")
Back to Journals » Clinical Ophthalmology » Volume 11
Single nucleotide polymorphism 1623 A/G (rs180195) in the promoter of the Thyroglobulin gene is associated with autoimmune thyroid disease but not with thyroid ophthalmopathy
Authors Lahooti H , Edirimanne S, Walsh JP, Delbridge L, Hibbert EJ , Wall JR
Received 3 March 2017
Accepted for publication 15 May 2017
Published 25 July 2017 Volume 2017:11 Pages 1337—1345
DOI https://doi.org/10.2147/OPTH.S136070
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Hooshang Lahooti,1,2 Senarath Edirimanne,1,2 John P Walsh,3,4 Leigh Delbridge,5,6 Emily J Hibbert,1,2 Jack R Wall1,2
1Thyroid Research Laboratory, Sydney Medical School – Nepean Clinical School, The University of Sydney, Kingswood, NSW, Australia; 2Nepean Blue Mountains Local Health District, Kingswood, NSW, Australia; 3Department of Endocrinology and Diabetes, Sir Charles Gairdner Hospital, Nedlands, WA, Australia; 4School of Medicine and Pharmacology, The University of Western Australia, Crawley, WA, Australia; 5Department of Surgery, Royal North Shore Hospital, St Leonards, NSW, Australia; 6Sydney Medical School – Northern Clinical School, The University of Sydney, NSW, Australia
Background: Our studies over recent years have focused on some new ideas concerning the pathogenesis for the orbital reaction that characterizes Graves’ ophthalmopathy namely, that there are antigens expressed by thyroid tissue and orbital tissue where they are targeted by autoantibodies and/or sensitized T cells, leading to orbital inflammation. While this has been well studied for the thyroid stimulating hormone-receptor, the possible role of another major thyroid antigen, Thyroglobulin (TG), has been largely ignored.
Methods: We identified novel variant 1623 A/G single nucleotide polymorphism (SNP) (rs180195) in the promoter of TG gene associated with autoimmune thyroid disorders. We genotyped the TG SNPs rs2069566, rs2076739, rs121912646, rs121912647, rs121912648, rs121912649, rs121912650, rs137854433, rs137854434, and rs180195 by MassARRAY SNP analysis using iPLEX technology in a cohort of 529 patients with thyroid autoimmunity with and without ophthalmopathy, and controls.
Results: We showed that variant 1623 A/G SNP (rs180195) in the promoter of TG gene is a marker for thyroid autoimmunity, but not for ophthalmopathy. We showed that there was a significant difference in the distribution of the major allele (G) vs minor allele (A) in patients with Hashimoto’s thyroiditis (HT). In HT the wild-type (GG) genotype was less common. We showed that the genotypes homozygous AA and heterozygous GA rs180195 SNP in the promoter of TG gene were more closely associated with thyroid autoimmunity than the wild-type (GG) polymorphism, and are thus, markers of autoimmunity.
Conclusion: rs180195 SNP was previously identified by Stefan et al independently of us, who showed that this TG SNP predisposed to autoimmune thyroid diseases. However, this is the first study to explore the association between TG SNPs and HT. Our findings support the notion that the thyroid and orbital disorders are not part of the same disease, ie, “Graves’ disease” or “Hashimoto’s disease”, but separate autoimmune disorders.
Keywords: hyperthyroid, Hashimoto’s, thyroid eye disease, orbital, homozygote, heterozygote, T-cells
Introduction
The etiology of thyroid eye disease is not well understood, although it is generally presumed to begin in the thyroid since the great majority of patients with Graves’ ophthalmopathy (GO) have active thyroid inflammation, ie, a thyroiditis, at the time they develop eye signs.1,2 A generally accepted hypothesis for the orbital reaction of this complex eye disorder is that there are antigens expressed by thyroid tissue and orbital tissue where they are targeted by autoantibodies and/or sensitized T cells, leading to orbital inflammation.3,4 This complex eye disorder has been well studied for the thyroid stimulating hormone-receptor (TSHr),5–7 but the possible role of the other major thyroid antigens, namely TG and TPO has been largely ignored.8
TG, a glycoprotein homodimer protein made by thyroid follicular cells with a molecular mass of 660 kDa, is the glycoprotein precursor of the thyroid hormones triiodothyronine (T3) and tetraiodothyronine (T4). On hydrolysis it yields only two to four molecules of T3 and T4. The protein contains a 19-amino acid signal peptide followed by 2,748 residues. TG has three functions; namely as a thyroid hormone precursor, storage of iodine, and storage of inactive thyroid hormones.9,10 The TG gene encodes an 8.7 kb mRNA, covers at least 300 kb of genomic DNA, and has 52 exons: 51 introns are as large as 64 kb and within intronic regions are highly conserved intergenic regions which are transposons and repetitive elements. The 5-prime and 3-prime parts of the gene are composed of two evolutionarily different regions. The first 30 kb of DNA encodes 3 kb of the mRNA and the remaining 270 kb encodes 5.7 kb of the mRNA. The TG gene maps to chromosome 8q24 region which has been shown to be strongly linked with autoimmune thyroid disease (AITD).11–17
In recent years, we have focused our research on some new ideas concerning the pathogenesis of GO. We showed that levels of serum TG were elevated in patients with Graves’ disease (GD) and ophthalmopathy compared to those with GD and no eye signs.18 It is possible that its release in the context of thyroid inflammation (“thyroiditis”) may lead to orbital inflammation, as has been suggested by us and others in the past.19,20 Mutations in the TG gene cause thyroid dyshormonogenesis,17,21–35 manifested as goiter, and are associated with moderate to severe congenital hypothyroidism. Polymorphisms in this gene are also associated with susceptibility to AITDs such as GD and Hashimoto’s thyroiditis (HT).36–42
The human TG gene contains 16,165 single nucleotide polymorphisms (SNPs) present (http://www.ncbi.nlm.nih.gov/snp/) – 800 of TG gene SNPs appeared to be highly conserved (http://ecrbrowser.dcode.org/). Thirty-two of these SNPs are of clinical significance and ten are pathogenic by virtue of being in germ cells. We hypothesized that these ten germ cell TG polymorphisms may be associated with thyroid disorders. We embarked upon genotyping ten evolutionary conserved SNPs within the TG gene in a cohort of patients with AITD and controls. We genotyped rs2069566, rs2076739, rs121912646, rs121912647, rs121912648, rs121912649, rs121912650, rs137854433, rs137854434, and rs180195 by MassARRAY SNP analysis using iPLEX technology of SEQUENOM (Agena Bioscience, San Diego, CA, USA); showing SNPs which show strong association with thyroid disorders, paving the way for future in-depth functional studies.
