")
Back to Journals » OncoTargets and Therapy » Volume 11
SIRT4 acts as a tumor suppressor in gastric cancer by inhibiting cell proliferation, migration, and invasion
Authors Sun H, Huang D, Liu G, Jian F, Zhu J, Zhang L
Received 5 November 2017
Accepted for publication 23 January 2018
Published 10 July 2018 Volume 2018:11 Pages 3959—3968
DOI https://doi.org/10.2147/OTT.S156143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Hongjie Sun,1 Dongli Huang,1 Guozheng Liu,1 Fengguo Jian,1 Jianfang Zhu,1 Lixia Zhang2
1Department of General Surgery, Changyi People’s Hospital, Changyi, Shandong, People’s Republic of China; 2Department of Nuclear Medicine, Zhejiang Provincial Hospital of Traditional Chinese Medicine (The First Affiliated Hospital of Zhejiang Chinese Medical University), Hangzhou, Zhejiang, People’s Republic of China
Background: Previous study has proven that SIRT4 is downregulated in gastric cancer (GC), but the role of SIRT4 has not been clearly understood. The aim of our work was to explore in detail the function and mechanism of SIRT4 in GC.
Methods: A total of 86 pairs of GC tumor tissues and adjacent normal tissues were collected, and quantitative real-time polymerase chain reaction and Western blotting analyses were used to determine the expression of SIRT4.
Results: Our study revealed that the expression of SIRT4 was downregulated in GC tissues and cells. In addition, the low expression of SIRT4 was negatively correlated with tumor size, pathological grade, and lymph node metastasis, which predicted a poor prognosis. Multiple functional experiments, including Cell Counting Kit-8 assay as well as colony formation assay, demonstrated SIRT4 suppressed cell proliferation. Moreover, we found epithelial–mesenchymal transition was regulated by SIRT4, thereby regulating cell migration and invasion.
Conclusion: Overall, our findings show that SIRT4 serves as a tumor suppressor in GC and might act as a novel biomarker and a therapeutic target of GC.
Keywords: sirtuin 4, proliferation, migration, invasion, epithelial–mesenchymal transition, EMT
Introduction
Gastric cancer (GC) has become one of the most common malignant cancers, with a high mortality rate.1 Almost 70% of GC cases and deaths occur in developing countries, and 50% of cases are seen in eastern Asian countries such as Japan and China.2 Unfortunately, there are no early signs of GC; most patients are diagnosed at a late stage, and therefore the effect of treatment is very poor in such cases.3,4 Thus, it is necessary to find effective markers and develop new therapies for GC. Epithelial–mesenchymal transition (EMT) plays a crucial role in cancer progression, such as in metastasis.5 The main character of EMT is the gain of mesenchymal properties and loss of epithelial properties, thereby promoting cell migration and invasion.6 However, the detailed mechanism of EMT in GC is still not clearly known.
Sirtuin family consists of SIRT1–SIRT7. Increasingly, studies have shown that sirtuin family exhibits ADP-ribosylase and ADP-deacetylase activities and is involved in several cellular processes, including longevity, stress response, and metabolism.7,8 Sirtuin family is found in different cellular locations: SIRT1, SIRT6, and SIRT7 have been found to locate in the nucleus, SIRT2 in the cytoplasm, and SIRT3, SIRT4, and SIRT5 in the mitochondria.9 Unlike other members of the sirtuin family, SIRT4 has ADP-ribosyltransferase activity on histones instead of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase activity.10 Several reports suggest that SIRT4 plays a key function in the regulation of cellular metabolism and maintenance of genomic stability.11–14 Many studies indicate SIRT4 serves as a tumor suppressor and inhibits cell growth and proliferation as well as transformation through suppressing glutamine anaplerosis.13,15–17 In addition, the inhibition of glutamine metabolism has been found to result in phosphorylation and activation of p53, thereby maintaining genomic stability.18 Moreover, a clinical meta-analysis reveals that the mRNA expression of SIRT4 is selectively downregulated in a variety of malignant carcinomas such as lung cancer.13 Downregulation of SIRT4 is closely correlated with several aggressive cancers, including breast cancer, colorectal cancer, and GC.13,19,20 However, the detailed mechanisms of SIRT4 in GC are not completely known.
In the present study, we found that SIRT4 was downregulated in GC tissues and cell lines, and its expression was associated with the tumor size, metastasis, and pathological grade. Interestingly, low expression of SIRT4 predicted a poor prognosis. Moreover, SIRT4, a tumor suppressor, inhibited cell proliferation. In addition, SIRT4 regulated E-cadherin expression, thereby suppressing EMT and cell migration as well as invasion of GC cells. In conclusion, we suggest SIRT4 as a novel therapeutic target for GC.
Materials and methods
Cell lines
Human GC cell lines, MKN-45 and HGC-27, and human normal gastric mucosal cell line GES-1 were purchased from the Cancer Research Institute of Central South University (Changsha, People’s Republic of China). All cells were incubated with RPMI-1640 medium supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin (Thermo Fisher Scientific, Waltham, MA, USA) as well as 20% fetal bovine serum (FBS; Thermo Fisher Scientific) at 37°C in a humidified incubator with 5% CO2. All cell lines were approved by the Clinical Research Ethics Committee of Changyi People’s Hospital.
Patients and specimens
A total of 86 pairs of GC tumor tissues and adjacent normal tissues were obtained from patients who were diagnosed with GC in Changyi People’s Hospital during 2013–2016. All the patients were informed about the experiments and signed informed consent. The tissues-associated experiments were approved by the Clinical Research Ethics Committee of Changyi People’s Hospital. The patients did not receive any therapy, such as radiotherapy or chemotherapy, before surgical operation. The clinicopathological characteristics of the tumor tissues, including TNM stage, tumor size, metastasis, and differentiation, were determined and diagnosed independently, according to the WHO guidelines of classification.
