanctly higher than other SGLTs, indicating that other SGLT2-6 may not play a main role in the glucose uptake of pancreatic cancers (Figure 2A) Then we further analyzed the mRNA expression of SGLT1 (SLC5A1) and SMIT1 (SLC5A3) in a panel of normal and cancerous pancreatic cells, and found that SLC5A1 expression has significant variation between normal and cancer cells, while expression of SMIT1 remains similar within the two cell types (Figure 2B). Our result suggests that SLC5A1 is over-expressed in pancreatic cancer cells and associated with poor prognosis in pancreatic cancer patients.
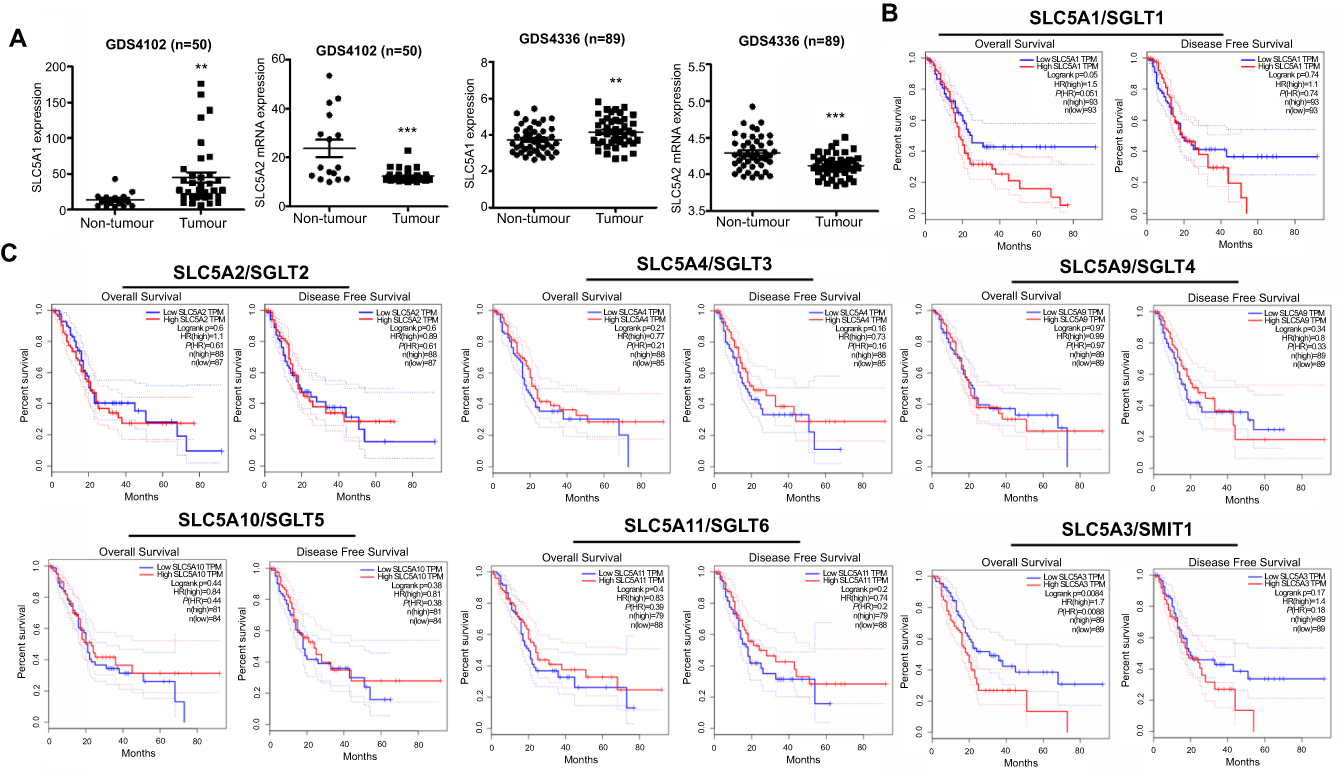 | Figure 1 SLC5A1 was over-expressed in human pancreatic cancer and associated with poor prognosis. (A) Comparison of mRNA expressions of SLC5A1 and SLC5A2 between human pancreatic tumor tissues and normal tissue. Increased SLC5A1 and reduced SLC5A2 were observed in pancreatic tumor tissue as compared to normal tissue. (B) Kaplan-Meier plots of overall and disease-free survival in pancreatic cancer patients with low SLC5A1 and high SLC5A1. Patients with high SLC5A1 is associated with reduced overall survival. (C) Kaplan-Meier plots of overall and disease-free survival in pancreatic cancer patients with other SGLTs expression. There is no significant correlation in SGLTs expression with patients’ overall and disease-free survival rate. **P<0.01, ***P<0.0001. |
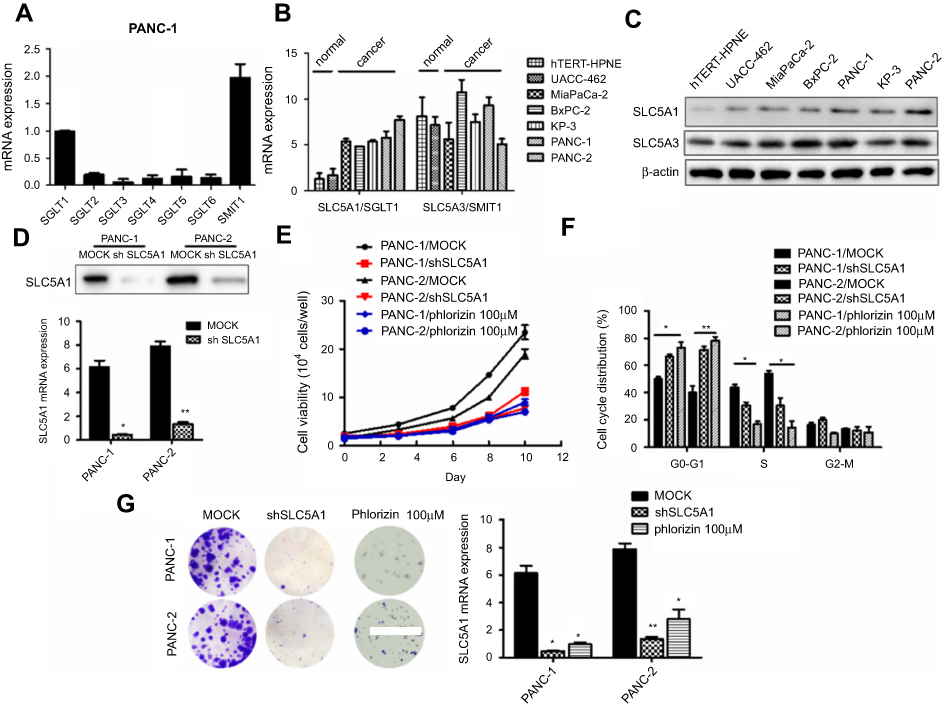 | Figure 2 SLC5A1 blockade suppressed pancreatic cell growth. (A) The mRNA of SGLTs in pancreatic cancer Panc-1 cells. Only the mRNA level of SGLT1 (SLC5A1) and SMIT1 (SLC5A3) showed relative high level of expression in Panc-1 cells. (B) the mRNA expression and (C) the protein expression of SLC5A1 and SLC5A3 in different pancreatic cancer cell lines and normal pancreatic cancer cells. SLC5A1 but not SLC5A3 was overexpressed in pancreatic cancer cells compared to normal cells. (D) Expressions of SLC5A1 in Panc-1 and Panc-2 cells were silenced by shRNA and validated by qPCR and immunoblotting. (E) Panc-1 and Panc-2 cells transfected with shSLC5A1 or scramble control were seeded, and cell number was counted every two days for 10 days. Inhibition of SLC5A1 reduced pancreatic cell growth. A non-selective SGLT1 inhibitor phlorizin, showed similar effects to SLC5A1 knockdown. (F) Cell cycle distribution analysis of 72 hours-cultured Panc-1 and Panc-2 cells with or without SLC5A1 knockdown. Inhibition of SLC5A1 arrested the pancreatic cancer cell lines at G0/G1 phase. A non-selective SGLT1 inhibitor phlorizin, showed similar effects to SLC5A1 knockdown. (G) SLC5A1 knockdown Panc-1 and Panc-2 cells showed reduced clonogenic survival compared to control cells. A non-selective SGLT1 inhibitor phlorizin, showed similar effects to SLC5A1 knockdown. *P<0.05, **P<0.01, ***P<0.0001. |
Inhibition of SLC5A1 interferes with pancreatic cancer cell growth