Clinical subjects
Comprehensive demographic, clinical details, and genotypes of SNP rs180195 for patients with GO, Graves’ hyperthyroidism (GH), HT, and control subjects without autoimmune diseases have been described in detail43 in an earlier publication, summarized in Table 1. Briefly, participants were recruited from outpatients’ clinics at Nepean and Royal North Shore Hospitals in New South Wales and the Sir Charles Gairdner Hospital in Western Australia. Previous treatments for hyperthyroidism (with particular reference to radioactive iodine therapy), sex distribution of patients with GD with or without GO, presence or absence of other autoimmune diseases, and presence or absence of ethnic differences in the different groups of patients are shown in Table 1. Patients’ recruitment criteria are described by Walsh et al.44 Nepean Blue Mountains Local Health District Human Research Ethics Committee approval was received for the study and informed consent of participating subjects was obtained.
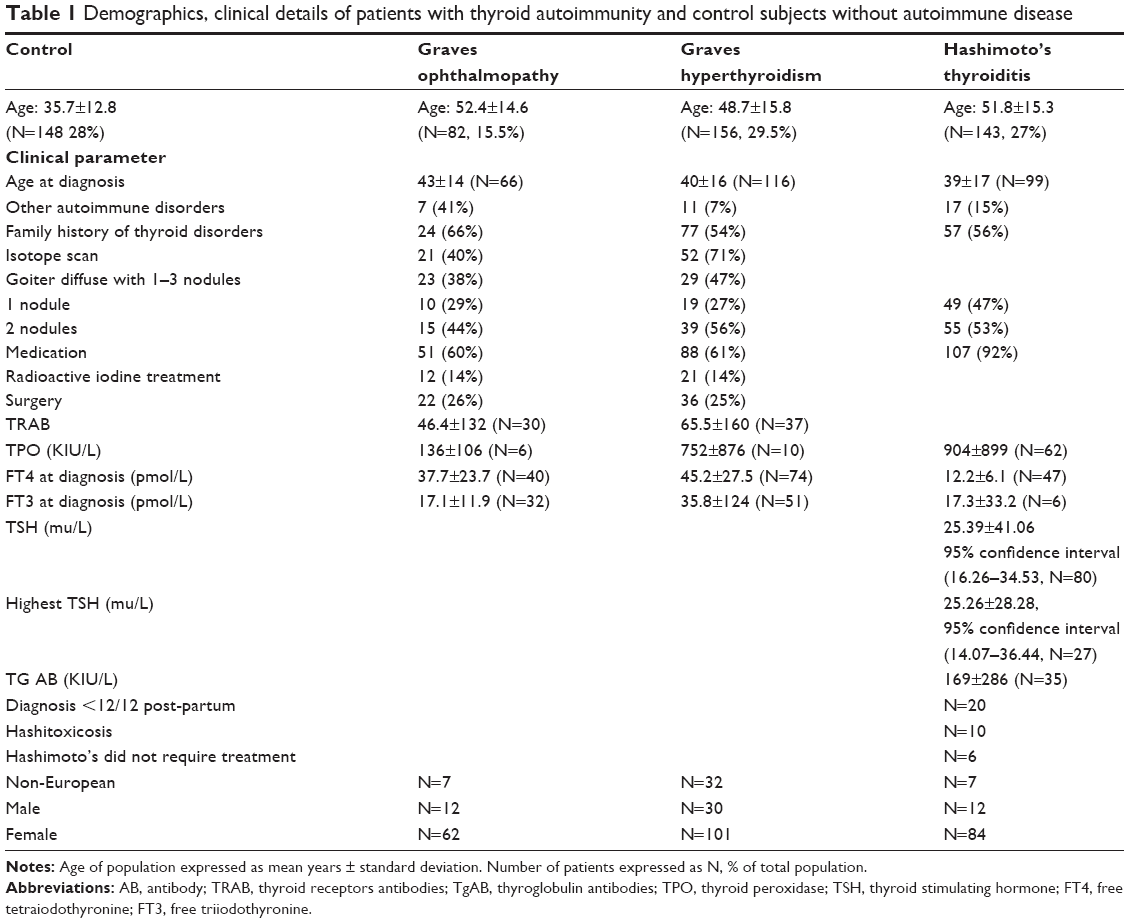 | Table 1 Demographics, clinical details of patients with thyroid autoimmunity and control subjects without autoimmune disease |
Methods
1) MassARRAY SNP analysis using iPLEX technology of SEQUENOM has been described in detail in previous publications of this laboratory.43,45,46 2) Functional analysis: T4, T3, thyroid receptors antibodies (TRAB), TSI, TSHr, TG antibody, and TPO antibody were measured by established radio immunoassay methods in pathology laboratories.