Plasmid and cell transfection
Empty vector (pCMV-Tag2B) and plasmids containing SIRT4 were purchased from OriGene (Rockville, MD, USA). Scramble siRNA (SCR) and SIRT4 siRNA (siSIRT4) were bought from Dharmacon (Lafayette, CO, USA). All transfections were performed using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. After transfection for 48 h, the cells were collected and used for further experiments (siSIRT4#1: 5′-AAGAGTTACAGCGCTTCATCACC-3′ and siSIRT4#2: 5′-CGATTGCAATACTGAACATTGG-3′).
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
TRIzol reagent (Invitrogen) was used to extract total RNA from tissues or cells according to the manufacturer’s instructions. For RNA extraction, 2 μg cDNA was reverse-transcribed using TransScript First-Strand cDNA Synthesis SuperMix (TransGen, Beijing, People’s Republic of China) according to the manufacturer’s instructions. Then, qRT-PCR was performed using TransStart Top Green qPCR SuperMix (TransGen). Relative mRNA expression was normalized against GAPDH. The relative expression was analyzed by the 2−ΔΔCt method.
Western blotting
RIPA buffer was used to extract total protein from the tissues and cells according to the manufacturer’s instructions. Protein concentration was measured using a protein assay kit (Thermo Fisher Scientific). Equal amounts of total protein (40 μg) were separated using 8% SDS-PAGE, and then transferred to nitrocellulose filter membranes (EMD Millipore, Billerica, MA, USA) and blocked in 5% skimmed milk in PBST (Beyotime Institute of Biotechnology, Haimen, People’s Republic of China) at room temperature for 1 h. The membrane was incubated with the indicated antibodies at 4°C overnight, followed by washing with PBST for three times and incubating with an HRP-conjugated secondary antibody (Abcam) at room temperature for 1 h. After washing with PBST three times, the blots were detected using the enhanced chemiluminescence system (EMD Millipore).
Colony formation assay
Colony formation assay was performed to determine the effect of SIRT4 on cell proliferation. In brief, approximately 5 × 103 transfected MKN-45 and HGC-27 cells with serum-free RPMI-1640 medium were plated in six-well plates and cultured for 16 days. Cells were fixed with 4% paraformaldehyde solution at room temperature for 15 min and stained with 0.5% (w/v) crystal violet (Sigma, St. Louis, MO, USA) at room temperature for 15 min.
Cell Counting Kit (CCK)-8 assay
CCK-8 assay was performed to detect the effect of SIRT4 on cell proliferation according to manufacturer’s instructions. In brief, after transfection for 48 h, cells were collected and counted. Approximately 4 × 103 cells with 200 μL RPMI-1640 medium were seeded in 96-well plates, followed by which 20 μL CCK-8 solution was added to each well and incubated for 30 min at 37°C with 5% CO2. The absorbance of optical density 450 was measured using a microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Wound-healing assay
After transfection, MKN-45 and HGC-27 cells were cultured in 12-well plates. A 20 μL pipette tip was used to scratch a line wound when cells had grown to 70%–80% confluence. Subsequently, the detached cells were washed with PBS for three times, and the cells were cultured in fresh RPMI-1640 medium. Cell wound areas were imaged under a light microscope at the set treatment time points (0 and 36 h). The width of the cell wound was measured by ImageJ software. Distance of migration was calculated as follows: (width of wound at 0 h − width of wound at 36 h)/width of wound at 0 h × 100%.
Transwell assay
Transwell assays were performed to determine the effect of SIRT4 on cell invasion according to the description given earlier.21 Briefly, invasion chamber with 8 μm pores (Corning, Corning, NY, USA) was coated with 80 μL Matrigel and incubated at 37°C for 30 min. Approximately 3 × 104 transfected MKN-45 and HGC-27 cells with 500 μL serum-free RPMI-1640 medium were added to the upper 24-well chambers, and RPMI-1640 supplemented with 10% FBS was added into the lower chamber. The chambers were incubated at 37°C for 24 h. After removing the noninvading cells on the upper surface of the membrane with a cotton swab, the invading cells on the lower surface of the membrane were stained with 0.5% (w/v) crystal violet and counted under a light microscope.
Soft agar assay
Soft agar was used to determine the effect of SIRT4 on cell anchorage-independent growth ability. Briefly, 2X RPMI-1640 media which contained 20% FBS and equal volumes of 1.2% agar were mixed at 37°C for 30 min, with 0.6% agar as base agar, in six-well plates. SIRT4 was overexpressed or knocked down in MKN-45 and HGC-27 cells, and 3 × 104 cells were suspended in 1.5 mL of 0.7% agar and 1.5 mL of 2X RPMI-1640 (with 20% FBS), and then were seeded into each well. Cells were incubated at 37°C with 5% CO2 for 16 days. Colonies were stained with 0.5% crystal violet, and the number of colonies was counted under a light microscope.
Enzyme-linked immunosorbent assay (ELISA)
MKN-45 and HGC-27 cells were collected and plated into six-well culture plates at a density of 2 × 105 cells/well. Cell supernatants in serum-free medium were homogenized and harvested 72 h later and centrifuged at 1,000 g for 20 min. The levels of MMP2 and MMP9 were subsequently determined using commercially available ELISA kits (USCN Life Science, Houston, TX, USA), according to the manufacturer’s instructions.
Statistical analysis
All data were analyzed by SPSS version 18.0 for Windows. The correlation between SIRT4 expression and clinicopathological factors was analyzed using χ2 test. The differences between the two groups were analyzed by Student’s t-test. Each experiment was done at least three independent times, and the results are shown as mean ± SD. p < 0.05 was considered as statistically significant.
