")
Back to Journals » Cancer Management and Research » Volume 13
siRNA Knockdown of REDD1 Facilitates Aspirin-Mediated Dephosphorylation of mTORC1 Target 4E-BP1 in MDA-MB-468 Human Breast Cancer Cell Line
Authors Savukaitytė A , Gudoitytė G, Bartnykaitė A, Ugenskienė R, Juozaitytė E
Received 17 June 2020
Accepted for publication 11 December 2020
Published 5 February 2021 Volume 2021:13 Pages 1123—1133
DOI https://doi.org/10.2147/CMAR.S264414
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Harikrishna Nakshatri
Aistė Savukaitytė,1 Greta Gudoitytė,1 Agnė Bartnykaitė,1 Rasa Ugenskienė,1,2 Elona Juozaitytė3
1Oncology Research Laboratory, Institute of Oncology, Lithuanian University of Health Sciences, Kaunas, Lithuania; 2Institute of Biology Systems and Genetic Research, Lithuanian University of Health Sciences, Kaunas, Lithuania; 3Department of Oncology and Hematology, Hospital of Lithuanian University of Health Sciences Kaunas Clinics, Kaunas, Lithuania
Correspondence: Aistė Savukaitytė Email [email protected]
Background: Mutations within genes encoding components of the PI3K/AKT/mTOR (phosphoinositide 3-kinase/protein kinase B/mechanistic target of rapamycin) signaling axis frequently activate the pathway in breast cancer, making it an attractive therapeutic target. Inhibition of mTORC1 (mechanistic target of rapamycin complex 1) activity upon aspirin treatment has been reported in breast cancer cells harboring PI3KCA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha) mutation and is considered to account for anticancer action.
Methods: MDA-MB-468 (harbors mutated PTEN (phosphatase and TENsin homolog)), MCF-7 (PI3KCA-mutated), MDA-MB-231 (no PI3K pathway mutations) cancer cell lines and MCF10A non-cancerous breast epithelial cells were employed for the assessment of modulation of mTORC1 signaling by aspirin. Targeted amplicon-based next-generation sequencing using the Ion Torrent technology was carried out to determine gene expression changes following drug treatment. Western blot was performed to analyze the expression and phosphorylation of proteins. Knockdown by siRNA approach was applied to assess the role of REDD1/DDIT4 (DNA damage-inducible transcript 4) in mTORC1 inhibition by aspirin.
Results: We show a decline in phosphorylation of mTORC1 downstream substrate 4E-BP1 (eukaryotic translation initiation factor 4E-binding protein 1) in response to treatment with aspirin and its metabolite salicylic acid in MDA-MB-468, MCF-7, MDA-MB-231, and MCF10A cell lines. We further demonstrate a novel molecular response to aspirin in breast cancer cells. Specifically, we found that aspirin and salicylic acid increase the expression of REDD1 protein, that is known for its suppressive function towards mTORC1. Unexpectedly, we observed that siRNA knockdown of REDD1 expression facilitated aspirin-mediated suppression of mTORC1 downstream substrate 4E-BP1 phosphorylation in the MDA-MB-468 cell line. REDD1 downregulation slightly encouraged reduction in 4E-BP1 phosphorylation by aspirin in MCF-7 cells but did not elicit a reproducible effect in the MDA-MB-231 cell line. siRNA knockdown of REDD1 did not affect the expression of phosphorylated form of 4E-BP1 following aspirin treatment in MCF10A non-cancerous breast epithelial cells.
Conclusion: The current findings suggest that REDD1 downregulation might improve the anticancer activity of aspirin in a subset of breast tumors.
Keywords: aspirin, breast cancer, REDD1, mTORC1 signaling, 4E-BP1
Introduction
While functioning in two distinct complexes, mTORC1 (mechanistic target of rapamycin complex 1) and mTORC2 (mechanistic target of rapamycin complex 2), mTOR kinase regulates cell growth through a number of cellular processes1 and the activation of mTOR signaling is involved in cancer development.2 mTOR signaling is often upregulated in breast cancer due to the genetic alterations of the genes encoding upstream components of the pathway, including PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha), PTEN (phosphatase and TENsin homolog), and AKT1 (AKT serine/threonine kinase 1).3 Recent studies4,5 have shown a prognostic synergy between Anillin-actin binding protein (ANLN), highly expressed in breast cancers and involved in PI3K/AKT signaling, and kinase insert domain receptor (KDR), that is a key receptor mediating cancer angiogenesis/metastasis switch. Activated mTOR signaling has been generally linked with poor prognosis in breast cancer2 and resistance to conventional therapies.6 However, frequent activation of the pathway makes it an attractive therapeutic target and research has shown that mTOR inhibition is a useful strategy in the treatment of cancers including breast cancer.2 Agents targeting the PI3K/AKT/mTOR signaling axis may improve the efficacy of endocrine, HER2-targeted and cytotoxic therapies due to their implication in resistance to these treatment strategies.6 Identification of biomarkers which could predict a response to the pathway inhibitors is warranted.6
Aspirin (acetylsalicylic acid) has been used in clinical practice for more than a century due to analgesic, antipyretic, and anti-inflammatory properties.7 These effects of aspirin are attributed to acetylation-mediated inhibition of COX (cyclooxygenase) enzymes and decreased production of prostaglandins.8 Recently, aspirin has attracted attention as an anticancer agent, which is strongly supported by epidemiological data.9 However, the underlying mechanisms by which aspirin exerts its antineoplastic effects are not clearly established.7 The effect of non-steroidal anti-inflammatory drugs (NSAIDs) is at least partly associated with inhibition of COX-2, which is overexpressed in cancer tissues and is implicated in tumorigenesis.10 However, since NSAIDs demonstrate antitumor activity in cancer cells, lacking expression of both COX-1 and COX-2, this mechanism of action remains questionable.11 An antithrombotic effect through inhibition of platelet COX-1 is also considered to be relevant.12 In terms of COX-independent mechanisms, inhibition of NF-kappaB13 and Wnt/β-catenin14 have been suggested to play a role.
Aspirin-mediated suppression of mTORC1 signaling in colorectal cancer cells has been reported by Din et al15 and Sun et al.16 Observational studies17,18 have shown a predictive role for PIK3CA mutations in colorectal tumors, which further supports the implication of the PI3K/AKT/mTORC1 pathway in the therapeutic effect of aspirin. Recently, Henry et al19 have demonstrated that growth suppression mediated by aspirin treatment was more pronounced in breast cancer cells harboring activating PIK3CA mutations in comparison with their wild-type counterparts. The anticancer effect was attributed to increased activation of mTORC1 repressor AMPK and the inhibition of mTORC1 signaling described in this report. A decline in mTORC1 signaling was reportedly both dependent and independent of AMPK (AMP-activated protein kinase),15,16,19 suggesting that alternative pathways may also inhibit mTORC1 upon aspirin treatment.