Statistical analyses
Genotypes and allelic frequencies of the TG SNP rs180195 in patients with GH, GO, HT, and control subjects were compared using chi-square test or Fisher’s exact test following Hardy–Weinberg equilibrium. Correlations were made between levels of the TG protein and parameters of the eye disease, and the presence of the genomic polymorphism of rs180195 with T4, T3, TRAB, TSI, TSHr, TG, and TPO antibody titers, in the three groups, and were analyzed using the Mann–Whitney test of GraphPad Prism Version 3.03 (San Diego, CA, USA). A P-value of <0.05 was taken as significant in all assessments.
Results
We studied the ten potential pathogenic TG SNPs occurring in germ cells in a cohort of 529 patients and controls. Only rs180195 was polymorphic. rs180195 SNP is in the promoter region of TG gene (Table 2), and the change in nucleotide is from G > A; therefore, genotypes are wild-type 189 (GG), heterozygote 238 (GA), and rare homozygote 102 (AA) with major allele frequency of 60% and minor allele frequency of 40%, following Hardy–Weinberg equilibrium. Statistical analysis suggested that rs180195 appeared to be informative and could be further studied for genotypic analysis.
 | Table 2 Characteristics of rs180195 informative polymorphism identified in TG gene |
We analyzed SNP rs180195 of TG gene across the three genotypes for GO, GH, HT, and control groups. rs180195 (Table 3) showed significant difference in the pattern of distribution of the genotypes between the different patient groups (P=0.038). We were interested to know if alleles G and A would show significant probability and odds ratio in GO, GH, HT, and control groups. Therefore, we performed pair-wise analysis to determine the probability and odds ratio for each allele separately shown in Table 4. Pair-wise analysis of alleles’ frequency distribution in GO, GH, HT vs control groups is shown in Table 4; rs180195 GO vs control showed odds ratio =0.875, 95% confidence interval =0.591–1.29, P=0.504; GH vs control showed odds ratio =0.734, 95% confidence interval =0.530–1.02, P=0.062; which did not reach significance; and HT vs control showed odds ratio =0.670, 95% confidence interval =0.481–0.93, P=0.018 which was significant.
 | Table 3 Genotype of TG rs180195 SNP distribution in patients with autoimmune thyroid disease with and without ophthalmopathy and controls |
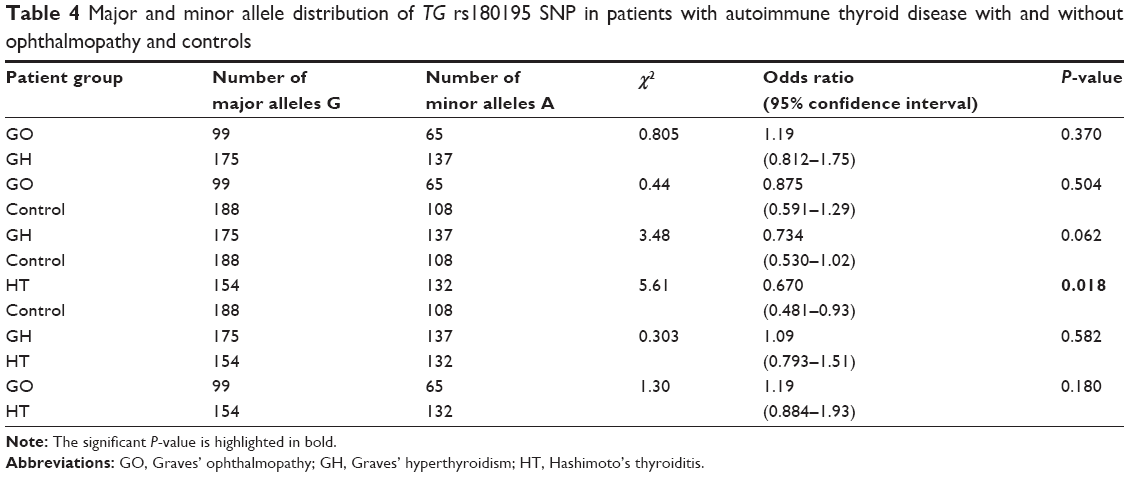 | Table 4 Major and minor allele distribution of TG rs180195 SNP in patients with autoimmune thyroid disease with and without ophthalmopathy and controls |
The difference in genotype frequencies for the wild-type GG (Table 5) observed in GO vs control was not significant with odds ratio 0.95, 95% confidence interval 0.55–1.64, P=0.856 while that for GO vs HT was significant with odds ratio 1.99, 95% confidence interval 1.12–3.52, P=0.018; it was also significant for GH vs control subjects with odds ratio 0.60, 95% confidence interval 0.38–0.96, P=0.033 and similarly, was significant for HT vs control with odds ratio 0.48, 95% confidence interval 0.29–0.78, P=0.003. No significant difference was observed for GH vs HT with odds ratio 1.26, 95% confidence interval 0.76–2.07, P=0.367; nor for GO vs GH with odds ratio 1.60, 95% confidence interval 0.90–2.74, P=0.104.