Observing the over-expression of SLC5A1 in most pancreatic cancer cell lines, we further examine if SLC5A1 contributes to the growth functions of pancreatic cancer. Due to the higher expressions of SLC5A1 on human Panc-1 and murine Panc-2 cell lines as compared to other pancreatic cancer cells line (Figures 2C), the two cell lines will be utilized throughout the study. To further understand how SLC5A1 regulates pancreatic cancer cell growth, we have stably transfected human Panc-1 and murine Panc-2 cells with shRNA plasmid targeting SLC5A1 (Figure 2D). Furthermore, similar analysis was conducted on GLUT1-4 proteins. We found that only GLUT1 expression was increased in pancreatic cancer cells, and was correlated with poor survival of the patients (Figure S1). However, knockdown of SGLT1 has no significant impact on the expression of GLUT1 in pancreatic cancer cells (Figure S2). This finding is consistent with the observation in another previous study.17 The inhibition of SLC5A1 reduced the viability of Panc-1 and Panc-2 cells as compared to their negative control counterparts (Figure 2E). Mechanistically, inhibition of SLC5A1 arrested the pancreatic cancer cell lines at G0/G1 phase, therefore leading to accumulation of cancer cells at G0/G1 phase and concomitant decrease of S phase (Figure 2F). Further clonogenic study confirmed that silencing of SLC5A1 decreased clonogenic survival in pancreatic cancer cells (Figure 2G). The effect of SLC5A1 knockdown on the proliferation and survival of pancreatic cancer cells was similar to a non-specific SGLT inhibitor, phlorizin. All this result proposes that SLC5A1 blockade may blunt the pancreatic cancer cell growth and proliferation.
Inactivation of SLC5A1 suppresses in vivo pancreatic cancer growth
To further evaluate if aiming at SLC5A1 suppresses pancreatic cancer growth in vivo, we established the orthotopic Panc-2 implantation mouse model in which luciferase-tagged Panc-2 cells expressing shRNA-control or shRNA-SLC5A1 were injected into the mice pancreas. The pancreatic tumor growth of mice was monitored weekly using non-invasive live animal imager. A significant decrease in tumor growth after the second week was observed in the mice with silencing of SLC5A1 (Figure 3A), as evidenced from the observation of reduced luciferase signal intensity. Further observation showed that inhibition of SLC5A1 in Panc-2 potently improved the survival of mice as compared to negative control group of mice (Figure 3B). Whereas there was no difference in body weight of mice between groups (Figure 3C), suggesting the safety of SLC5A1 blockade in pancreatic cancer cells. By the end of the experiment, the mice were sacrificed, and pancreatic tumor were harvested and weighed. Consistently, the pancreatic tumor weight was significantly reduced in the SLC5A1 silencing group of mice (Figure 3D), suggesting that there was reduced tumor growth after SLC5A1 inhibition. Taken together, the in vitro and in vivo results postulate that inhibition of SLC5A1 in pancreatic cancer cells interfered with the cancer cell growth and progression.