In the present study we further explored the modulation of mTORC1 signaling by aspirin in breast cancer cells. We analyzed the expression of genes encoding components of the PI3K/AKT/mTORC1 pathway and found three differentially expressed genes, including DDIT4/REDD1 (DNA damage-inducible transcript 4; herein after referred as REDD1) following aspirin and salicylic acid treatment. We then demonstrated the drug-induced decrease in phosphorylation of mTORC1 target 4E-BP1 (eukaryotic translation initiation factor 4E-binding protein 1) in various cell lines. Given the ability of REDD1 to suppress mTORC1 signaling,20,21 and the lack of data in the literature on the involvement of REDD1 in aspirin anticancer action, we aimed to test the implication of REDD1 in cell response to aspirin treatment. Unexpectedly, we revealed that REDD1 downregulation using siRNA promotes aspirin-mediated dephosphorylation of 4E-BP1 in the MDA-MB-468 cell line. These findings suggest that REDD1 downregulation might improve the anticancer activity of aspirin in certain breast tumors.
Materials and Methods
Cell Lines and Culture Conditions
Human breast cancer cell lines MDA-MB-468 (PTEN-mutated), MCF-7 (PI3KCA-mutated), and MDA-MB-231 (does not harbor PI3K pathway mutations) were purchased from CLS Cell Line Service (Eppelheim, Germany) and grown as monolayers in Dulbecco’s Modified Eagle’s medium (DMEM; Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (Gibco, Gaithersburg, MD, USA), 100 U/mL penicillin with 100 µg/mL streptomycin (Gibco) and 2 mM L-glutamine (Gibco) at 37°C in humidified 5% CO2. The MCF10A cell line was kindly provided by dr. Valeryia Mikalayeva (Lithuanian University of Health Sciences, Kaunas, Lithuania). MCF10A cells were cultured in DMEM/F-12 (Gibco) with 2.5 mM L-glutamine and 15 mM HEPES supplemented with 100 ng/mL cholera toxin (Sigma-Aldrich), 20 ng/mL epidermal growth factor (Gibco), 10 µg/mL insulin (Gibco), 500 ng/mL hydrocortisone (Sigma-Aldrich), 5% horse serum (Gibco), and 100 U/mL penicillin with 100 µg/mL streptomycin (Gibco) at 37°C in humidified 5% CO2.
Chemicals and Antibodies
Aspirin and salicylic acid were purchased from Sigma-Aldrich, and 0.5 M stock solutions of both compounds were prepared in water (pH 7) and frozen at −20°C in small quantities to prevent freeze-thaw cycles. Working solutions were prepared before each experiment.
Antibody against 4E-BP1 (#9644) was purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-phospho-4E-BP1 Ser65 antibody (#MA5-14948) and anti-β-actin (#AM4302) were obtained from Invitrogen (Waltham, MA, USA), while anti-REDD1 (#ab191871) was from Abcam (Cambridge, UK). Horseradish peroxidase-conjugated anti-rabbit (#65-6120) and alkaline phosphatase conjugated anti-mouse (#WP20006) secondary antibodies were bought from Invitrogen.
Cell Treatment
Exponentially growing cells were plated into 40 mm diameter Petri dishes (for RNA expression analysis) or 25 cm2 flasks (for protein expression analysis) for 24 hours and then treated with indicated concentrations of aspirin or salicylic acid for 1, 17, or 24 hours. We used 0.5 and 2 mM concentrations of the tested drugs, since these are achievable salicylate plasma concentrations obtained from hydrolysis of anti-inflammatory doses of aspirin.22 Since we23 and others24,25 had previously suggested that salicylic acid contributes to aspirin anticancer action, we performed most of the experiments with salicylic acid in parallel.
RNA Extraction and Quantification
Total RNA was extracted from cell lines using the PureLinkTM RNA Mini Kit (Invitrogen) according to manufacturer’s instructions. RNA quantity was assessed using a Qubit® 3.0 fluorometer (Life Technologies, Carlsbad, CA, USA) and the QubitTM RNA HS Assay Kit (Invitrogen) following manufacturer’s recommendations. RNA integrity was assessed by agarose gel electrophoresis.
cDNA Library Preparation
RNA (10 ng) was reverse transcribed using SuperScriptTM IV VILOTM Master Mix with ezDNaseTM Enzyme (Invitrogen), as per manufacturer’s recommendations. Construction of libraries was carried out using Ion AmpliseqTM Library Kit Plus (Ion Torrent, Guilford, CT, USA) according to manufacturer’s instructions. A custom Ion AmpliSeq™ RNA Panel (Ion Torrent) was used to generate 66 amplicons representing unique targeted genes (the list of genes included in the panel is given in Supplementary Table 1). Each library was barcoded with the Ion XpressTM Barcode Adapters 1–16 Kit (Ion Torrent) and subsequently purified using Agencourt™ AMPure™ XP Reagent (Beckman Coulter, Brea, CA, USA) followed by a dilution in Low TE. Library quantification was performed as per Ion Library TaqManTM Quantitation Kit (Ion Torrent) protocol by quantitative PCR on ABI Fast 7500 System (Applied Biosystems, Foster City, CA, USA). Libraries were then diluted to 30 pM and combined with 12 samples per pool for further processing.
Ion Torrent Sequencing and DEG Analysis
Template preparation and chip loading was carried out using Ion 520™ and Ion 530™ Kit-Chef (Ion Torrent) on Ion ChefTM Instrument (Ion Torrent). Sequencing was performed using Ion 520TM Chip Kit (Ion Torrent) on Ion S5TM System (Ion Torrent). Primary analysis of the sequencing data was performed using ampliSeqRNA plugin in the Torrent Suite™ Software v.5.10.1, as was published before.26–28 Further analysis was performed on the EdgeR package from the open-source Bioconductor project. A matrix of gene-wise read counts was used as input. Normalization by trimmed mean of M values (TMM) was performed to adjust for different RNA compositions between libraries and library size. Genes from MCF-7 and MDA-MB-468 cells with less than 21.7 and 36.6 counts per million (CPM) (in any treatment or control group) were excluded from further analysis, respectively. This step was followed by differentially expressed gene (DEG) analysis under the GLM framework. Adjusted P-values for multiple testing, using Benjamini-Hochberg to estimate the false discovery rate (FDR), were calculated for the final estimation of DEG significance.26 Genes with a fold change ≥1.5 or ≤0.67 and an adjusted P<0.05 were considered to be differentially expressed.
