")
Back to Journals » OncoTargets and Therapy » Volume 13
Simvastatin Inhibits the Malignant Behaviors of Gastric Cancer Cells by Simultaneously Suppressing YAP and β-Catenin Signaling
Authors Liu Q , Xia H, Zhou S, Tang Q, Zhou J, Ren M, Bi F
Received 7 November 2019
Accepted for publication 25 February 2020
Published 9 March 2020 Volume 2020:13 Pages 2057—2066
DOI https://doi.org/10.2147/OTT.S237693
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Qing Liu,1,2,* Hongwei Xia,2,* Sheng Zhou,1,2 Qiulin Tang,1,2 Jitao Zhou,1 Min Ren,1 Feng Bi1,2
1Department of Abdominal Oncology, Cancer Center, West China Hospital, Sichuan University, Chengdu 610041, People’s Republic of China; 2Laboratory of Molecular Targeted Therapy in Oncology, West China Hospital, Sichuan University, Chengdu 610041, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Feng Bi
Department of Abdominal Oncology, Cancer Center, West China Hospital, Sichuan University, Chengdu 610041, People’s Republic of China
Tel +86 189 8060 1771
Email [email protected]
Background: Statins, which are used to lower blood cholesterol levels by inhibiting HMG-CoA reductase, have shown anticancer effects in many cancer cells. However, the role of statins in gastric cancer remains unclear. This study aims to investigate whether the statins could antagonize progression of gastric cancer cells and tried to find the molecule mechanism.
Methods: Effects of simvastatin on the morphology, proliferation, migration, apoptosis, and invasion of gastric cancer cells were detected and compared. Western blotting, cell viability assay, fluorescence, and transfection were employed to study the molecule mechanism of the effects and the interaction between YAP and β-catenin signaling.
Results: Simvastatin could inhibit proliferation, migration and invasion, and promote the apoptosis in gastric cancer cells. Mechanistic studies showed that simvastatin treatment could inhibit the expression of β-catenin and the activity of YAP and the downstream targets of YAP and β-catenin in gastric cancer cells. Moreover, we found that YAP and β-catenin could form a positive feedback loop in gastric cancer cells. Further investigation revealed that simvastatin mainly acted through by inhibiting the activity of RhoA to inhibit YAP and β-catenin, and the geranylgeranyl pyrophosphate pathway mediated this regulation.
Conclusion: Statins represent a promising therapeutic option for gastric cancer by simultaneously targeting YAP and β-catenin signaling.
Keywords: gastric cancer, simvastatin, YAP, β-catenin, RhoA
Introduction
Gastric cancer (GC), ranking fifth in incidence among all cancers, is one of the most deadly cancers, especially in developing countries.1 Due to the lack of specific symptoms and tendencies toward tumor invasion and metastasis, most patients are diagnosed with GC at an advanced stage. Perioperative treatment might be curative for patients with early-stage gastric cancer, but chemotherapy constitutes the main current therapy for gastric cancer patients.2 Therefore, it is critical to understand the molecular and cellular mechanisms underlying gastric cancer progression and metastasis and to identify new medicines or therapies for the treatment of GC.
Statins, specific inhibitors of 3-hydroxy-methylglutaryl CoA reductase (HMGCR), are widely used to treat hypercholesterolemia and prevent cardiovascular diseases.3 3-Hydroxy-methylglutaryl CoA reductase (HMGCR) catalyzes the rate-limiting, mevalonate production step in the mevalonate pathway for the biosynthesis of isoprenoids and downstream products, such as cholesterol, dolichol, and ubiquinone.4 HMGCR expression was found to be upregulated in gastric cancer and promotes the growth and migration of cancer cells.5 Remarkably, recent studies in vitro and in vivo have found that in addition to their cholesterol reduction effects, statins also have multiple anticancer effects, such as antiproliferative, proapoptotic, antiangiogenic, immunomodulatory, and anti–invasive effects, which make them promising therapeutic agents against many cancers, such as liver cancer, colon cancer, and breast cancer.4,6 However, the role of statins in gastric cancer remains elusive.