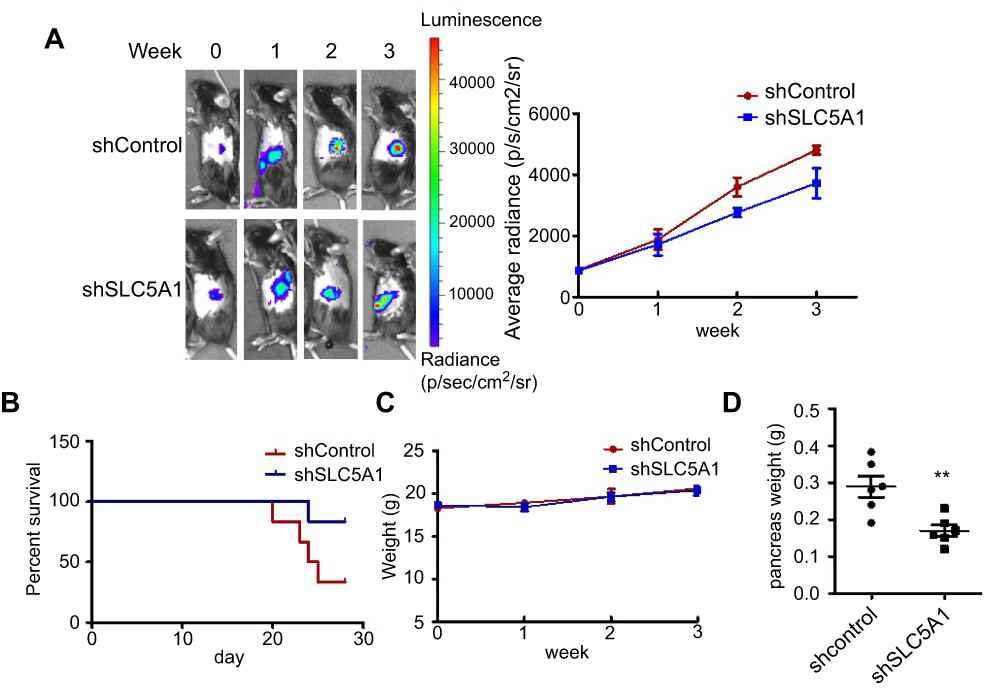 | Figure 3 Silencing of SLC5A1 reduced in vivo pancreatic cancer growth. (A) Representative images of Panc-2-implanted mice captured every week throughout 3 weeks of experiment. Knockdown of SLC5A1 decreased in vivo tumor growth. (B) Kaplan-Meier survival plots of mice implanted with Panc-2 with or without SLC5A1 knockdown. Knockdown of SLC5A1 enhanced mice survival. (C) Weekly monitoring of body weight of mice. There were no significant changes in body weight throughout the study period. (D) The weight of tumour-bearing pancreas was reduced in mice implanted with SLC5A1 knockout Panc-2. **P<0.01. |
Reduced SLC5A1 mediated glucose transport suppresses pancreatic cancer growth
Since glucose is the key energy source of all cells especially tumor cells and SLC5A1 is one of the glucose carriers for transporting glucose into tumors, we further examined if inhibition of SLC5A1 interrupts the glucose uptake ability of cancer cells. Notably, inhibition of SLC5A1 resulted in reduced 2-NBDG, a fluorescent-labeled deoxy-glucose analog in both Panc-1 and Panc-2 (Figure 4A). Observing glucose is the major substrate regulated by SLC5A1 in pancreatic cancer cells, we further assessed if glucose replenishment in cancer cells rescues the cellular growth. It was observed that reduced cell viability because of SLC5A1 inhibition could be recovered by increasing the glucose concentration to 50 mM in culture medium (Figure 4B), whereas low glucose (0.5 mM) content triggered marked cellular death in both SLC5A1 knockout and negative control cells. All these suggest that the cancer growth suppression resulting from the SLC5A1 blockade contributed to the reduced glucose transportation into tumor cells.
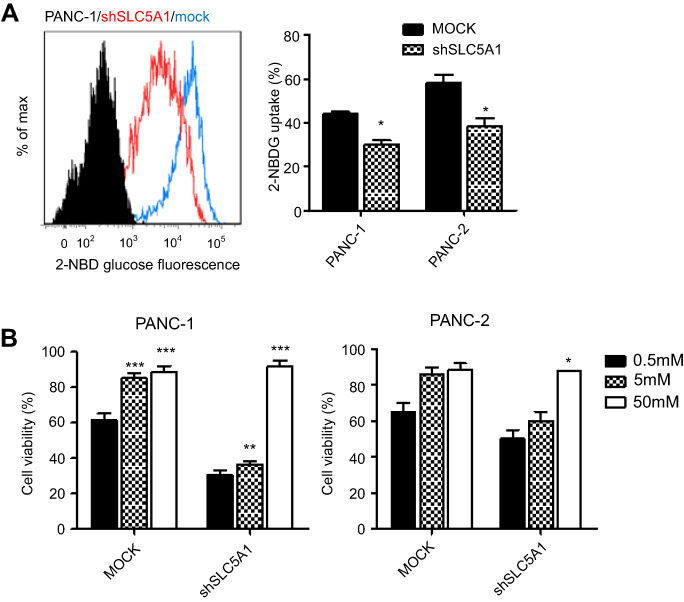 | Figure 4 Inhibition of SLC5A1 suppressed glucose uptake of pancreatic cancer cells. (A) Comparison of histogram from flow cytometry analysis. Knockdown of SLC5A1 reduced uptake of 2-NBDG by cancer cells. (B) Cells with or without SLC5A1 inhibition were cultured with 0.5 mM, 5 mM and 50 mM of glucose and cell viability was determined after 72 hours. 50 mM of glucose culture recovered the cell death induced by SLC5A1 inhibition. *P<0.05, **P<0.01, ***P<0.0001. |
SLC5A1-regulated cancer cell growth is dependent on AMPK/mTOR signaling
Many studies have postulated that the activation of AMPK/mTOR signaling in mediating cellular energy homeostasis in response to cellular stress or low cellular energy.18 We therefore examined the effect of SLC5A1 blockade on AMPK/mTOR signaling in Panc-1 cells. The cells cultured in low glucose content (0.5 mmol/L) are used as positive control cells. Notably, the expressions of phosphorylated-AMPK were increased while phosphorylated-MTOR was significantly decreased in SLC5A1 blockade cells but not in its negative control counterpart (Figure 5A). A similar trend in AMPK/mTOR expression was observed in cells cultured in low glucose content. To further confirm the role of AMPK activation in SLC5A1 knockout cells, we pretreated the cells with compound C, the chemical inhibitor of AMPK and the cell viability was again assessed. It was observed that compound C treatment rescued the pancreatic cancer cell viability after SLC5A1 inhibition (Figure 5B). Similarly, the prolonged G0/G1 phase decreased clonogenic survival because SLC5A1 inhibition were diminished after compound C intervention (Figure 5C and D). In addition, we treated the pancreatic cancer cells with Torin 1, a mTOR inhibitor, to confirm the essential role of mTOR phosphorylation in glucose-induced pancreatic cancer cells proliferation and survival, and we found that Torin 1 can significantly reduce the growth of pancreatic cancer cells. All these suggested that SLC5A1-mediated pancreatic cancer cell growth is regulated by AMPK/mTOR signaling.