Raw sequencing data has been deposited in publicly available Sequence Read Archive (SRA) repository with links to BioProject accession number PRJNA675979 in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/).
Western Blot Analysis
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Abcam), supplemented with protease (Sigma-Aldrich) and phosphatase inhibitor cocktails (Sigma-Aldrich). Protein concentration was measured using PierceTM BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA), as per manufacturer’s instructions. Fifty micrograms of protein were loaded in each lane for electrophoresis in 12% or 4–12% gradient SDS-PAGE. The resolved proteins were electrophoretically transferred onto PVDF (polyvinylidene fluoride) membranes. Blots were blocked in optimal blocking buffer for each antibody for 45 minutes and incubated overnight at 4°C with phospho-4E-BP1 Ser65 (1:250), 4E-BP-1 (1:1,000), REDD1 (1:1,000), or β-actin (1:2,000) primary antibody. After washing three times using TBST (Tris buffered saline with tween 20) the blots were incubated with HRP (for probing p-4E-BP1, 4E-BP1, and REDD1) or AP-conjugated secondary antibodies (for β-actin). The blots were washed again with TBST three times and the proteins were detected using Super SignalTM West Pico PLUS Chemiluminescent Substrate, SuperSignalTM West Atto Ultimate Sensitivity Substrate, or CDP-Star® chemiluminescent substrate from WesternBreeze Chemiluminescent Kit (Invitrogen), respectively. Chemiluminescent reaction was visualized using a CCD imager ChemiDoc XRS+ System (Bio-Rad Laboratories, Hercules, CA, USA). Signal intensity of the protein bands was quantified by the ImageLab 6.0.1 Software (Bio-Rad Laboratories, USA). β-actin was used as a loading control.
Cell Transfection
MDA-MB-468, MCF-7, MDA-MB-231, and MCF10A cells were reverse transfected using LipofectamineTM RNAiMAX transfection reagent (Invitrogen), according to the manufacturer’s instructions, with Silencer Select siRNA at 8 nM, 8 nM, 35 nM, and 50 nM concentrations, respectively. The following siRNAs were employed: siRNA targeting REDD1 (#s29166; Ambion, Austin, TX, USA), non-targeting siRNA (#4390843; Invitrogen), and positive control siRNA against GAPDH (#4390849; Invitrogen). Gene knockdown efficiency was evaluated by quantitative reverse transcription-PCR on ABI Fast 7500 System (Applied Biosystems). For this purpose RNA was extracted and quantified as described above. cDNA synthesis was performed using the High-Capacity RNA-to-cDNA kit (Applied Biosystems), according to the manufacturer’s instructions. PCR reaction was performed with TaqmanTM Gene Expression Assays (Hs99999905_m1 for GAPDH and Hs01111686_g1 for REDD1 quantification) and TaqmanTM Universal Master Mix II with UNG from Applied Biosystems, following the manufacturer’s instructions. Relative gene expression was normalized to β-actin and determined by the 2−ΔΔCt method.
Statistical Analysis
All experiments were repeated at least three times unless otherwise noted. All data are presented as the means±standard deviation (SD). The statistical significance was tested via Student’s t-test using SPSS software (SPSS Inc, Chicago, IL, USA). The results were considered statistically significant when P<0.05.
Results
Aspirin and Salicylic Acid Induce the Expression of REDD1 in Breast Cancer Cell Lines