The Hippo pathway is an evolutionarily conserved regulator of tissue growth and cell fate, and an increasing number of studies have observed mutations and altered expression patterns in a subset of Hippo pathway genes in human cancers.7 Moreover, deregulation of the Hippo pathway is strongly associated with initiation, development and distant metastasis in human gastric cancer.8,9
The Wnt/β-catenin pathway plays a vital role in orchestrating complex cellular behaviors during development, tissue homeostasis, and cancer.10,11 Approximately 10–30% of human gastric tumors displayed deregulated Wnt signaling,12 and a significant number of Wnt pathway mutations were confirmed in GC by the latest TCGA study.13 As resistance to current chemotherapy is common, targeting the Wnt pathway may be a potential strategy for the treatment of GC.14
In our study, we investigated whether statins could antagonize the progression of gastric cancer cells and tried to determine whether the molecular mechanism depended on YAP and β-catenin signaling.
Materials and Methods
Cell Culture
The gastric cancer cell lines MKN45 and MGC803 were purchased from ATCC (American Type Culture Collection). All cell lines were maintained in DMEM (Gibco Laboratories, Grand Island, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco, Gaithersburg, USA). Cell cultures were maintained at 37°C in a humidified 5% CO2 and 95% air incubator.
Transfection
The siRNAs targeting YAP, RhoA, β-Catenin and the control were designed and synthesized by RiboBio (Guangzhou, China). The sequences of the siRNAs used in this study were as follows: siYAP: 5ʹ-GACAUCUUCUGGUCAGAGA-3ʹ, siRhoA: 5ʹ-GACAUGCUUGCUCAUAGUCTT-3ʹ, sicontrol: 5ʹ-AAUUCUCCGAACGUGUCACGUUU-3ʹ, and siβ-catenin: 5ʹ-GGUGGUGGUUAAUAAGGCU-3ʹ. The plasmids pcDNA3.1, pcDNA3.1YAP, pcDNA3.1β-Catenin and pcDNA3.1RhoA-V14 were generated in our laboratory. The siRNAs and plasmids were transfected into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
Western Blot Analysis
Cells were lysed with RIPA buffer containing a protease inhibitor cocktail (Sigma-Aldrich) and then quantified with a BCA protein assay (Pierce Chemical Co, USA). Approximately 20 μg total protein was loaded on a 10% or 12% SDS-PAGE gel, separated electrophoretically and then transferred to NC membranes (Millipore, Bedford, USA). Finally, protein expression was detected using an ODYSSEY Infrared Imaging System (LI-OR Biosciences, Lincoln, USA). Antibody against CYR61 was purchased from Sangon Biotech. Antibodies against RhoA, GAPDH, and β-catenin were purchased from Santa Cruz Biotechnology. Antibodies against YAP, p-YAP (Ser127), c-Myc, and cyclin D1 were purchased from Abcam.
Cell Proliferation Assay
Cells were seeded (4×103 cells per well in 100 μL) in a 96-well flat-bottomed plate 24 h before treatment. After an incubation for 48–72 h, cell growth was measured using a Cell Counting Kit-8 assay (Dojingdo, Kumamoto, Japan) according to the manufacturer’s instructions.
Crystal Violet Assay
Equal number of control cells and experimental cells were seeded in 12-well plates and cultured in medium supplemented with 10% FBS at a density of 1000 cells/well. The medium was changed every other day. After ten days of culture under the standard conditions, the medium was removed and the cells were stained with 0.5% crystal violet solution in 20% methanol. After staining for 10 mins, the fixed cells were washed with phosphate-buffered saline (PBS) and photographed.