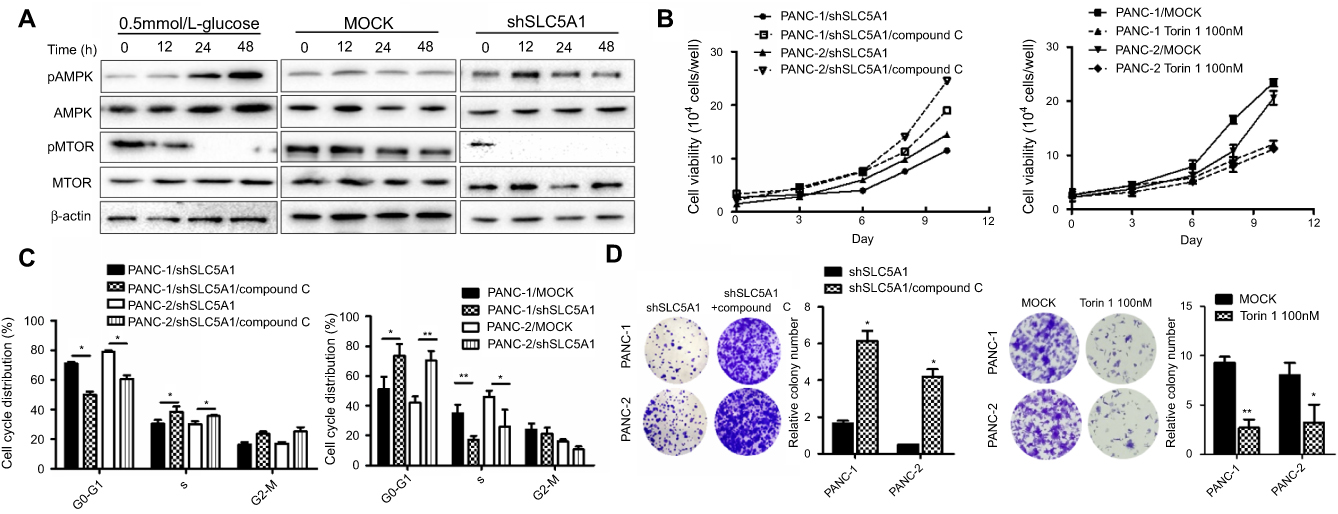 | Figure 5 SLC5A1-regulated cancer cell growth is dependent on AMPK/mTOR signaling. (A) Immunoblotting analysis of phosphorylated-AMPK and mTOR with cell lysates from Panc-2 with or without SLC5A1 blockade and cultured with 0.5 mmol/L-glucose. Protein expression of phosphorylated-AMPK was increased while phosphorylated-MTOR was decreased in SLC5A1 blockade cells. (B) Pretreatment with compound C (1 μM) recovered the viability of SLC5A1 knockdown pancreatic cancer cells. (C) Compound C diminished the prolonged G0/G1 arrest induced in SLC5A1 knockdown Panc-1 and Panc-2 cells. (D) Compound C recovered the clonogenic survival in SLC5A1 knockdown Panc-1 and Panc-2 cells. *P<0.05. |
The association of SLC5A with EGFR mediates pancreatic cancer cell growth
Furthermore, we assessed the potential interaction of SLC5A1 with other proteins by analyzing the InBio Mapo database. It was noted that ATPases (ATP1A3, ATP1A2, and ATP1A1), HSP70 members (HSPA1A and HSPA1B), PAWR and EGFR have possible protein-protein interaction with SGLT1 encoded by SLC5A1 are the target ligands of SLC5A1 (Figure 6A). To further examine the clinical correlation of SLC5A1 and EGFR, we analyzed the mRNA expressions of SLC5A1 and EGFR from 149 pairs of patients extracted from the TCGA database. It was confirmed that SLC5A1 and EGFR are positively correlated (**P=0.0035) (Figure 6B). A previous study showed that EGFR may interact with SGLT1 encoded by SLC5A1 to suppress its expression.19 Consistently, we observed by co-immunoprecipitation that EGFR can bind with SGLT1 protein in pancreatic cancer cells (Figure 6C). We thereby transiently transfected human Panc-1 and murine Panc-2 cells with siRNA plasmid targeting EGFR. In accordance with the silence of EGFR protein, it was observed that the SLC5A1 protein expression was also correspondingly reduced (Figure 6D). Based on the analysis of expression pattern and clinical significance of SGLTs and GLUTs proteins as aforementioned, we found that only SGLT1 and GLUT1 may have aberrant expression which is correlated with the clinical outcome of pancreatic cancer. However, it had minimal effect on the protein level of GLUT1 in pancreatic cancer cells. This was similarly observed in another previous study.19 Consistently, inhibition of EGFR in pancreatic cancer cells also resulted in reduction of 2-NBDG uptake (Figure 6E), followed by cancer cell death (Figure 6F). As expected, silencing of EGFR also activated AMPK/mTOR signaling as evidenced from the observation of increased phosphorylated-AMPK and decrease of phosphorylated-mTOR (Figure 6G). In sum, our findings propose the association of SLC5A1 with EGFR-regulated pancreatic cancer cell growth through AMPK/mTOR signaling.