Since the literature suggested the mTORC1 pathway to be implicated in aspirin anticancer action, we aimed to further analyze the modulation of mTORC1 signaling. Firstly, we assessed the expression of pathway-related molecules in response to aspirin and its metabolite salicylic acid treatment, using targeted amplicon-based next-generation sequencing. Analysis revealed three differentially expressed genes upon 24-hour exposure to the drugs (Table 1). While a decrease in IRS1 mRNA and an increase in DEPTOR expression were found in the MDA-MB-468 cell line, these changes were not detected in the MCF-7 cell line treated with aspirin or salicylic acid. However, treatment with 2 mM of either drugs elevated the expression of DDIT4 in both cell lines. All the observed expression changes implied inhibition of mTORC1 signaling by aspirin and salicylic acid, in accordance with the biological function of genes that were differentially expressed upon drug treatment.
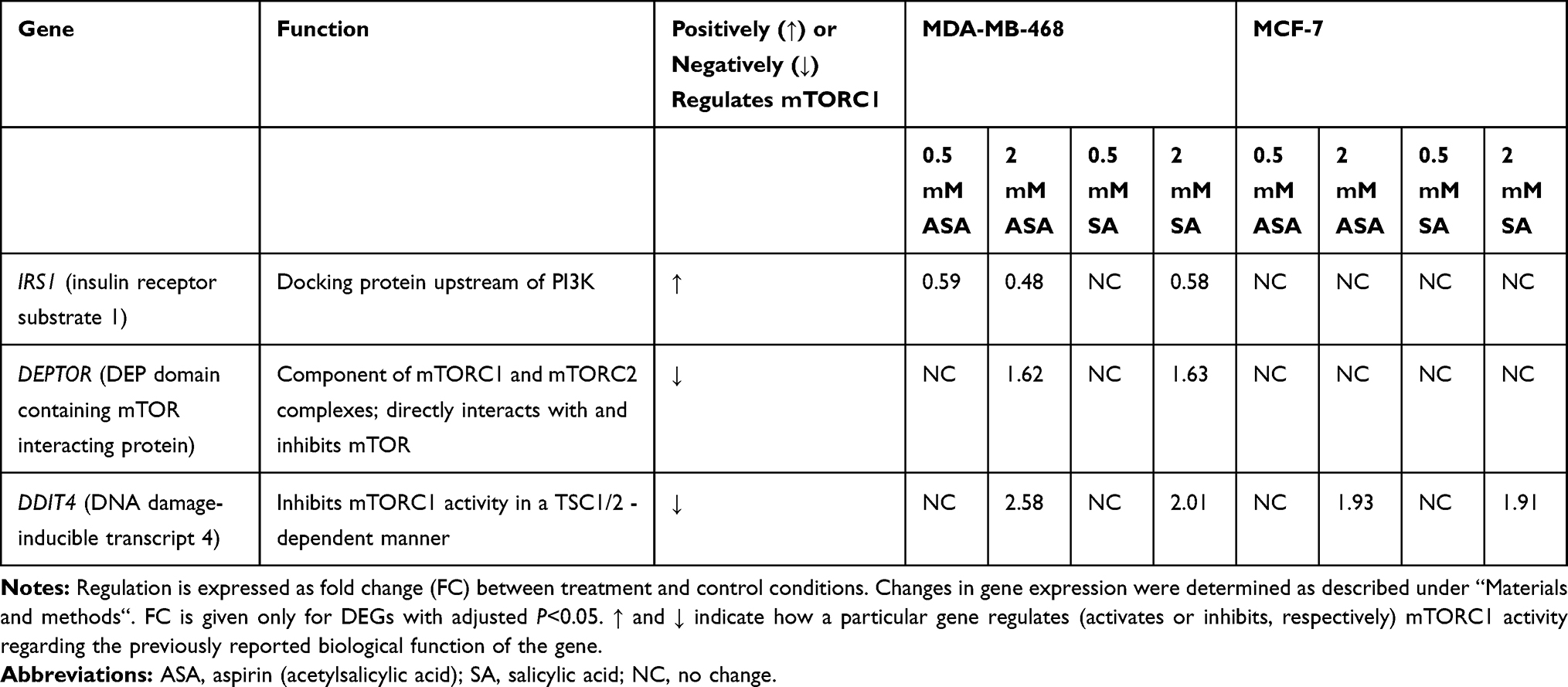 |
Table 1 Regulation of Gene Expression After 24-Hour Treatment with Aspirin and Salicylic Acid |
Since DDIT4 was differentially expressed in both cell lines we next examined whether the increase in DDIT4 mRNA following aspirin and salicylic acid treatment corresponded with elevated protein level in these cell lines. As expected, our data has shown an increase in REDD1 (encoded by DDIT4 gene) protein amount in both MDA-MB-468 and MCF-7 (Figure 1A) cell lines. We further tested the effect in MDA-MB-231 cancer cell line and MCF10A non-cancerous breast epithelial cells. We found an upregulation of REDD1 in MDA-MB-231 cells and a downregulation in MCF10A cells (Figure 1A). Of note, relatively higher baseline expression of REDD1 was observed in non-cancerous breast epithelial cells in comparison to breast cancer cells (Figure 1B).
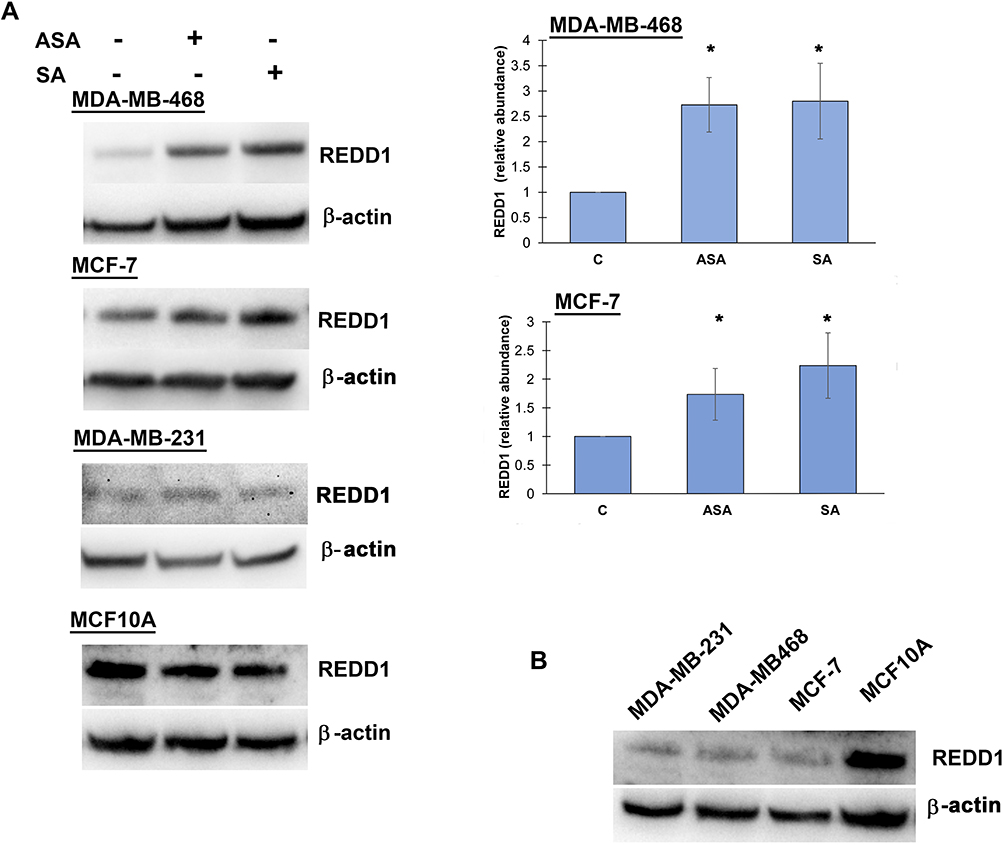 |
Figure 1 REDD1 expression in various cell lines. (A) Effect of aspirin and salicylic acid treatment on REDD1 expression in MDA-MB-468, MCF-7, MDA-MB-231, and MCF10A cell lines. Cells were exposed to 2 mM of aspirin (ASA), salicylic acid (SA), or vehicle control (C) for 24 hours. Equal amounts of proteins (50 µg) from vehicle or drug-treated cells were loaded on each lane of SDS-PAGE gel for electrophoresis, followed by transfer onto PVDF membranes, which were then probed with anti-REDD1 antibody. β-actin was used as loading control. Densitometric quantification of REDD1 levels were normalized to β-actin for fold change calculations. Data are expressed as means±SD of at least three independent experiments. *P<0.05 (vs vehicle control) by Student’s t-test. (B) Baseline levels of REDD1 in non-treated cells. |
Aspirin and Salicylic Acid Treatment Attenuate mTORC1 Signaling
We then investigated whether the observed expression changes upon drug exposure coincide with the expected attenuation of mTORC1 activity. We chose the PTEN-mutant MDA-MB-468 cell line over PIK3CA-mutated MCF-7 cells for subsequent analysis, primarily because the effects of aspirin on mTOR signaling have not been reported in breast cancer cell lines with this mutation of the pathway. In addition, most gene expression changes were determined in this cell line in the present study. Although eukaryotic initiation factor 4E binding protein 1 (4E-BP1) and the ribosomal protein S6 kinase (S6K1) are two main targets of mTORC1, we did not detect baseline S6K1 phosphorylation in MDA-MB-468 cells. We used phosphorylation of translational repressor 4E-BP1 as a marker of mTORC1 activity as it is generally accepted to reflect activation of mTORC129 and predict a response to AKT/mTOR inhibitors.30 As shown in Figure 2A, aspirin and salicylic acid treatment attenuated the activity of mTORC1. Similarly, aspirin and salicylic acid exposure caused a decrease in 4E-BP1 phosphorylation in MCF-7, MDA-MB-231, and MCF10A cell lines (Figure 2B).