Results
Downregulation of SIRT4 expression correlates with clinicopathological factors and poor prognosis in GC patients
Previous study has found that SIRT4 is downregulated in GC, but the detailed functions of SIRT4 have not been known. Here, we first detected the expression of SIRT4 in 86 GC tissues using qRT-PCR and Western blotting analysis, respectively. Adjacent normal tissues were used as control. As shown in Figure 1A, the expression of SIRT4 was significantly decreased in tumor tissues compared with the adjacent normal tissues (Figure 1A). Subsequently, we detected the expression of SIRT4 in human GC cell lines, including MKN-45 and HGC-27, and human normal gastric mucosal cell line GES-1. The results showed that the SIRT4 expression in MKN-45 and HGC-27 cells was less than that of GES-1 cells (Figure 1B). To further investigate the clinical significance of SIRT4 protein in GC tissues, we analyzed the relationship between clinicopathological parameters and SIRT4 expression. We found that SIRT4 expression had no correlation with age (p = 0.639) and gender (p = 0.618). However, the expression of SIRT4 was negatively correlated with the tumor size (p < 0.001), pathological grade (p = 0.044), and lymph node metastasis (p = 0.034), suggesting that SIRT4 might play a crucial role in tumor proliferation and invasion in GC (Table 1). In addition, analysis of survival data using Kaplan–Meier assay suggested the patients with a low expression of SIRT4 had a significantly lower survival rate than those with a high expression of SIRT4 (p < 0.05, log-rank test; Figure 1C). Taken together, downregulation of SIRT4 correlates with clinicopathological factors and poor prognosis in GC patients.
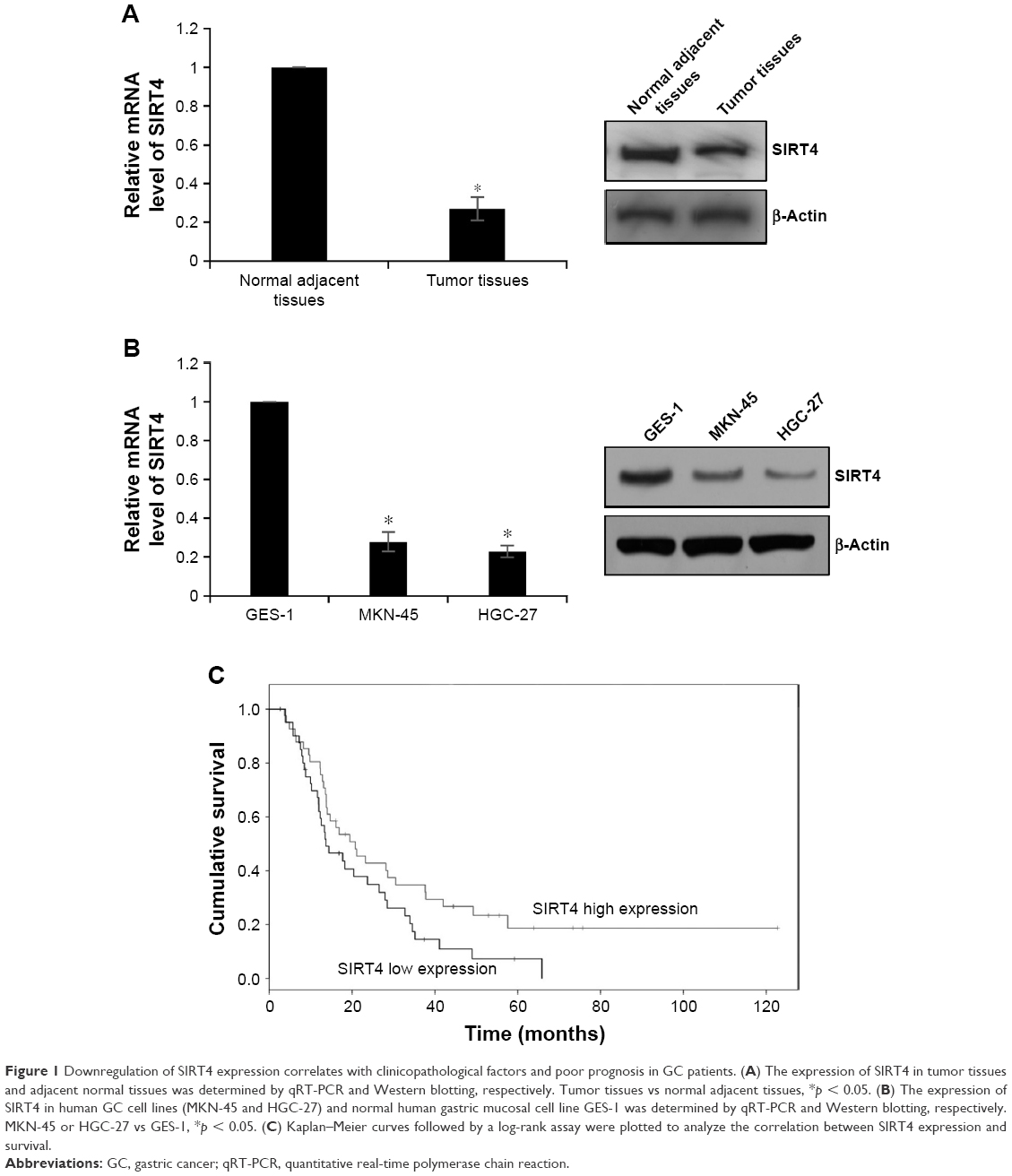 | Figure 1 Downregulation of SIRT4 expression correlates with clinicopathological factors and poor prognosis in GC patients. (A) The expression of SIRT4 in tumor tissues and adjacent normal tissues was determined by qRT-PCR and Western blotting, respectively. Tumor tissues vs normal adjacent tissues, *p < 0.05. (B) The expression of SIRT4 in human GC cell lines (MKN-45 and HGC-27) and normal human gastric mucosal cell line GES-1 was determined by qRT-PCR and Western blotting, respectively. MKN-45 or HGC-27 vs GES-1, *p < 0.05. (C) Kaplan–Meier curves followed by a log-rank assay were plotted to analyze the correlation between SIRT4 expression and survival. |