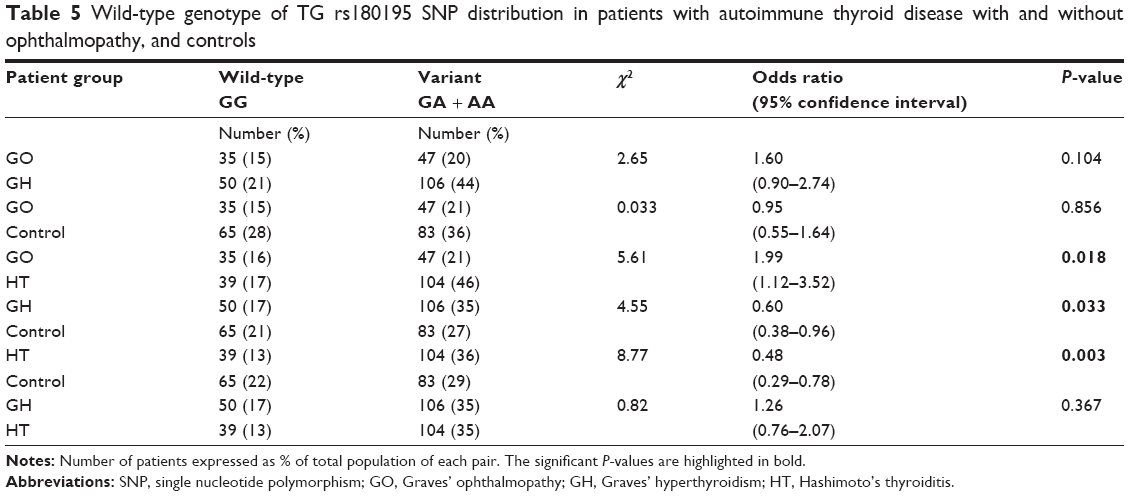 | Table 5 Wild-type genotype of TG rs180195 SNP distribution in patients with autoimmune thyroid disease with and without ophthalmopathy, and controls |
A significant difference in frequency for heterozygote GA (Table 6) was observed for GO vs HT with odds ratio 0.48, 95% confidence interval 0.27–0.84, P=0.010 and HT vs control with odds ratio 1.76, 95% confidence interval 1.10–2.80, P=0.017, but not for GO vs GH with odds ratio 0.59, 95% confidence interval 0.34–1.02, P=0.060.
 | Table 6 Heterozygous genotype of TG rs180195 SNP distribution in patients with autoimmune thyroid disease with and without ophthalmopathy, and controls |
No difference in frequency for homozygote AA (Table 7) was observed for GO vs control with odds ratio 1.38, 95% confidence interval 0.75–2.72, P=0.346 or GH vs control with odds ratio 1.22, confidence interval 0.68–2.18, P=0.501 or HT vs control with odds ratio 1.20, confidence interval 0.66–2.17, P=0.552. Wild-type and heterozygote genotypic changes were most significant in HT patients.
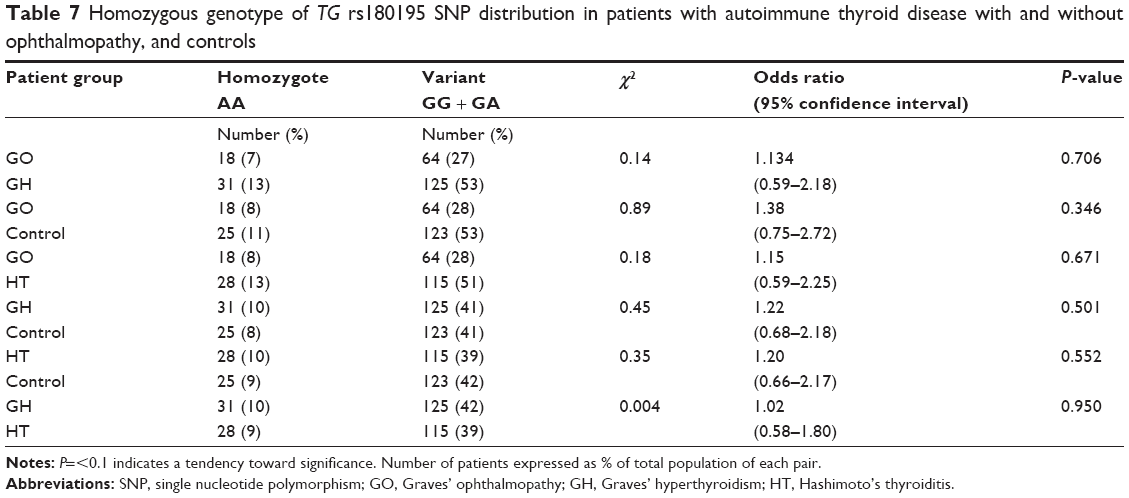 | Table 7 Homozygous genotype of TG rs180195 SNP distribution in patients with autoimmune thyroid disease with and without ophthalmopathy, and controls |
We measured the serum levels of TSH, free T4 (FT4), TPO antibody, TG antibody, and the age at the onset of disease in each of the wild-type, heterozygote and rare homozygote genotypes (Figure 1A–E). There was a significant difference in levels of TSH between the wild-type GG and heterozygote GA genotypes of TG (P=0.037) but the difference in levels of TSH between wild-type and rare homozygote AA genotype (P=0.065) was not significant. There were no differences in levels of TPO antibodies, FT4, TG antibodies or the age at the onset of disease between wild-type GG and heterozygote GA or wild-type GG and rare homozygote AA.