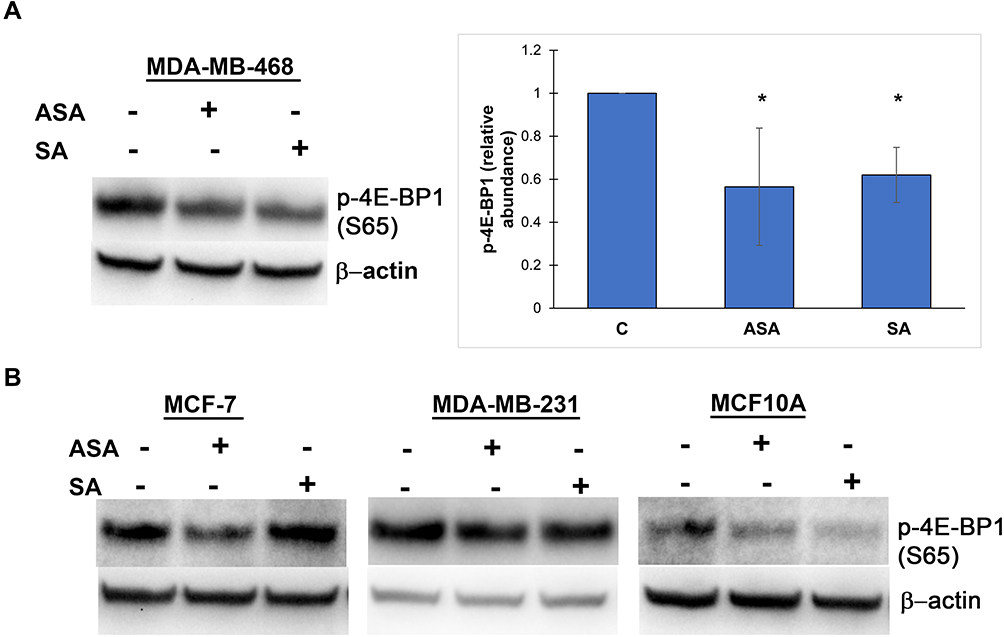 |
Figure 2 Aspirin and salicylic acid attenuate mTORC1 signaling in breast cancer and non-cancerous breast epithelial cells. Cells were exposed to 2 mM of aspirin (ASA), salicylic acid (SA), or vehicle control (C) for 24 (MDA-MB-468, MCF10A) or 17 (MCF-7, MDA-MB-231) hours. Equal amounts of proteins (50 µg) from vehicle or drug-treated cells were loaded on each lane of SDS-PAGE gel for electrophoresis, followed by transfer onto PVDF membranes, which were then probed with respective antibody. β-actin was used as loading control. (A) Data for MDA-MB-468 cell line are expressed as means±SD of four independent experiments. *P<0.05 (vs vehicle control) by Student’s t-test. (B) One experiment was performed in MCF-7, MDA-MB-231, and MCF10A cell lines. |
siRNA Knockdown of REDD1 Triggers Aspirin-Mediated Dephosphorylation of 4E-BP1 in the MDA-MB-468 Cell Line
Observation of REDD1 induction in all tested cancer cell lines led us to test what role REDD1 plays in cell response to aspirin treatment. In regards to the above-mentioned REDD1 function in inhibiting mTORC1 signaling, we originally hypothesized that mTORC1 signaling is suppressed through REDD1 induction upon aspirin treatment. To test this hypothesis we used siRNA interference. In consistence with the previously reported ability of REDD1 to inactivate mTORC1, REDD1 downregulation elicited a slight increase in 4E-BP1 phosphorylation, indicating enhancement in mTORC1 activity (Figure 3). Unexpectedly, we found that REDD1 downregulation using siRNA transfection prior to incubation with aspirin promoted the drug-induced dephosphorylation of 4E-BP1 (Figure 3). We next performed analogical experiments in MCF-7, MDA-MB-231, and MCF10A cell lines. Knockdown of REDD1 alone led to a higher increase in phosphorylation of 4E-BP1 in MDA-MB-231 and MCF-7 cells compared to MDA-MB-468 and had no effect in MCF10A cell line (Figure 3). REDD1 downregulation only slightly encouraged reduction in 4E-BP1 phosphorylation by aspirin in MCF-7 cells and did not elicit a repetitive effect in MDA-MB-231 cell line (Figure 3). siRNA knockdown of REDD1 did not affect the expression of phosphorylated form of 4E-BP1 following aspirin treatment in MCF10A cells (Figure 3).