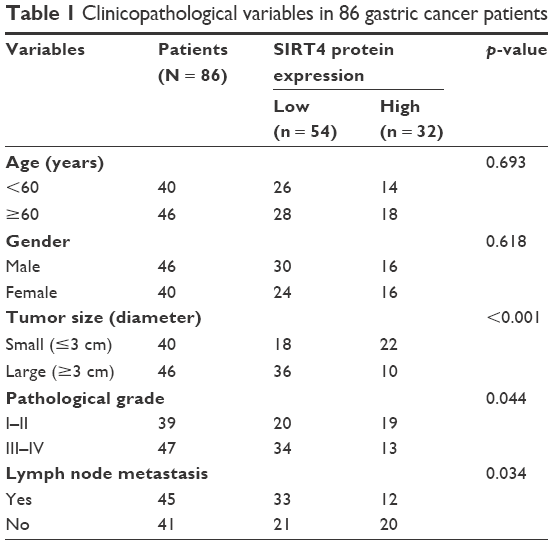 | Table 1 Clinicopathological variables in 86 gastric cancer patients |
SIRT4 suppresses GC cell proliferation
To explore the role of SIRT4 in human GC, we performed gain-of-function and loss-of-function studies by transfecting SIRT4 plasmid or SIRT4 siRNA into MKN-45 or HGC-27 cells. The transfection efficiency was determined by qRT-PCR and Western blotting analysis (Figure 2A and B). The efficiency of SIRT4 siRNA#1 was higher than SIRT4 siRNA#2. Because of the correlation between SIRT4 expression and tumor size, we detected the effect of SIRT4 on cell proliferation using CCK-8 and colony formation assays. The results of CCK-8 assay indicated that overexpression of SIRT4 significantly led to decreasing the growth rate of MKN-45 and HGC-27 cells as compared with that of the vector-transfected cells (Figure 2C). On the contrary, the inhibition of SIRT4 obviously increased the growth rate of MKN-45 and HGC-27 cells as compared with that of the scramble siRNA (SCR)-transfected cells (Figure 2C). Similar results were observed in colony formation analysis, which showed that ectopic expression of SIRT4 decreased the number of colonies in MKN-45 and HGC-27 cells, but the inhibition of SIRT4 increased the number of colonies in MKN-45 and HGC-27 cells (Figure 2D). In conclusion, SIRT4 serves as a tumor suppressor and suppresses GC cell proliferation.
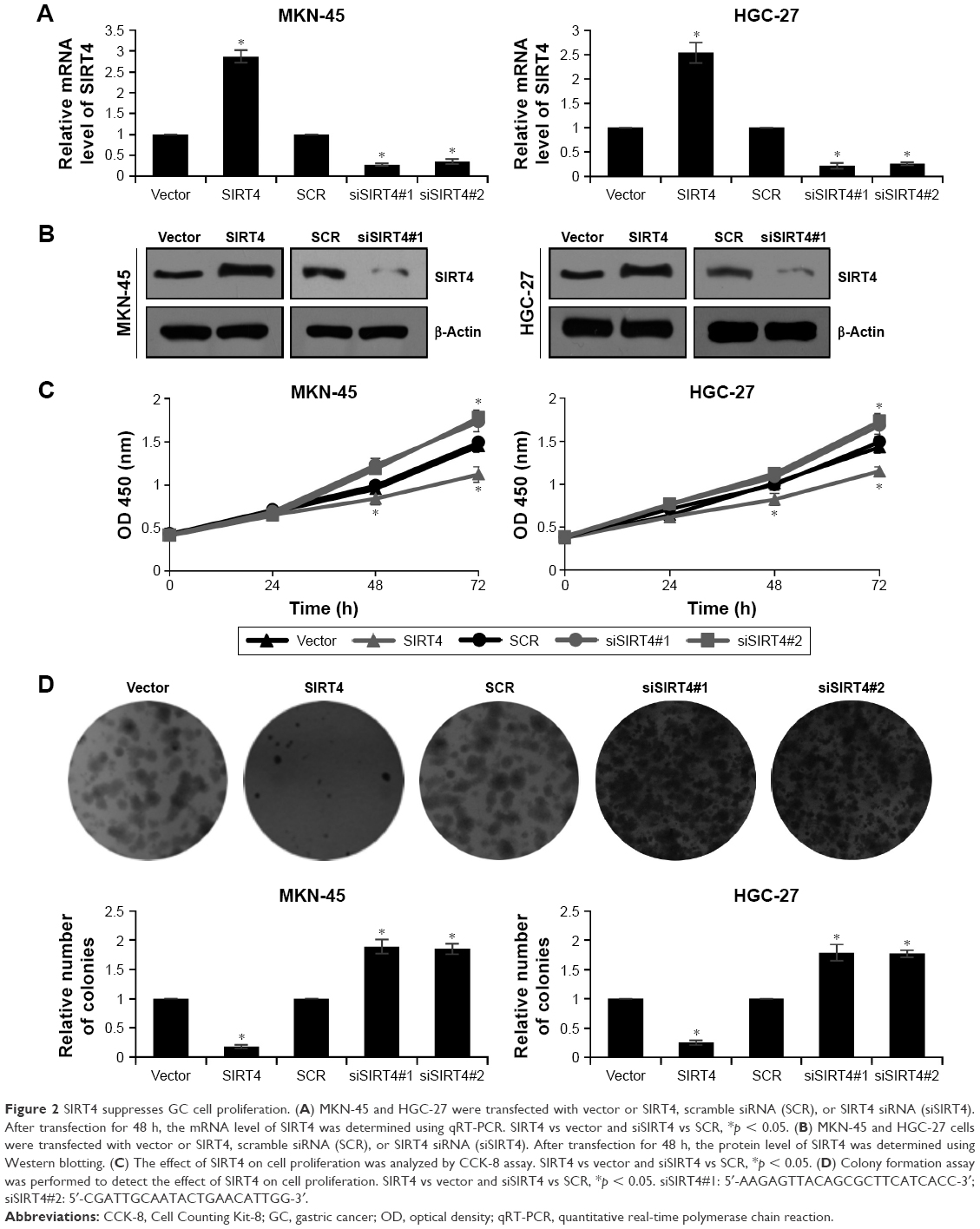 | Figure 2 SIRT4 suppresses GC cell proliferation. (A) MKN-45 and HGC-27 were transfected with vector or SIRT4, scramble siRNA (SCR), or SIRT4 siRNA (siSIRT4). After transfection for 48 h, the mRNA level of SIRT4 was determined using qRT-PCR. SIRT4 vs vector and siSIRT4 vs SCR, *p < 0.05. (B) MKN-45 and HGC-27 cells were transfected with vector or SIRT4, scramble siRNA (SCR), or SIRT4 siRNA (siSIRT4). After transfection for 48 h, the protein level of SIRT4 was determined using Western blotting. (C) The effect