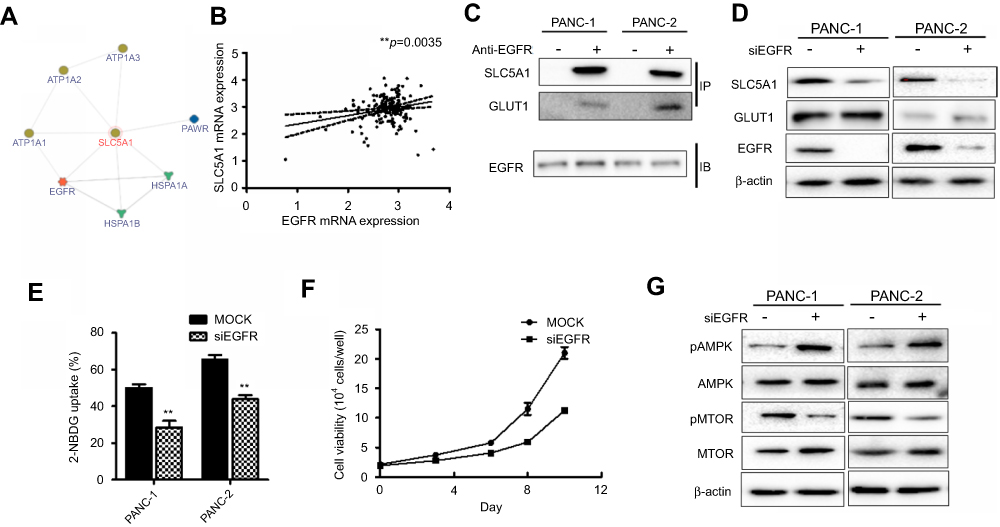 | Figure 6 The association of SLC5A with EGFR mediates pancreatic cancer cell growth. (A) SLC5A1 is closely co-related with EGFR with a confidence score of 0.38. (B) The positive correlation between SLC5A1 and EGFR mRNA expressions. (C) co-immunoprecipitation assay showing the interaction between SLC5A1 and EGFR protein. (D) Immunoblotting analysis of SLC5A1 with cell lysates from cells transfected with siEGFR or scrambles negative control. Protein expression of SLC5A1 was reduced consistently in EGFR knockdown cells. (E) Knockdown of EGFR decreased uptake of 2-NBDG by cancer cells. (F) Knockdown of EGFR reduced cancer cell viability as compared to mock cells. Torin 1, a mTOR inhibitor, showed similar effect to EGFR knockdown on cell viability. (G) Immunoblotting analysis of phosphorylated-AMPK and mTOR with cell lysates from EGFR knockdown cells. Protein expression of p-AMPK was increased while p-mTOR was reduced in EGFR knockdown cells. **P<0.01. |
Discussion
The need of tumor cells for energy for their uncontrolled growth is predatory.20 Several studies have reported that pancreatic tumor cells applied several special pathways to grab resources of energy from the tumor microenvironment.21–24 This does not only includes the overexpression of glycolysis and oxidative phosphorylation-associated enzymes,25 but also the forced expression of transporters of essential substrates that directly or indirectly contribute to the energy production. As the direct source of energy production, glucose was transported across the plasma membrane of tumor cells.26 While the members in the GLUT family transport glucose at the basolateral membrane of the cells,27 SGLT1 encoded by SLC5A1 and SGLT2 encoded by SLC5A2, facilitate the glucose transport at the apical membrane.28 The aberrant expression of the GLUT family has been widely reported in pancreatic cancers,29 however, transporters at the apical membrane were less studied. In our study, we found that pancreatic tumour cells required a forced expression of SLC5A1 as well as its coding protein SGLT1. Although under physiological contribution SGLT2 contributes to around 90% of glucose absorption at the apical side,30 its expression was not up-regulated in pancreatic tumor and showed no significance with patients’ survival rate. Our study also further showed overexpression of SLC5A1 may promote the tumor progression of pancreatic cancer. The fact of this oncogenic effect of SLC5A1 was dependent on glucose has suggested that SLC5A1 overexpression in cancer cells is to cater for the need of cells on glucose for energy production. Suppression of SLC5A1 or deprivation of glucose stopped the tumor progression by inducing an AMPK-dependent mTOR suppression. As the mTOR pathway controls several critical energy-requiring biological processes involving cell proliferation and survival,31 our findings have depicted an important mechanism underlying the oncogenic protein expression to fulfill the requirement of tumor progression, which include a specific overexpression of non-classical glucose transporter SGLT1 to facilitate as much glucose influx as possible for energy production.
Previous studies have shown that high glucose concentration can increase the expression of SGLT1 in intestine cells, and overexpression of SGLT1 but not GLUT2 which predominantly mediates the transportation and absorption of glucose.32 In animal models of obese diabetes, hyperglycemia could also increase the tissue expression of SGLT1.33 This kind of expression pattern can also be observed in other SGLT family proteins such as SMIT1.34 In our study, we found that the pro-tumoral function of SGLT1 in pancreatic cancer was associated with its function as a glucose transporter. Knockdown of SGLT1 activated glucose deprivation-associated proliferation inhibition. This finding combined with the previous report of glucose-dependent SGLT1 expression suggested that SGLT1 may be an essential mediator in glucose-initiated proliferative signaling in pancreatic cancer cells. SLGT1 expression could be initiated in response to glucose, which leads to more glucose transport to fuel the proliferation of pancreatic cancer cells.