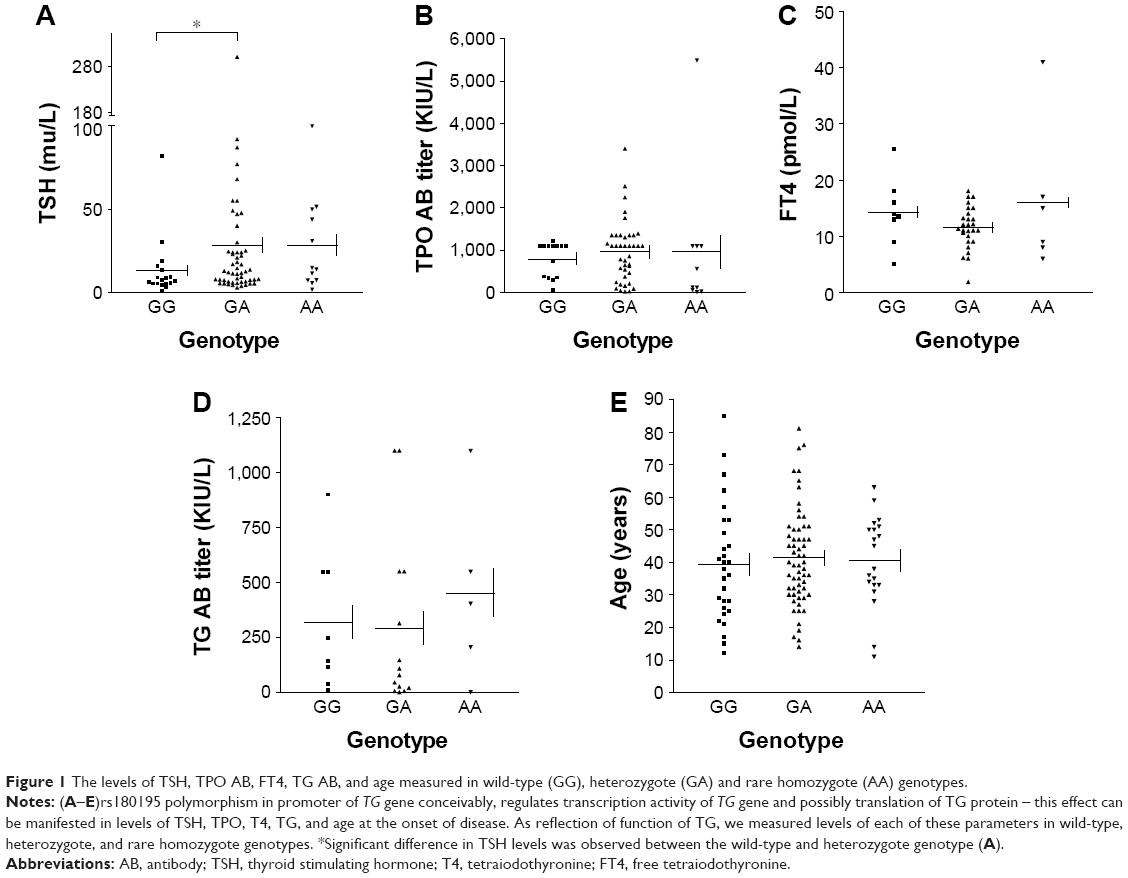 | Figure 1 The levels of TSH, TPO AB, FT4, TG AB, and age measured in wild-type (GG), heterozygote (GA) and rare homozygote (AA) genotypes. |
Levels of the TG protein in the heterozygote GA genotype showed a negative correlation with FT4 (Spearman r=−0.695, confidence interval −0.899 to −0.244, P=0.0058). Finally, we showed there is a positive correlation between the levels of TG protein and the levels of TSH (Spearman r=0.874, P=0.007) and a negative correlation with FT4 (Spearman r=−0.718, P=0.058) in wild-type GG genotype (Table 8).
 | Table 8 Assessments of correlation between levels of TG protein and other biochemical parameters affected by TG protein in wild-type GG and heterozygote GA genotypes of HT patients |
Discussion
rs180195 SNP was previously identified by Stefan et al,40 independently of us. They showed that this TG SNP predisposes to AITDs. However, this is the first study to explore the association between TG SNPs and HT. We showed that there was a significant difference in the distribution of major allele (G) vs minor allele (A) in patients with HT. In HT, the wild-type (GG) genotype was less common. In this study we showed that the genotypes homozygous AA and heterozygous GA rs180195 SNP in the promoter of the TG gene were more associated with thyroid autoimmunity than the wild-type (GG) polymorphism.
rs180195 is located in the promoter region of the TG gene, identified previously by a series of studies40,47–49 to be a cis-regulatory element – a binding site for IRF-1. In thyroid cells activation of transcription by INF-α takes place through binding of IRF-1 to the rs180195 SNP, which results in induction of the TG gene which may influence development of AITD. Stefan et al40 showed that the changes in nucleotides in the promoter region of the TG gene modified a binding site for IRF-1, a major interferon-induced transcription factor. The same group showed that the polymorphism 1623 A/G SNP (rs180195) increased transcription of the TG gene through a genetic/epigenetic mechanism, which we postulate could lead to the development of AITD. These findings support the notion that the thyroid and orbital disorders are not part of the same disease, ie, “Graves’ disease”, “Hashimoto’s disease”, but separate autoimmune disorders.