of SIRT4 on cell proliferation was analyzed by CCK-8 assay. SIRT4 vs vector and siSIRT4 vs SCR, *p < 0.05. (D) Colony formation assay was performed to detect the effect of SIRT4 on cell proliferation. SIRT4 vs vector and siSIRT4 vs SCR, *p < 0.05. siSIRT4#1: 5′-AAGAGTTACAGCGCTTCATCACC-3′; siSIRT4#2: 5′-CGATTGCAATACTGAACATTGG-3′. |
SIRT4 inhibits GC cell migration and invasion
Metastatic disease is still a major problem in the management of GC. The effects of SIRT4 on invasion of GC cell lines were detected using a wound-healing assay and transwell assay. As shown in Figure 3A, the ectopic expression of SIRT4 markedly suppressed GC cells’ migration ability; however, the knockdown of SIRT4 enhanced the GC cells’ migration ability. Transwell assay was performed to determine the effect of SIRT4 on cell invasion, which revealed the overexpression of SIRT4 inhibited cell invasion, but SIRT4 knockdown promoted cell invasion (Figure 3B). MMP2 and MMP9 have been found to be strongly associated with tumor aggressiveness and poor prognosis in several human cancers.22,23 Subsequently, we detected whether SIRT4 also regulated MMP2 or MMP9 secretion using ELISA, which revealed ectopic expression of SIRT4 led to decreasing MMP2 or MMP9 secretion; however, inhibition of SIRT4 promoted MMP2 or MMP9 secretion (Figure 3C). These data demonstrate that SIRT4 inhibits GC cell migration and invasion through regulation of MMP2 or MMP9 secretion. To further investigate whether overexpressing or knocking down SIRT4 could reduce or increase “transformation”, we performed soft agar assays. The result suggested that overexpression of SIRT4 reduced MKN-45 cell transformation, and knocking down SIRT4 increased MKN-45 cell transformation (Figure 3D).
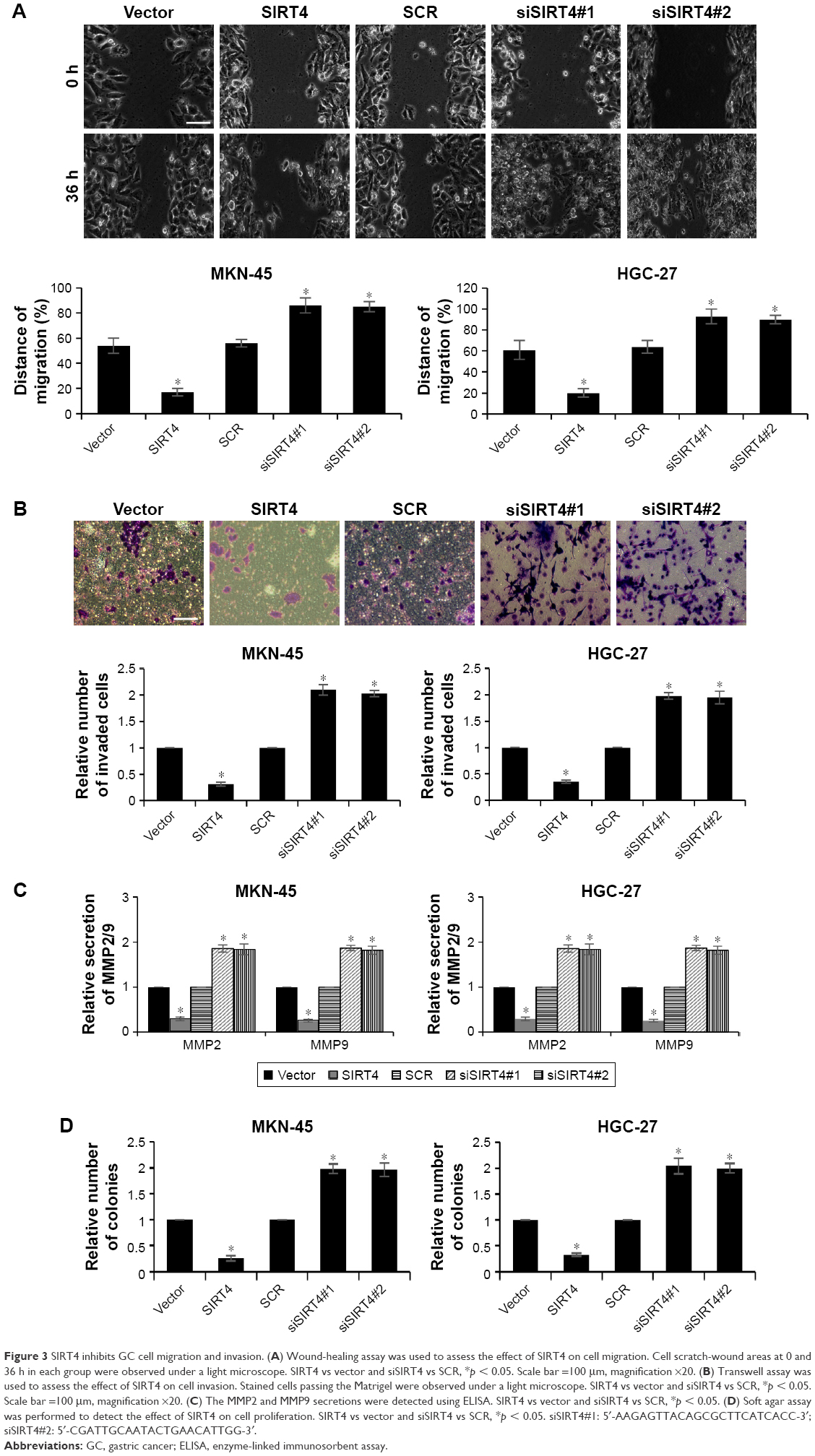 | Figure 3 SIRT4 inhibits GC cell migration and invasion. (A) Wound-healing assay was used to assess the effect of SIRT4 on cell migration. Cell scratch-wound areas at 0 and 36 h in each group were observed under a light microscope. SIRT4 vs vector and siSIRT4 vs SCR, *p < 0.05. Scale bar =100 μm, magnification ×20. (B) Transwell assay was used to assess the effect of SIRT4 on cell invasion. Stained