It was noticed that expression and function of SLC5A1 was associated with EGFR expression in pancreatic cancer cells. Co-expression of EGFR with SLC5A1 mRNA in pancreatic tumour tissue was observed and suppressing EGFR expression in PAAD cells by RNA interference significantly blocked expression and function of SLC5A1 protein. It was first identified in 2008 by Weihua et al19 that EGFR expression in human cancer cells was critical for the maintenance of intracellular glucose levels through interaction and stabilization of SGLT1 protein, while this function of EGFR was independent to its kinase activity. The regulation of EGFR on SGLT1 protein expression was found to be independent of the transcription-related mechanism, as in oral squamous cell carcinoma cells mRNA and protein expression of SGLT1 was not compatible.35 However, it was also found that EGFR may regulate the phosphorylation of CREB in the presence of EGF, which in turn bound to SGLT1 gene promoter to induce its transcription.36 These findings suggested EGFR-dependent and -independent roles in SGLT1 expression.37 Further study suggested that co-expression of EGFR and SGLT1 indicated a poor prognosis in human colorectal cancer, while SGLT1 alone had no prognostic value. This again suggested that the oncogenic role of SGLT1 may be highly associated with EGFR expression in cancer.8 The upstream of EGFR in SGLT1 expression in cancers was indirectly proved by the observation that HDACs inhibitor synchronously suppressed EGFR and SGLT1 in the cells,38 while EGFR mediated the transmissible gastroenteritis virus (TGEV)-induced SGLT1 expression and glucose uptake. In our study, we observed that RNA interference against EGFR inhibited the expression of SGLT1 as well as pancreatic cell survival. This gives a relatively direct evidence that EGFR stands as the upstream signaling of SGLT1 in pancreatic cancer. Inhibition of EGFR in our study led to PAAD cell death with activation of AMPK signaling (Figure 7). It was previously found that in cancer cell undergoing ionizing radiation, co-expression of EGFR and SGLT1 facilitated the chromatin remodeling to increase glucose uptake, so that cancer cells can produce more energy to counteract the ATP crisis induced by radiotherapy.39 The loss of ATP in cancer cells with suppression of EGFR in our study activated its downstream AMPK signaling, which in turn inhibited mTOR pathway. Our observation together with these previous findings have suggested that EGFR/SGLT1 protein pair should be a potential target for cancer treatment. Indeed, attempts to suppress cancer cells by targeting this protein pair have been made. It was found that some compounds from a natural anti-cancer medicinal plant Panax ginseng concurrently suppressed EGFR and SGLT1 in a CREB-associated mechanism.40,41 Co-inhibition of EGFR and SGLT1 obtained an optimal outcome in suppressing prostate cancer compared with EGFR inhibitor alone. More systematic evaluation on the efficacy and safety of co-inhibition on EGFR/SGLT1 protein in cancer treatment should be performed in the future.
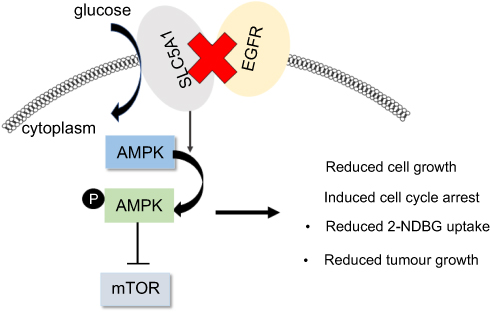 | Figure 7 Reduced co-expressions of SLC5A1 with EGFR suppressed pancreatic cancer cell growth through AMPK/mTOR signaling. |
We noticed that the orthotopic pancreatic cancer mice experienced better survival without significant body weight loss. The presence of cachexia and loss of body weight in cancer patients during tumor progression is one of the major reasons that lead to poor survival, but unfortunately, it is not easy to observe similar cachexia in animal model of cancers. The animal models of cancer, both xenografted and orthotopic ones, are unsatisfactory for assessing cachexia-related outcomes, such as physical activity and quality of life, because of the larger proportional size and aggressive doubling of the tumor rate, which lead to different biological behavior from the clinical setting.42 Although a few of the cancer-associated cachexia models have been reported, pancreatic cancer models have not been fully addressed with observational cachexia.43 In addition, we actually observed that ascites in our animal models. The presence of ascites may cause fluctuation on the body weight data in this animal model. More ascites were observed in mice with wild type Panc2 cells, while mice implanted with SLC5A1-knockdowned Panc-2 cells due to the smaller tumor burden and progression (data not shown). A previous study has shown that ascites was a significant factor leading to poor survival of pancreatic cancer animals.44 Although we cannot fully address the reason of better survival in the knockdown group, it is possible that less ascites are associated with better prognosis of mice with SCL5A1-knockdowned Panc-2 cells.
Conclusion
In conclusion, our study reported a novel finding in the aberrant expression and oncogenic function of SLC5A1 gene in human pancreatic cancer. The aberrant expression of SLC5A1 is observed in pancreatic cancer patients as compared to normal tissue and associated with reduced overall patient survival. Blockade of SLC5A1 suppressed in vitro and in vivo pancreatic cancer cell survival, and the effect was dependent on reduced glucose uptake by cancer cells. Suppression of SLC5A1 or glucose deprivation further induced an AMPK-dependent mTOR suppression. We have further demonstrated that SLC5A1 is positively correlated and dependent on EGFR. Our current findings identified that SLC5A1/EGFR is involved in pancreatic cancer growth and a potential therapeutic candidate for pancreatic cancer patients.
Acknowledgment
This work was financially supported by the National Natural Science Foundation of China (81503405 and 81603444).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Luo J, Xiao L, Wu C, et al. The incidence and survival rate of population-based pancreatic cancer patients: Shanghai cancer registry 2004–2009. PLoS One. 2013;8(10): e76052. doi: 10.1371/journal.pone.0076052.