Fluorescence
To analyze the F-actin cytoskeleton, cells were washed with phosphate-buffered saline (PBS) and then fixed for 5 mins in 3.7% formaldehyde solution in PBS. Then, the cells were washed extensively in PBS. The gastric cancer cells were dehydrated with acetone, permeabilized with 0.1% Triton X-100 in PBS and washed again in PBS. Then, the cells were stained with a 50 mg/mL fluorescent isothiocyanate (FITC)-conjugated phalloidin (Sigma-Aldrich, P5282) solution in PBS for 40 mins at room temperature, and then washed several times with PBS to remove unbound phalloidin conjugate. The gastric cancer cells were rinsed and mounted on slides in glycerol. The fluorescence was detected using a fluorescence microscope (Eclipse TE300, Nikon, Japan), and image analysis was completed with Photoshop software (Adobe).
GST Pull-Down Assay
Inoculate 5 mL of LB broth with ampicillin (100 μg/mL) in a culture vial with a single colony of BL21 strain of E. coli (transformed with Rhotekin plasmid) and incubate overnight at 37°C with shaking at 200 rpm until O. D 600 of 0.8–0.9 is achieved. Induce the culture with 0.6 mM conc. of IPTG for 6 hrs at 33.5°C. Harvest the cells by centrifugation and wash the cells twice with ice-cold 1X PBS. Resuspend the pellet in Cell Lysis Buffer and broke the cells by a sonic oscillator (15 s for 20 times at 1 min intervals). Spin at 12,000 rpm at 4°C for 3 min. Transfer the supernatant carefully to a new tube and add 50 μL GSH beads (Sigma). Then mix well by a mixer at 4°C for 2 hrs. Use the same cell Lysis Buffer lysate cancer cells, mix well and then spin at 12,000 rpm at 4°C for 15 min. After detecting the concentration of protein, add a same amount of protein to beads for each sample. Add ice-cold 1X PBS to make the whole volume 1mL, and mix well by the mixer at 4°C for 2 hrs. Spin at 12000rpm at 4°C for 5 min, decant the supernatant carefully and wash the beads three times with 1mL ice-cold 1X PBS. Add 50 μL 2X loading buffer, boil the samples for 10 min. Spin at 12000rpm at 4°C for 5 min, and use the supernatant to do Western blot.
Data Analysis
Statistical analyses were performed using GraphPad Prism 6 software. Comparisons between the values from two groups were performed using Student’s t-tests and comparisons between multiple groups were performed by analysis of variance. The significance level was set at P < 0.05.
Results
Effects of Simvastatin on the Morphology, Proliferation, Migration, Apoptosis, and Invasion of Gastric Cancer Cells
To determine the effect of simvastatin on gastric cancer cells, MKN45 and MGC803 cells were treated with simvastatin at different concentrations varying from 0 to 60 μM for 24 or 48 h. As indicated in Figure 1A, after treatment with simvastatin for 48 h, the morphology of the two cell lines changed significantly from a fusiform shape to a round shape. The proliferative activity of the two cell lines was detected by a Cell Counting Kit-8 assay (Figure 1C) and clone formation assay (Figure 1B), which both showed that simvastatin retarded the growth of gastric cancer cells in a concentration-dependent manner. Moreover, cell apoptosis was assessed with flow cytometry, and the result showed that simvastatin induced both early and late apoptosis in the two gastric cancer cell lines (Figure 1D). In addition, a scratch assay showed that simvastatin inhibited the migration of gastric cancer cells (Figure 1E), and the Matrigel-coated Transwell assay further confirmed that simvastatin suppressed the invasiveness of gastric cancer cells (Figure 1F).