In previous publications43,45,46 we suggested that CASQ1 gene SNPs rs3838216, rs74123279, and rs2275703 are possible genetic markers for GO and HT, in addition to those that are already known. They are potentially pathogenic genetic markers for the eye muscle component of GO. However, pathogenic manifestations of CASQ1 SNP could be secondary to a primary target gene such as the TG gene. The protein product TG has three important functions; namely as a thyroid hormone precursor, storage site for iodine, and a storage site for inactive thyroid hormones. A small genetic variation in this vital gene would be expected to result in profound effects on the function of the TG protein. Recently, Yin et al,50 found no difference in HLA, CTLA4, IL23R or TSHr genotype in GD patients with or without GO, but they did not examine the TG promoter SNP.
The main thyroid antigens are: TPO in the follicular cell membrane, TG in the colloid and the TSHr in the thyroid follicular cell membranes. TSHr antibody is associated with hyperthyroidism while serum antibodies against TPO and TG are markers for the thyroid autoimmune processes of HT and GD. In our previous study we showed there was not a significant relationship between ophthalmopathy and these thyroid antibodies in patients with GD, HT or transient (sub-acute, silent, post-partum) thyroiditis.18
In HT patients, at allelic level the A polymorphisms occurred more commonly than the G polymorphism in HT than in controls, in GH than controls, and more commonly in HT than in GO. In HT and GH patients compared with controls, wild-type GG genotypic level showed significant probability with low odds ratio for both groups. Interestingly, when we compared wild-type GG genotype of GO vs wild-type GG genotype of HT, a significantly high probability with high odds ratio was observed, which possibly indicates that there are more common characteristics between these two diseases. It is possible that GO is a separate disease from AITD. A similar picture was observed with the heterozygote genotype GA of 180195 SNP for GO vs HT and in contrast to wild-type genotype, heterozygote genotype of HT vs control showed significant prevalence with high odds ratio (Table 6).
To demonstrate a functional role of rs180195, we measured levels of each of these parameters in wild-type GG, heterozygote GA and rare homozygote AA genotypes. A significant difference in TSH levels was observed between the wild-type and heterozygote genotype. In the wild-type genotype in HT patients there is a significant positive correlation between TSH and TG levels, indicating TSH as a primary stimulant for thyroid cells and subsequent effects of TG synthesis and function, and negative correlation with FT4. In heterozygote genotypes in HT patients, a significant negative correlation was observed between FT4 and TG indicating that with lower levels of TG, there will be lower levels of thyroid hormones in HT patients.
Conclusion
We identified rs180195 TG gene polymorphisms in a panel of TG SNPs and showed that the wild-type and heterozygote genotypes of TG are significantly associated with HT and GH patients, ie, patients with thyroid autoimmunity. This polymorphism appears not to be associated with GO, raising the possibility that the thyroid and orbital disorders are not part of the same disease, ie, “Graves’ disease” or “Hashimoto’s disease”. In future studies, a promoter construct of this polymorphism could be used to study the effect of INF-α. Panels of candidate drugs could be used to control the transcription and translation of the TG gene in order to characterize the detailed mechanisms of the action of TG in HT and GH in AITD.
Acknowledgments
We are grateful to Nepean Medical Research Foundation and Nepean Blue Mountains Local Health District for grants supporting this study. It is with sadness that we acknowledge that Professor Patrick Cregan, a co-investigator during the course of this project, recently passed away. We gratefully acknowledge Professor Cregan’s contributions.
Disclosure
The authors report no conflicts of interest in this work.