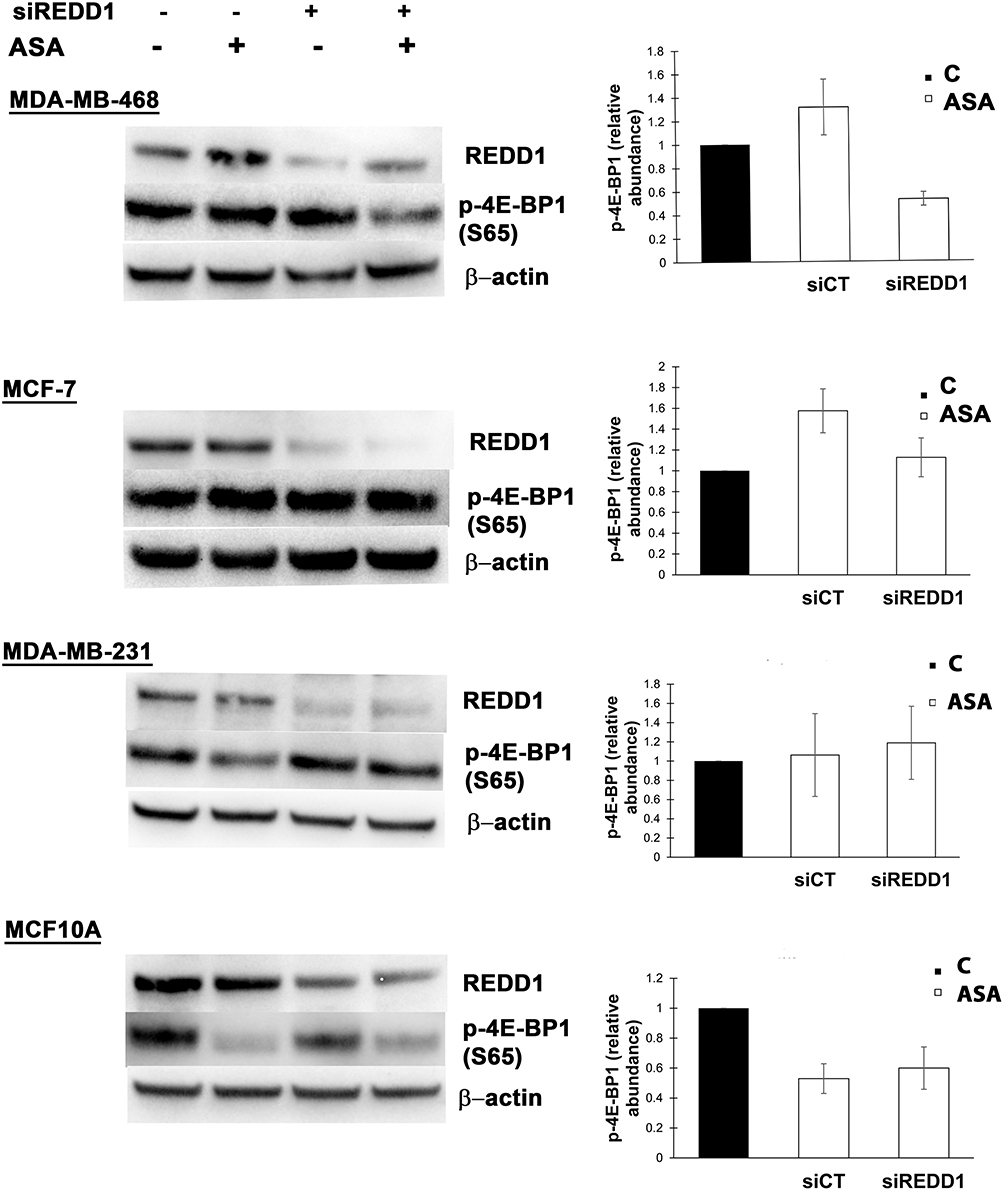 |
Figure 3 Different effects of REDD1 silencing on aspirin-mediated expression of phosphorylated 4E-BP1. Cells were transfected with non-targeting (siCT) or REDD1 siRNA (siREDD1) for 24 (MDA-MB-468, MCF-7, MDA-MB-231 cell lines) or 48 hours (MCF10A) and treated with vehicle control (C) or 2 mM of aspirin (ASA) for the next 24 hours before cell lysis. Equal amounts of proteins (50 µg) were loaded on each lane of SDS-PAGE gel for electrophoresis, followed by transfer onto PVDF membranes, which were then probed with respective antibody. β-actin was used as loading control. REDD1 was probed on a different blot than p-4E-BP1. Data are expressed as means±SD from three independent experiments for MDA-MB-468, MCF-7, MDA-MB-231, and from two independent experiments for MCF10A. |
Discussion
In the present study we explored the modulation of mTORC1 signaling by aspirin in breast cancer cells. Here, we show that aspirin and salicylic acid induce REDD1 expression. REDD1 is encoded by DDIT4 gene. DDIT4 located on chromosome 10 (10q22.1) is ubiquitously expressed at low levels in most adult tissues31 but is downregulated in a subset of human cancers.32,33 REDD1 is induced by various stress conditions, including hypoxia,31 energy depletion,34 oxidative stress,35 and endoplasmic reticulum stress.36 REDD1 has been shown to inhibit mTOR signaling20,21 in a TSC1/2-dependent manner.37
Little is currently known about the role of REDD1 in breast cancer pathogenesis, and existing studies report contradictory findings. For example, Horak et al38 demonstrated that REDD1 suppressed tumorigenesis in breast cancer. Consistently, Koo and Jung39 reported that REDD1 is downregulated in triple-negative and HER2 overexpression breast cancer subtypes. On the contrary, a study by Pinto et al40 linked high DDIT4 expression with tumor aggressiveness in triple-negative breast cancer resistant to neoadjuvant chemotherapy. Our observation of the relatively higher REDD1 expression in non-cancerous breast epithelial cells in comparison to breast cancer cells supports the tumor suppressor role of REDD1.
While REDD1 is known to negatively regulate mTORC1 signaling, the involvement of REDD1 function in anticancer activity of aspirin has not been reported. REDD1 has been implicated in mTORC1 inhibition by metformin,41 N-butylidenephthalide,42 and a combination of melatonin with arsenic trioxide.43 Given the increased expression of REDD1 following aspirin and salicylic acid treatment we raised a hypothesis that REDD1 mediates mTORC1 inhibition upon drug treatment. Surprisingly, siRNA knockdown of REDD1 assisted the aspirin-mediated dephosphorylation of mTORC1 target 4E-BP1 in MDA-MB-468 breast cancer cells. Cap-dependent translational control driven by phosphorylated 4E-BP1 and active downstream eiF4F is essential for tumorigenicity of breast carcinoma cells.44 However, REDD1 downregulation showed a relatively weak benefit in dephosphorylating 4E-BP1 by aspirin in the MCF-7 cell line and did not elicit a reproducible effect in MDA-MB-231 cells. Additionally, REDD1 downregulation itself substantially elevated 4E-BP1 phosphorylation in MCF-7 and MDA-MB-231 cell lines which may abolish the beneficial effect of REDD1 downregulation. Altogether, our results are in contrast with the observations of REDD1 induction-mediated mTORC1 inhibition, where REDD1 downregulation restored dephosphorylation of mTORC1 downstream substrates caused by metformin, N-butylidenephthalide, and a combination of melatonin with arsenic trioxide.