2. Veisani Y, Jenabi E, Khazaei S, Nematollahi S. Global incidence and mortality rates in pancreatic cancer and the association with the Human Development Index: decomposition approach. Public Health. 2018;156:87–91. doi:10.1016/j.puhe.2017.12.015
3. Castellanos JA, Merchant NB. Intensity of follow-up after pancreatic cancer resection. Ann Surg Oncol. 2014;21(3):747–751. doi:10.1245/s10434-013-3289-7
4. Kim MP, Gallick GE. Gemcitabine resistance in pancreatic cancer: picking the key players. Clin Cancer Res. 2008;14(5):1284–1285. doi:10.1158/1078-0432.CCR-07-2247
5. Ancey P-B, Contat C, Meylan E. Glucose transporters in cancer – from tumor cells to the tumor microenvironment. FEBS J. 2018;285(16):2926–2943. doi:10.1111/febs.2018.285.issue-16
6. Harada N, Inagaki N. Role of sodium‐glucose transporters in glucose uptake of the intestine and kidney. J Diabetes Investig. 2012;3(4):352–353. doi:10.1111/j.2040-1124.2012.00227.x
7. Mojica L, Luna-Vital DA, Gonzalez de Mejia E. Black bean peptides inhibit glucose uptake in Caco-2 adenocarcinoma cells by blocking the expression and translocation pathway of glucose transporters. Toxicol Rep. 2018;5:552–560. doi:10.1016/j.toxrep.2018.04.007
8. Guo GF, Cai YC, Zhang B, et al. Overexpression of SGLT1 and EGFR in colorectal cancer showing a correlation with the prognosis. Med Oncol. 2011;28(Suppl 1):S197–203. doi:10.1007/s12032-010-9696-8
9. Lei S, Yang J, Chen C, et al. FLIP(L) is critical for aerobic glycolysis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2016;35:79. doi:10.1186/s13046-016-0358-3
10. Ren J, Bollu LR, Su F, et al. EGFR-SGLT1 interaction does not respond to EGFR modulators, but inhibition of SGLT1 sensitizes prostate cancer cells to EGFR tyrosine kinase inhibitors. Prostate. 2013;73(13):1453–1461. doi:10.1002/pros.22692
11. Perez M, Praena-Fernandez JM, Felipe-Abrio B, et al. MAP17 and SGLT1 protein expression levels as prognostic markers for cervical tumor patient survival. PLoS One. 2013;8(2):e56169. doi:10.1371/journal.pone.0056169
12. Lai B, Xiao Y, Pu H, Cao Q, Jing H, Liu X. Overexpression of SGLT1 is correlated with tumor development and poor prognosis of ovarian carcinoma. Arch Gynecol Obstet. 2012;285(5):1455–1461. doi:10.1007/s00404-011-2166-5
13. Hanabata Y, Nakajima Y, Morita K, Kayamori K, Omura K. Coexpression of SGLT1 and EGFR is associated with tumor differentiation in oral squamous cell carcinoma. Odontology. 2012;100(2):156–163. doi:10.1007/s10266-011-0033-2
14. Casneuf VF, Fonteyne P, Van Damme N, et al. Expression of SGLT1, Bcl-2 and p53 in primary pancreatic cancer related to survival. Cancer Invest. 2008;26(8):852–859. doi:10.1080/07357900801956363
15. Scafoglio C, Hirayama BA, Kepe V, et al. Functional expression of sodium-glucose transporters in cancer. Proc Natl Acad Sci U S A. 2015;112(30):E4111–4119. doi:10.1073/pnas.1511698112
16. Qiu W, Su GH. Development of orthotopic pancreatic tumor mouse models. Methods Mol Biol. 2013;980:215–223. doi:10.1007/978-1-62703-287-2_11
17. Ramratnam M, Sharma RK, D’Auria S, et al. Transgenic knockdown of cardiac sodium/glucose cotransporter 1 (SGLT1) attenuates PRKAG2 cardiomyopathy, whereas transgenic overexpression of cardiac SGLT1 causes pathologic hypertrophy and dysfunction in mice. J Am Heart Assoc. 2014;3:4. doi:10.1161/JAHA.114.000844
18. Hardie DG, Ashford MLJ. AMPK: regulating energy balance at the cellular and whole body levels. Physiology. 2014;29(2):99–107. doi:10.1152/physiol.00050.2013
19. Weihua Z, Tsan R, Huang WC, et al. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13(5):385–393. doi:10.1016/j.ccr.2008.03.015
20. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
21. Sousa CM, Kimmelman AC. The complex landscape of pancreatic cancer metabolism. Carcinogenesis. 2014;35(7):1441–1450. doi:10.1093/carcin/bgu097
22. Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi:10.1038/nature12040
23. Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149(3):656–670. doi:10.1016/j.cell.2012.01.058
24. Sousa CM, Biancur DE, Wang X, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536(7617):479–483. doi:10.1038/nature19084
25. Coller HA. Is cancer a metabolic disease? Am J Pathol. 2014;184(1):4–17. doi:10.1016/j.ajpath.2013.07.035
26. Ooi AT, Gomperts BN. Molecular pathways: targeting cellular energy metabolism in cancer via inhibition of SLC2A1 and LDHA. Clin Cancer Res. 2015;21(11):2440–2444. doi:10.1158/1078-0432.CCR-14-1209
27. Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med. 2013;34(2–3):121–138. doi:10.1016/j.mam.2012.07.001
28. Roder PV, Geillinger KE, Zietek TS, Thorens B, Koepsell H, Daniel H. The role of SGLT1 and GLUT2 in intestinal glucose transport and sensing. PLoS One. 2014;9(2):e89977. doi:10.1371/journal.pone.0089977
29. Chen X, Lu P, Zhou S, Zhang L, Zhao JH, Tang JH. Predictive value of glucose transporter-1 and glucose transporter-3 for survival of cancer patients: a meta-analysis. Oncotarget. 2017;8(8):13206–13213. doi:10.18632/oncotarget.14570
30. Abdul-Ghani MA, DeFronzo RA, Norton L. Novel hypothesis to explain why SGLT2 inhibitors inhibit only 30–50% of filtered glucose load in humans. Diabetes. 2013;62(10):3324–3328. doi:10.2337/db13-0604
31. Alayev A, Holz MK. mTOR signaling for biological control and cancer. J Cell Physiol. 2013;228(8):1658–1664. doi:10.1002/jcp.24351
32. Gorboulev V, Schurmann A, Vallon V, et al. Na(+)-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes. 2012;61(1):187–196. doi:10.2337/db11-1029
33. Szablewski L. Distribution of glucose transporters in renal diseases. J Biomed Sci. 2017;24(1):64. doi:10.1186/s12929-017-0371-7
34. Li SYT, Cheng STW, Zhang D, Leung PS. Identification and functional implications of sodium/myo-inositol cotransporter 1 in pancreatic beta-cells and Type 2 diabetes. Diabetes. 2017;66(5):1258–1271. doi:10.2337/db16-0880
35. Hanabata Y. Expression analysis of EGFR and SGLT1 in oral squamous cell carcinoma. Kokubyo Gakkai Zasshi. 2011;78(1):12–18.
36. Wang CW, Chang WL, Huang YC, et al. An essential role of cAMP response element-binding protein in epidermal growth factor-mediated induction of sodium/glucose cotransporter 1 gene expression and intestinal glucose uptake. Int J Biochem Cell Biol. 2015;64:239–251. doi:10.1016/j.biocel.2015.04.006
37. Cossu-Rocca P, Muroni MR, Sanges F, et al. EGFR kinase-dependent and kinase-independent roles in clear cell renal cell carcinoma. Am J Cancer Res. 2016;6(1):71–83.