cells passing the Matrigel were observed under a light microscope. SIRT4 vs vector and siSIRT4 vs SCR, *p < 0.05. Scale bar =100 μm, magnification ×20. (C) The MMP2 and MMP9 secretions were detected using ELISA. SIRT4 vs vector and siSIRT4 vs SCR, *p < 0.05. (D) Soft agar assay was performed to detect the effect of SIRT4 on cell proliferation. SIRT4 vs vector and siSIRT4 vs SCR, *p < 0.05. siSIRT4#1: 5′-AAGAGTTACAGCGCTTCATCACC-3′; siSIRT4#2: 5′-CGATTGCAATACTGAACATTGG-3′. |
SIRT4 suppresses EMT through promoting E-cadherin expression
Tumor invasion and metastasis are complex cellular processes and regulated by several cellular processes, such as loss of cell–cell adhesion which is frequently accompanied by downregulation of E-cadherin.24 Previous work has indicated that SIRT4 regulates E-cadherin expression.20 To further investigate the mechanism by which SIRT4 regulates cell invasion and migration in GC, we assessed the association between E-cadherin expression and SIRT4 in MKN-45 and HGC-27 cells by qRT-PCR and Western blotting analyses. We found that the mRNA and protein levels of E-cadherin were significantly upregulated with SIRT4 overexpression and downregulated with SIRT4 knockdown (Figure 4A and B). Moreover, we also detected other EMT-associated proteins, including N-cadherin and vimentin. As shown in Figure 4C and D, ectopic expression or knockdown of SIRT4 had no effect on the mRNA levels of N-cadherin and vimentin; however, the protein levels of N-cadherin and vimentin were significantly downregulated with SIRT4 overexpression and upregulated with SIRT4 knockdown, suggesting that SIRT4 might suppress EMT through regulation of E-cadherin. In addition, we also detected whether SIRT4 regulated EMT-associated transcription factors, such as slug, snail, and twist. As shown in Figure 4E, the result indicated that the ectopic expression of SIRT4 decreased the expression of slug, and the inhibition of SIRT4 increased the expression of slug. However, the expression of SIRT4 had no effect on the expression of snail and twist (Figure 4E). Whether SIRT4 regulates E-cadherin expression through regulation of slug still needs to be further investigated.