Distinct roles in modulation of mTORC1 activity by REDD1 may depend on the cellular context. Our results in the MDA-MB-468 cell line suggest that REDD1 upregulation might interfere with mTORC1 inhibition by aspirin which contradicts the known biological function of REDD1 to suppress mTORC1 signaling. It is therefore possible that REDD1 induction is a self-defense mechanism of these cancer cells that induces resistance to aspirin treatment. This suggests a modulation of REDD1 activity in cancer treatment strategy, which is supported by several other reports. Vadysirisack et al45 demonstrated an increase in sensitivity to doxorubicin and UV radiation determined by REDD1 loss. In a report by Molitoris et al46 REDD1 knockdown also elevated dexamethasone-induced cell death in murine lymphocytes. Unlike in our study, downregulation of REDD1 abrogated treatment-related inhibition of mTORC1 activity in these reports. While REDD1 downregulation did not improve dephosphorylation of 4E-BP1 by aspirin in MDA-MB-231 cell line and showed little effect in MCF-7 cells in our study, elucidation of the mechanism through which REDD1 downregulation facilitates aspirin-mediated mTORC1 inhibition in MDA-MB-468 cells is required. Of note, REDD1 downregulation had no effect on aspirin activity towards dephosphorylation of 4E-BP1 in non-cancerous breast epithelial cells. Taken together, downregulation of REDD1 in combination with aspirin use might show specific activity against certain breast cancers while not affecting healthy cells.
As mentioned previously, REDD1 is downregulated in triple negative and HER2 breast tumors39 which may also enhance the efficacy of aspirin in a subset of breast cancers. Thus, a clarification of the molecular basis which determines improved aspirin-induced dephosphorylation of 4E-BP1 by REDD1 downregulation appears promising for the identification of biomarkers that, in combination with intrinsic REDD1 downregulation in tumors, could predict a response to aspirin treatment.
Conclusion
Our results uncover a novel molecular response in breast cancer cells exposed to aspirin. The findings suggest that REDD1 downregulation might improve the anticancer activity of aspirin in certain types of breast tumors.
Abbreviations
PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; mTORC, mechanistic target of rapamycin; mTORC1, mechanistic target of rapamycin complex 1; mTORC2, mechanistic target of rapamycin complex 2; PTEN, phosphatase and TENsin homolog; REDD1/DDIT4, DNA damage-inducible transcript 4; 4E-BP1, eukaryotic translation initiation factor 4E-binding protein 1; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; AKT1, AKT serine/threonine kinase 1; ANLN, anillin-actin binding protein; KDR, kinase insert domain receptor; COX, cyclooxygenase; NSAIDs, non-steroidal anti-inflammatory drugs; AMPK, AMP-activated protein kinase; DEG, differentially expressed gene.
Acknowledgments
We thank dr. Valeryia Mikalayeva from Lithuanian University of Health Sciences for providing MCF10A cell line. This research was funded by a grant (No. S-MIP-17-56) from the Research Council of Lithuania. The funder played no role in the study design, data analyses, the decision to publish, or manuscript preparation.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Betz C, Hall MN. Where is mTOR and what is it doing there? J Cell Biol. 2013;203(4):563–574. doi:10.1083/jcb.201306041
2. Hare SH, Harvey AJ. mTOR function and therapeutic targeting in breast cancer. Am J Cancer Res. 2017;7(3):383–404.
3. Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19 784 diverse solid tumors. JAMA Oncol. 2016;2(12):1565–1573. doi:10.1001/jamaoncol.2016.0891
4. Dai X, Mei Y, Chen X, Anln CD, Are Jointly KDR. Prognostic of breast cancer survival and can be modulated for triple negative breast cancer control. Front Genet. 2019;10(October):1–11. doi:10.3389/fgene.2019.00790
5. Dai X, Chen X, Hakizimana O, Mei Y. Genetic interactions between ANLN and KDR are prognostic for breast cancer survival. Oncol Rep. 2019;42(6):2255–2266. doi:10.3892/or.2019.7332
6. Paplomata E, O’regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol. 2014;6(4):154–166. doi:10.1177/1758834014530023
7. Wu KK. Aspirin and Salicylate. Circulation. 2000;102(17):2022–2023. doi:10.1161/01.CIR.102.17.2022
8. Vane JR, Botting RM. The mechanism of action of aspirin. Thrombosis res. 2003. 110(5). 225. doi:10.1016/S0049-3848(03)00379-7
9. Alfonso L, Ai G, Spitale RC, Bhat GJ. Molecular targets of aspirin and cancer prevention. Br J Cancer. 2014;111(1):61–67. doi:10.1038/bjc.2014.271
10. Zhang Z, Chen F, Shang L. Advances in antitumor effects of NSAIDs. Cancer Manag Res. 2018;10:4631–4640. doi:10.2147/CMAR.S175212
11. Chen J, Stark LA. Aspirin Prevention of Colorectal Cancer: focus on NF-κB Signalling and the Nucleolus. Biomedicines. 2017;5:43. doi:10.3390/biomedicines5030043
12. Xu XR, Yousef GM, Ni H. Cancer and platelet crosstalk: opportunities and challenges of aspirin and other antiplatelet agents. Blood. 2018;131(16):1777–1789. doi:10.1182/blood-2017-05-743187
13. Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of IκB kinase-β. Nature. 1998;396(6706):77–80. doi:10.1038/23948
14. Bos CL, Kodach LL, Van Den Brink GR, et al. Effect of aspirin on the Wnt/β-catenin pathway is mediated via protein phosphatase 2A. Oncogene. 2006;25(49):6447–6456. doi:10.1038/sj.onc.1209658
15. Din FVN, Valanciute A, Houde VP, et al. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology. 2012;142(7):7. doi:10.1053/j.gastro.2012.02.050
16. Sun D, Liu H, Dai X, et al. Aspirin disrupts the mTOR-Raptor complex and potentiates the anti-cancer activities of sorafenib via mTORC1 inhibition. Cancer Lett. 2017;406:105–115. doi:10.1016/j.canlet.2017.06.029
17. Liao X, Lochhead P, Nishihara R, et al. Aspirin Use, Tumor PIK3CA Mutation, and Colorectal-Cancer Survival. N Engl J Med. 2012;367(17):1596–1606. doi:10.1056/NEJMoa1207756
18. Domingo E, Church DN, Sieber O, et al. Evaluation of PIK3CA Mutation As a Predictor of Benefit From Nonsteroidal Anti-Inflammatory Drug Therapy in Colorectal Cancer. J Clin Oncol. 2013;31(34):4297–4305. doi:10.1200/JCO.2013.50.0322
19. Henry WS, Laszewski T, Tsang T, et al. Aspirin suppresses growth in PI3K-mutant breast cancer by activating AMPK and inhibiting mTORC1 signaling. Cancer Res. 2018;77(3):790–801. doi:10.1158/0008-5472.CAN-16-2400
20. Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18(23):2893–2904. doi:10.1101/gad.1256804
21. Corradetti MN, Inoki K, Guan KL. The stress-inducted proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. J Biol Chem. 2005;280(11):9769–9772. doi:10.1074/jbc.C400557200
22. Dovizio M, Tacconelli S, Sostres C, Ricciotti E, Patrignani P. Mechanistic and pharmacological issues of aspirin as an anticancer agent. Pharmaceuticals. 2012;5(12):1346–1371. doi:10.3390/ph5121346
23. Savukaitytė A, Vadeikienė R, Laukaitienė D, Ugenskienė R, Juozaitytė E. Salicylic acid plays a major role in aspirin anticancer action in breast cancer cells. Liet Bendr Prakt Gydyt. 2019;23(9):584–588.