38. Chou CW, Wu MS, Huang WC, Chen CC. HDAC inhibition decreases the expression of EGFR in colorectal cancer cells. PLoS One. 2011;6(3):e18087. doi:10.1371/journal.pone.0018087
39. Dittmann K, Mayer C, Rodemann HP, Huber SM. EGFR cooperates with glucose transporter SGLT1 to enable chromatin remodeling in response to ionizing radiation. Radiother Oncol. 2013;107(2):247–251. doi:10.1016/j.radonc.2013.03.016
40. Wang CW, Su SC, Huang SF, et al. An essential role of cAMP response element binding protein in ginsenoside Rg1-mediated inhibition of Na+/Glucose cotransporter 1 gene expression. Mol Pharmacol. 2015;88(6):1072–1083. doi:10.1124/mol.114.097352
41. Wang CW, Huang YC, Chan FN, et al. A gut microbial metabolite of ginsenosides, compound K, induces intestinal glucose absorption and Na(+)/glucose cotransporter 1 gene expression through activation of cAMP response element binding protein. Mol Nutr Food Res. 2015;59(4):670–684. doi:10.1002/mnfr.201400688
42. Donohoe CL, Ryan AM, Reynolds JV. Cancer cachexia: mechanisms and clinical implications. Gastroenterol Res Pract. 2011;2011:601434. doi:10.1155/2011/601434
43. Deboer MD. Animal models of anorexia and cachexia. Expert Opin Drug Discov. 2009;4(11):1145–1155. doi:10.1517/17460440903300842
44. Akladios C, Ignat M, Mutter D, Aprahamian M. Survival variability of controls and definition of imaging endpoints for longitudinal follow-up of pancreatic ductal adenocarcinoma in rats. J Cancer Res Clin Oncol. 2017;143(1):29–34. doi:10.1007/s00432-016-2265-6
Supplementary materials
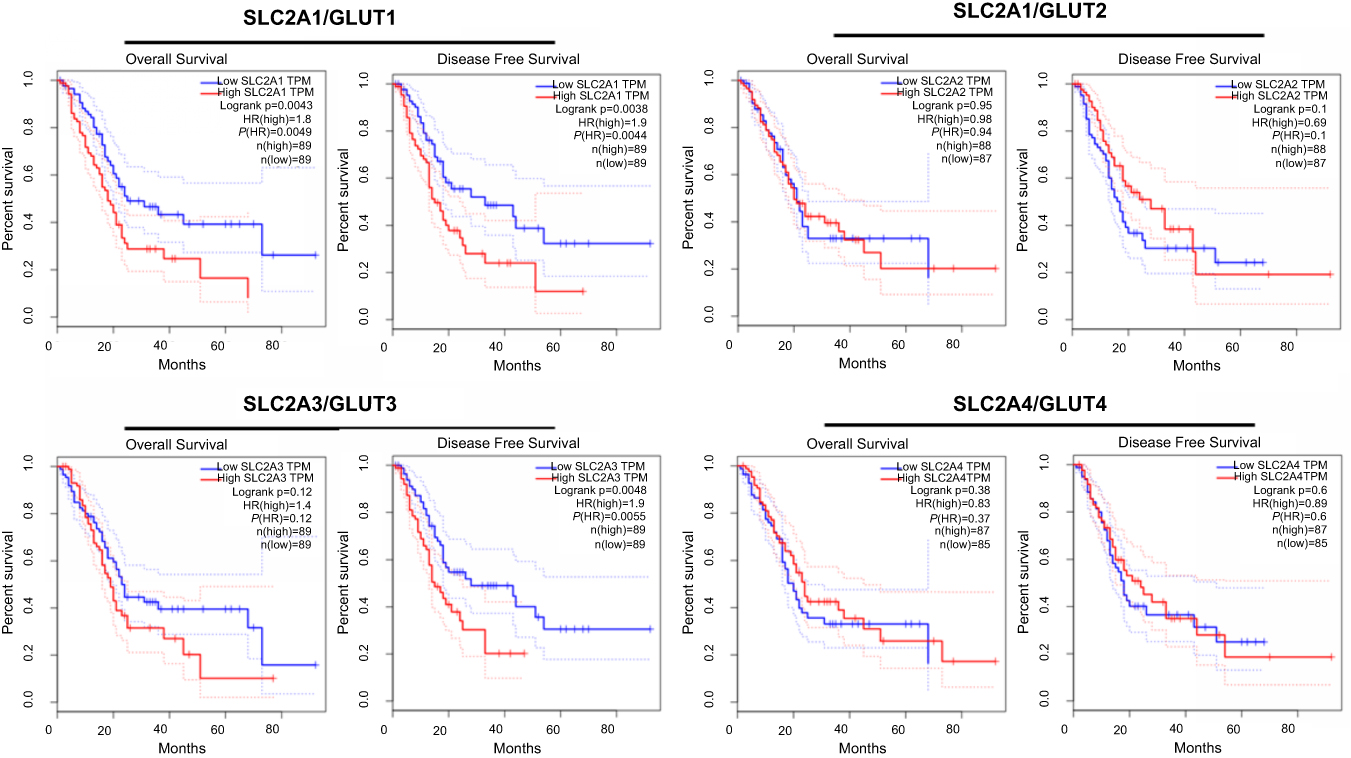 | Figure S1 Kaplan-Meier plots of overall and disease-free survival in pancreatic cancer patients with low GLUTs and high GLUTs. Patients with high GLUT1 but not GLUT2-4 is associated with reduced overall survival. |
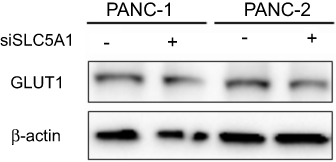 | Figure S2 Knockdown of SLC5A1 has no significant effect on GLUT1 expression. Panc-1 and Panc-2 cells expressing siRNA against SLC5A1 was collected for immunoblotting. No significant change on GLUT1 expression in both cells was observed after SLC5A1 knockdown. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.