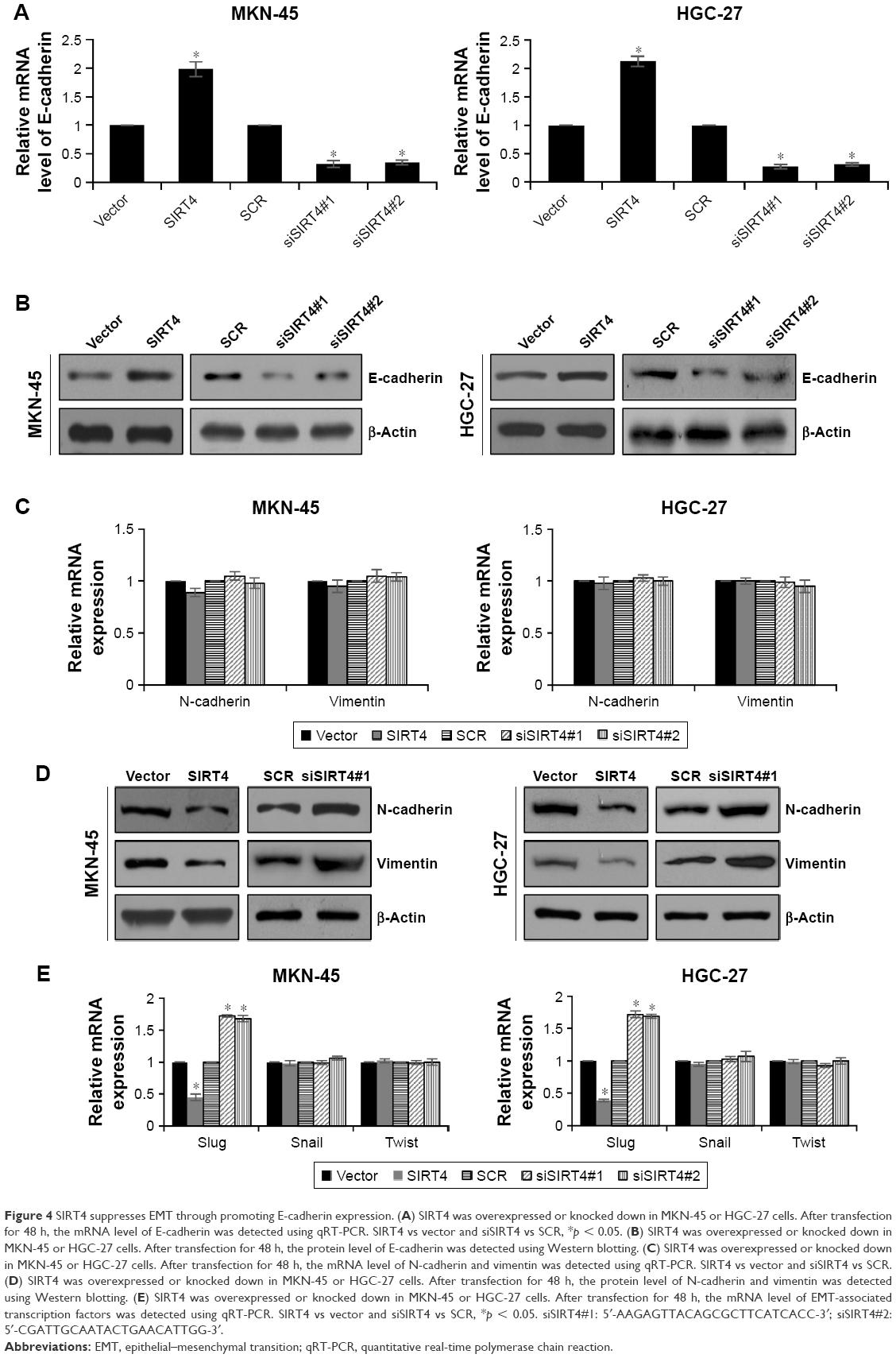 | Figure 4 SIRT4 suppresses EMT through promoting E-cadherin expression. (A) SIRT4 was overexpressed or knocked down in MKN-45 or HGC-27 cells. After transfection for 48 h, the mRNA level of E-cadherin was detected using qRT-PCR. SIRT4 vs vector and siSIRT4 vs SCR, *p < 0.05. (B) SIRT4 was overexpressed or knocked down in MKN-45 or HGC-27 cells. After transfection for 48 h, the protein level of E-cadherin was detected using Western blotting. (C) SIRT4 was overexpressed or knocked down in MKN-45 or HGC-27 cells. After transfection for 48 h, the mRNA level of N-cadherin and vimentin was detected using qRT-PCR. SIRT4 vs vector and siSIRT4 vs SCR. (D) SIRT4 was overexpressed or knocked down in MKN-45 or HGC-27 cells. After transfection for 48 h, the protein level of N-cadherin and vimentin was detected using Western blotting. (E) SIRT4 was overexpressed or knocked down in MKN-45 or HGC-27 cells. After transfection for 48 h, the mRNA level of EMT-associated transcription factors was detected using qRT-PCR. SIRT4 vs vector and siSIRT4 vs SCR, *p < 0.05. siSIRT4#1: 5′-AAGAGTTACAGCGCTTCATCACC-3′; siSIRT4#2: 5′-CGATTGCAATACTGAACATTGG-3′. |
Discussion
Aberrant expression of SIRT4 has been observed in GC and is correlated with pathological grade, depth of tumor invasion, and positive lymph node.19 However, the detailed mechanism of SIRT4 in GC continues to be unknown. In our work, qRT-PCR and Western blotting analyses revealed that the expression of SIRT4 was obviously downregulated both in GC tumor tissues and cell lines. Moreover, ectopic expression of SIRT4 suppressed cell proliferation, migration, and invasion. These results highlight SIRT4 serves as a tumor suppressor in GC.
EMT plays a crucial role in cancer progression, such as metastasis,5,25 during which epithelial cells lose polarity and cell–cell junctions, resulting in a mesenchymal cell phenotype.26 Loss of expression of epithelial cell markers, such as E-cadherin, and gain of expression of mesenchymal genes, such as N-cadherin and vimentin, are the main characteristics of EMT.27,28 In the present study, SIRT4 suppressed cell migration and invasion through increasing the expression of E-cadherin and decreasing the expression of N-cadherin and vimentin. This finding suggested that SIRT4 suppressed EMT process. Interestingly, both mRNA and protein levels of E-cadherin were regulated by SIRT4, but only protein levels of N-cadherin and vimentin were regulated by SIRT4; SIRT4 had no effect on mRNA levels of N-cadherin and vimentin. The detailed mechanism of SIRT4 in EMT has not been clearly known. We hypothesize that SIRT4 works as a mitochondrial localization protein, and it might take part in several metabolic signaling pathways, regulating some metabolites, thereby suppressing EMT. Meanwhile, MMP2 and MMP9 have been found to be strongly associated with tumor aggressiveness and poor prognosis in several human cancers.22,23 We found that SIRT4 also suppressed MMP2 and MMP9 secretions. Several transcription factors play key roles in EMT, including slug, snail, and twist. Here, we found the ectopic expression of SIRT4 decreased the expression of slug, and inhibition of SIRT4 increased the expression of slug. However, the expression of SIRT4 had no effect on the expression of snail and twist. Whether SIRT4 regulated E-cadherin expression through the regulation of slug still needs to be further investigated.