24. Borthwick GM, Sarah Johnson A, Partington M, Burn J, Wilson R, Arthur HM. Therapeutic levels of aspirin and salicylate directly inhibit a model of angiogenesis through a Cox- independent mechanism. FASEB J. 2006;20(12):2009–2016. doi:10.1096/fj.06-5987com
25. Dachineni R, Ai G, Kumar DR, Sadhu SS, Tummala H, Bhat GJ. Cyclin A2 and CDK2 as novel targets of aspirin and salicylic acid: A potential role in cancer prevention. Mol Cancer Res. 2016;14(3):241–252. doi:10.1158/1541-7786.MCR-15-0360
26. Osman A, Hitzler WE, Ameur A, Provost P, Schubert M. Differential expression analysis by RNA-seq reveals perturbations in the platelet mRNA transcriptome triggered by pathogen reduction systems. PLoS One. 2015;10:7. doi:10.1371/journal.pone.0133070
27. Li W, Turner A, Aggarwal P, et al. Comprehensive evaluation of AmpliSeq transcriptome, a novel targeted whole transcriptome RNA sequencing methodology for global gene expression analysis. BMC Genomics. 2015;16(1):1–13. doi:10.1186/s12864-015-2270-1
28. Fornecker LM, Muller L, Bertrand F, et al. Multi-omics dataset to decipher the complexity of drug resistance in diffuse large B-cell lymphoma. Sci Rep. 2019;9(1):1–9. doi:10.1038/s41598-018-37273-4
29. Qin X, Jiang B, Zhang Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle. 2016;15. doi:10.1080/15384101.2016.1151581
30. Mi W, Ye Q, Liu S, She Q-B. AKT Inhibition Overcomes Rapamycin Resistance by Enhancing the Repressive Function of PRAS40 on MTORC1/4E-BP1 Axis. Oncotarget. 2015;6(16):13962–13977. doi:10.18632/oncotarget.3920
31. Shoshani T, Faerman A, Mett I, et al. Identification of a Novel Hypoxia-Inducible Factor 1-Responsive Gene, RTP801, Involved in Apoptosis. Mol Cell Biol. 2002;22(7):2283–2293. doi:10.1128/mcb.22.7.2283-2293.2002
32. Deyoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22(2):239–251. doi:10.1101/gad.1617608
33. Lapointe J, Li C, Higgins JP, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci U S A. 2004;101(3):811–816. doi:10.1073/pnas.0304146101
34. Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and Cell Growth in Response to Energy Stress by REDD1. Mol Cell Biol. 2005;25(14):5834–5845. doi:10.1128/mcb.25.14.5834-5845.2005
35. Jin HO, Seo SK, Woo SH, et al. Activating transcription factor 4 and CCAAT/enhancer-binding protein-β negatively regulate the mammalian target of rapamycin via Redd1 expression in response to oxidative and endoplasmic reticulum stress. Free Radic Biol Med. 2009;46(8):1158–1167. doi:10.1016/j.freeradbiomed.2009.01.015
36. Whitney ML, Jefferson LS, Kimball SR. ATF4 is necessary and sufficient for ER stress-induced upregulation of REDD1 expression. Biochem Biophys Res Commun. 2009;379(2):451–455. doi:10.1016/j.bbrc.2008.12.079
37. Gordon BS, Steiner JL, Williamson DL, Lang CH, Kimball SR. Emerging role for regulated in development and DNA damage 1 (REDD1) in the regulation of skeletal muscle metabolism. Am J Physiol Endocrinol Metab. 2016;311(1):E157–E174. doi:10.1152/ajpendo.00059.2016
38. Horak P, Crawford AR, Vadysirisack DD, et al. Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci U S A. 2010;107(10):4675–4680. doi:10.1073/pnas.0907705107
39. Koo JS, Jung W. Alteration of REDD1-mediated mammalian target of rapamycin pathway and hypoxia-inducible factor-1α regulation in human breast cancer. Pathobiology. 2011;77(6):289–300. doi:10.1159/000320936
40. Pinto JA, Araujo J, Cardenas NK, et al. A prognostic signature based on three-genes expression in triple-negative breast tumours with residual disease. Npj Genomic Med. 2016;1(May):2015. doi:10.1038/npjgenmed.2015.15
41. Ben SI, Regazzetti C, Robert G, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71(13):4366–4372. doi:10.1158/0008-5472.CAN-10-1769
42. Liao KF, Chiu TL, Huang SY, et al. Anti-Cancer effects of radix angelica sinensis (Danggui) and N-Butylidenephthalide on Gastric Cancer: implications for REDD1 activation and mTOR inhibition. Cell Physiol Biochem. 2018;48(6):2231–2236. doi:10.1159/000492641
43. Yun SM, Woo SH, Oh ST, et al. Melatonin enhances arsenic trioxide-induced cell death via sustained upregulation of Redd1 expression in breast cancer cells. Mol Cell Endocrinol. 2016;422:64–73. doi:10.1016/j.mce.2015.11.016
44. Avdulov S, Li S, Michalek V, et al. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell. 2004;5(6):553–563. doi:10.1016/j.ccr.2004.05.024
45. Vadysirisack DD, Baenke F, Ory B, Lei K, Ellisen LW. Feedback control of p53 translation by REDD1 and mTORC1 Limits the p53-Dependent DNA Damage Response. Mol Cell Biol. 2011;31(21):4356–4365. doi:10.1128/mcb.05541-11
46. Molitoris JK, McColl KS, Swerdlow S, et al. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J Biol Chem. 2011;286(34):30181–30189. doi:10.1074/jbc.M111.245423
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.