References
Wall JR. Thyroid function. Pathogenesis of Graves ophthalmopathy – a role for TSH-R? Nat Rev Endocrinol. 2014;10(5):256–258. | ||
Girgis CM, Champion BL, Wall JR. Current concepts in Graves’ disease. Ther Adv Endocrinol Metab. 2011;2(3):135–144. | ||
Wiersinga WM, Bartalena L. Epidemiology and prevention of Graves’ ophthalmopathy. Thyroid. 2002;12(10):855–860. | ||
Weetman AP. Graves’ disease. N Engl J Med. 2000;343(17):1236–1248. | ||
Gerding MN, van der Meer JW, Broenink M, Bakker O, Wiersinga WM, Prummel MF. Association of thyrotropin receptor antibodies with the clinical features of Graves’ ophthalmopathy. Clin Endocrinol (Oxf). 2000;52(3):267–271. | ||
Eckstein AK, Plicht M, Lax H, et al. Thyrotropin receptor autoantibodies are independent risk factors for Graves’ ophthalmopathy and help to predict severity and outcome of the disease. J Clin Endocrinol Metab. 2006;91(9):3464–3470. | ||
Paschke R, Vassart G, Ludgate M. Current evidence for and against the TSH receptor being the common antigen in Graves’ disease and thyroid associated ophthalmopathy. Clin Endocrinol (Oxf). 1995;42(6):565–569. | ||
Shanmuganathan T, Girgis C, Lahooti H, Champion B, Wall JR. Does autoimmunity against thyroglobulin play a role in the pathogenesis of Graves ophthalmopathy: a review. Clin Ophthalmol. 2015;9:2271–2276. | ||
Dumont JE, Vassart G, Refetoff S. Thyroid Disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic Basis of Inherited Disease. Vol. II. 6th ed. New York: McGraw Hill; 1989:1854–1861. | ||
Di Jeso B, Arvan P. Thyroglobulin from molecular and cellular biology to clinical endocrinology. Endocr Rev. 2016;37(1):2–36. | ||
Brocas H, Christophe D, Pohl V, Vassart G. Cloning of human thyroglobulin complementary DNA. FEBS Lett. 1982;137(2):189–192. | ||
Baas F, van Ommen GJ, Bikker H, Arnberg AC, de Vijlder JJ. The human thyroglobulin gene is over 300 kb long and contains introns of up to 64 kb. Nucleic Acids Res. 1986;14(13):5171–5186. | ||
Malthiéry Y, Lissitzky S. Primary structure of human thyroglobulin deduced from the sequence of its 8448-base complementary DNA. Eur J Biochem. 1987;165(3):491–498. | ||
Parma J, Christophe D, Pohl V, Vassart G. Structural organization of the 5′ region of the thyroglobulin gene. Evidence for intron loss and “exonization” during evolution. J Mol Biol. 1987;196(4):769–779. | ||
Mendive FM, Rivolta CM, Moya CM, Vassart G, Targovnik HM. Genomic organization of the human thyroglobulin gene: the complete intron-exon structure. Eur J Endocrinol. 2001;145(4):485–496. | ||
Park SM, Chatterjee VK. Genetics of congenital hypothyroidism. J Med Genet. 2005;42(5):379–389. | ||
Vono-Toniolo J, Rivolta CM, Targovnik HM, Medeiros-Neto G, Kopp P. Naturally occurring mutations in the thyroglobulin gene. Thyroid. 2005;15(9):1021–1033. | ||
Lahooti H, Shanmuganathan T, Champion B, Wall JR. Serum thyroglobulin levels in patients with thyroid autoimmunity with and without ophthalmopathy or isolated upper eyelid retraction. Int Trends Immun. 2015;3:22–27. | ||
Konishi J, Herman MN, Kriss JP. Binding of thyroglobulin and thyroglobulin-antithyroglobulin immune complex to extraocular muscle membrane. Endocrinology. 1974;95(2):434–466. | ||
McDougall IR, Kriss JP. New thoughts about the cause and treatment of the severe ocular manifestations of Graves’ disease. Scott Med J. 1974;19(4):165–169. | ||
Ieiri T, Cochaux P, Targovnik HM, et al. A 3′ splice site mutation in the thyroglobulin gene responsible for congenital goiter with hypothyroidism. J Clin Invest. 1991;88(6):1901–1905. | ||
Targovnik H, Propato F, Varela V, et al. Low levels of thyroglobulin messenger ribonucleic acid in congenital goitrous hypothyroidism with defective thyroglobulin synthesis. J Clin Endocrinol Metab. 1989;69(6):1137–1147. | ||
Targovnik HM, Frechtel GD, Mendive FM, et al. Evidence for the segregation of three different mutated alleles of the thyroglobulin gene in a Brazilian family with congenital goiter and hypothyroidism. Thyroid. 1998;8(4):291–297. | ||
Targovnik HM, Medeiros-Neto G, Varela V, Cochaux P, Wajchenberg BL, Vassart G. A nonsense mutation causes human hereditary congenital goiter with preferential production of a 171-nucleotide-deleted thyroglobulin ribonucleic acid messenger. J Clin Endocrinol Metab. 1993;77(1):210–215. | ||
Targovnik HM, Rivolta CM, Mendive FM, Moya CM, Vono J, Medeiros-Neto G. Congenital goiter with hypothyroidism caused by a 5′ splice site mutation in the thyroglobulin gene. Thyroid. 2001;11(7):685–690. | ||
Targovnik HM, Vono J, Billerbeck AE, et al. A 138-nucleotide deletion in the thyroglobulin ribonucleic acid messenger in a congenital goiter with defective thyroglobulin synthesis. J Clin Endocrinol Metab. 1995;80(11):3356–3360. | ||
Hishinuma A, Fukata S, Kakudo K, Murata Y, Ieiri T. High incidence of thyroid cancer in long-standing goiters with thyroglobulin mutations. Thyroid. 2005;15(9):1079–1084. | ||