Previous works have found that SIRT4 is downregulated in lung cancer, breast cancer, colorectal cancer, and GC.13,19,20 In our present work, we found SIRT4 was downregulated in GC. We merely investigated the downstream regulation of SRT4, but the upstream regulation mechanism of SIRT4 is not yet clear. Previous work has indicated that microRNA-497 inhibits cardiac hypertrophy by targeting SIRT4,29 suggesting microRNAs might regulate SIRT4 expression in GC. We thought it is necessary to find out the upstream regulation mechanism of SIRT4 in GC. MicroRNAs might be a good breakthrough point.
Our findings indicate that SIRT4 is downregulated in GC tissues and cell lines and acts as a tumor suppressor. SIRT4 inhibits cell proliferation, migration, and invasion. In conclusion, we suggest SIRT4 works as a novel therapeutic target of GC.
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | ||
Lansdorp-Vogelaar I, Kuipers EJ. Screening for gastric cancer in Western countries. Gut. 2016;65(4):543–544. | ||
Thrumurthy SG, Chaudry MA, Chau I, Allum W. Does surgery have a role in managing incurable gastric cancer? Nat Rev Clin Oncol. 2015;12(11):676–682. | ||
Wu Y, Zhou BP. New insights of epithelial-mesenchymal transition in cancer metastasis. Acta Biochim Biophys Sin (Shanghai). 2008;40(7):643–650. | ||
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | ||
Haigis MC, Mostoslavsky R, Haigis KM, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126(5):941–954. | ||
Liu B, Che W, Xue J, et al. SIRT4 prevents hypoxia-induced apoptosis in H9c2 cardiomyoblast cells. Cell Physiol Biochem. 2013;32(3):655–662. | ||
Laurent G, German NJ, Saha AK, et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol Cell. 2013;50(5):686–698. | ||
Mahlknecht U, Voelter-Mahlknecht S. Fluorescence in situ hybridization and chromosomal organization of the sirtuin 4 gene (Sirt4) in the mouse. Biochem Biophys Res Commun. 2009;382(4):685–690. | ||
Ahuja N, Schwer B, Carobbio S, et al. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem. 2007;282(46):33583–33592. | ||
Argmann C, Auwerx J. Insulin secretion: SIRT4 gets in on the act. Cell. 2006;126(5):837–839. | ||
Jeong SM, Xiao C, Finley LW, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell. 2013;23(4):450–463. | ||
Nasrin N, Wu X, Fortier E, et al. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J Biol Chem. 2010;285(42):31995–32002. | ||
Csibi A, Fendt SM, Li C, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell. 2013;153(4):840–854. | ||
Jeong SM, Lee A, Lee J, Haigis MC. SIRT4 protein suppresses tumor formation in genetic models of Myc-induced B cell lymphoma. J Biol Chem. 2014;289(7):4135–4144. | ||
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. | ||
Reid MA, Wang WI, Rosales KR, Welliver MX, Pan M, Kong M. The B55alpha subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol Cell. 2013;50(2):200–211. | ||
Huang G, Cui F, Yu F, et al. Sirtuin-4 (SIRT4) is downregulated and associated with some clinicopathological features in gastric adenocarcinoma. Biomed Pharmacother. 2015;72:135–139. | ||
Miyo M, Yamamoto H, Konno M, et al. Tumour-suppressive function of SIRT4 in human colorectal cancer. Br J Cancer. 2015;113(3):492–499. | ||
Hu H, Zhang H, Ge W, et al. Secreted protein acidic and rich in cysteines-like 1 suppresses aggressiveness and predicts better survival in colorectal cancers. Clin Cancer Res. 2012;18(19):5438–5448. | ||
Cai X, Zhu H, Li Y. PKCzeta, MMP2 and MMP9 expression in lung adenocarcinoma and association with a metastatic phenotype. Mol Med Rep. 2017;16(6):8301–8306. | ||
Zhao Y, Zhou FL, Li WP, Wang J, Wang LJ. Slit2Robo1 signaling promotes the adhesion, invasion and migration of tongue carcinoma cells via upregulating matrix metalloproteinases 2 and 9, and downregulating Ecadherin. Mol Med Rep. 2016;14(3):1901–1906. | ||
Rachagani S, Senapati S, Chakraborty S, et al. Activated KrasG(1)(2)D is associated with invasion and metastasis of pancreatic cancer cells through inhibition of E-cadherin. Br J Cancer. 2011;104(6):1038–1048. | ||
Fuxe J, Vincent T, Garcia de Herreros A. Transcriptional crosstalk between TGF-beta and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle. 2010;9(12):2363–2374. | ||
Pozharskaya V, Torres-Gonzalez E, Rojas M, et al. Twist: a regulator of epithelial-mesenchymal transition in lung fibrosis. PLoS One. 2009;4(10):e7559. | ||
Wang Y, Yao B, Wang Y, et al. Increased FoxM1 expression is a target for metformin in the suppression of EMT in prostate cancer. Int J Mol Med. 2014;33(6):1514–1522. | ||
Hugo H, Ackland ML, Blick T, et al. Epithelial–mesenchymal and mesenchymal–epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213(2):374–383. | ||
Xiao Y, Zhang X, Fan S, Cui G, Shen Z. MicroRNA-497 inhibits cardiac hypertrophy by targeting Sirt4. PLoS One. 2016;11(12):e0168078. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.