Hishinuma A, Fukata S, Nishiyama S, et al. Haplotype analysis reveals founder effects of thyroglobulin gene mutations C1058R and C1977S in Japan. J Clin Endocrinol Metab. 2006;91(8):3100–3104. | ||
Hishinuma A, Takamatsu J, Ohyama Y, et al. Two novel cysteine substitutions (C1263R and C1995S) of thyroglobulin cause a defect in intracellular transport of thyroglobulin in patients with congenital goiter and the variant type of adenomatous goiter. J Clin Endocrinol Metab. 1999;84(4):1438–1444. | ||
Gutnisky VJ, Moya CM, Rivolta CM, et al. Two distinct compound heterozygous constellations (R277X/IVS34-1G-C and R277X/R1511X) in the thyroglobulin (TG) gene in affected individuals of a Brazilian kindred with congenital goiter and defective TG synthesis. J Clin Endocrinol Metab. 2004;89(2):646–657. | ||
van de Graaf SA, Ris-Stalpers C, Veenboer GJ, et al. A premature stop codon in thyroglobulin messenger RNA results in familial goiter moderate hypothyroidism. J Clin Endocrinol Metab. 1999;84(7):2537–2542. | ||
Kitanaka S, Takeda A, Sato U, et al. A novel compound heterozygous mutation in the thyroglobulin gene resulting in congenital goitrous hypothyroidism with high serum triiodothyronine levels. J Hum Genet. 2006;51(4):379–382. | ||
Kanou Y, Hishinuma A, Tsunekawa K, et al. Thyroglobulin gene mutations producing defective intracellular transport of thyroglobulin are associated with increased thyroidal type 2 iodothyronine deiodinase activity. J Clin Endocrinol Metab. 2007;92(4):1451–1457. | ||
Caron P, Moya CM, Malet D, et al. Compound heterozygous mutations in the thyroglobulin gene (1143delC and 6725G-A [R2223H]) resulting in fetal goitrous hypothyroidism. J Clin Endocrinol Metab. 2003;88(8):3546–3553. | ||
Alzahrani AS, Baitei EY, Zou M, Shi Y. Clinical case seminar: metastatic follicular thyroid carcinoma arising from congenital goiter as a result of a novel splice donor site mutation in the thyroglobulin gene. J Clin Endocrinol Metab. 2006;91(3):740–746. | ||
Ban Y, Greenberg DA, Concepcion E, Skrabanek L, Villanueva R, Tomer Y. Amino acid substitutions in the thyroglobulin gene are associated with susceptibility to human and murine autoimmune thyroid disease. Proc Natl Acad Sci U S A. 2003;100(25):15119–15124. | ||
Ban Y, Tozaki T, Taniyama M, Tomita M, Ban Y. Association of a thyroglobulin gene polymorphism with Hashimoto’s thyroiditis in the Japanese population. Clin Endocrinol (Oxf). 2004;61(2):263–268. | ||
Collins JE, Heward JM, Howson JM, et al. Common allelic variants of exons 10, 12, and 33 of the thyroglobulin gene are not associated with autoimmune thyroid disease in the United Kingdom. J Clin Endocrinol Metab. 2004;89(12):6336–6339. | ||
Belguith-Maalej S, Hadj Kacem H, Rebai A, Mnif M, Abid M, Ayadi H. Thyroglobulin polymorphisms in Tunisian patients with autoimmune thyroid diseases (AITD). Immunobiology. 2008;213(7):577–583. | ||
Stefan M, Jacobson EM, Huber AK, et al. Novel variant of thyroglobulin promoter triggers thyroid autoimmunity through an epigenetic interferon alpha-modulated mechanism. J Biol Chem. 2011;286(36):31168–31179. | ||
Ban Y, Tozaki T, Taniyama M, et al. Multiple SNPs in intron 41 of thyroglobulin gene are associated with autoimmune thyroid disease in the Japanese population. PLoS One. 2012;7(5):e37501. | ||
Wang LQ, Wang TY, Sun QL, Qie YQ. Correlation between thyroglobulin gene polymorphisms and autoimmune thyroid disease. Mol Med Rep. 2015;12(3):4469–4475. | ||
Lahooti H, Cultrone D, Edirimanne S, et al. Novel single nucleotide polymorphisms in the calsequestrin-1 gene are associated with Graves’ ophthalmopathy and Hashimoto’s thyroiditis. Clin Ophthalmol. 2015;9:1731–1740. | ||
Walsh JP, Berry J, Liu S, et al. The clinical presentation of autoimmune thyroid disease in men is associated with IL12B genotype. Clin Endocrinol (Oxf). 2011;74(4):508–512. | ||
Cultrone D, Lahooti H, Edirimanne S, Delbridge S, Champion B, Wall JR. Calsequestrin is decreased in the thyroid gland of patients with Graves’ disease – further evidence for a role of autoimmunity against this protein in Graves’ ophthalmopathy. British Journal of Medicine and Medical Research. 2014;4(23):4065–4075. | ||
Lahooti H, Cultrone D, Edirimanne S, et al. Association of the CASQ1 gene SNP rs3838216 with Graves’ ophthalmopathy and Hashimoto’s thyroiditis in patients with thyroid autoimmunity. Ophthalmology Research: An International Journal. 2014;2(6):281–293. | ||
Hasham A, Tomer Y. Genetic and epigenetic mechanisms in thyroid autoimmunity. Immunol Res. 2012;54(1–3):204–213. | ||
Tomer Y. Mechanisms of autoimmune thyroid diseases: from genetics to epigenetics. Annu Rev Pathol. 2014;9:147–156. | ||
Wang Y, Smith TJ. Current concepts in the molecular pathogenesis of thyroid-associated ophthalmopathy. Invest Ophthalmol Vis Sci. 2014;55(3):1735–1748. | ||
Yin X, Latif R, Bahn R, Davies TF. Genetic profiling in Graves’ disease: further evidence for lack of a distinct genetic contribution to Graves’ ophthalmopathy. Thyroid. 2012;22(7):730–736. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.