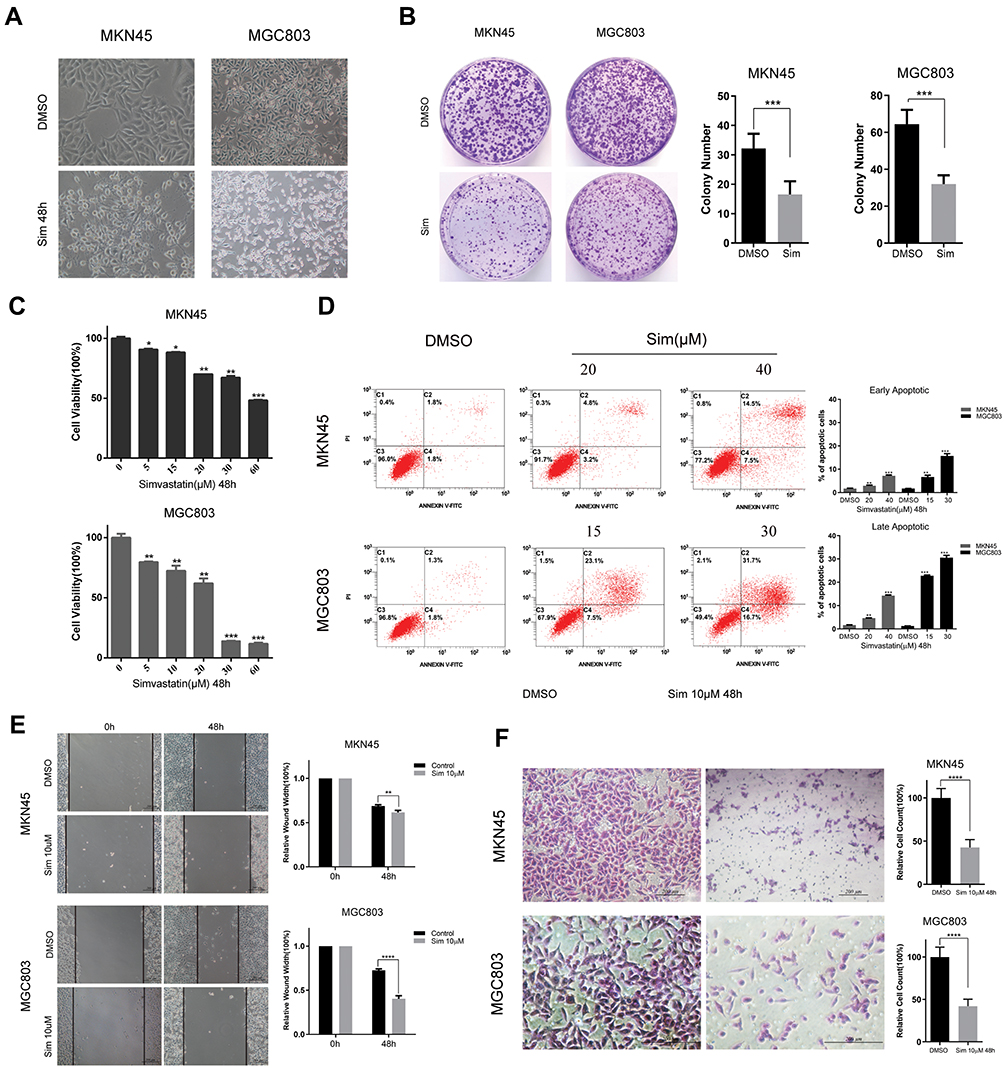 |
Figure 1 Effects of simvastatin on the morphology, proliferation, migration, apoptosis, and invasion of gastric cancer cells. (A) The morphologies of MKN45 and MGC803 cells changed from a fusiform shape to a round shape after treatment with simvastatin for 48 h. (B) MKN45 and MGC803 cells were treated with simvastatin for approximately 7 days at concentrations of 5 µM and 7.5 µM, respectively. (C) The effects of simvastatin on the proliferation of MKN45 and MGC803 cells were detected by a CCK8 assay. (D) The effects of simvastatin on apoptosis in MKN45 and MGC803 cells were detected by flow cytometry, and the data on the early and late apoptotic cells were counted. (E) Cell migration abilities were determined by wound healing assay. The relative wound width = wound width at 0 h, or 48 h/wound width at 0 h. (F) The migratory abilities of MKN45 and MGC803 cells after treatment with simvastatin were assayed with a Matrigel-coated Transwell assay. The cells that successfully migrated into the lower chamber were quantified at 48 h after plating. The data are shown as the mean ± SD of three independent experiments. A Student’s two-tailed t test was used for the statistical analysis (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). |
Simvastatin Treatment Inhibits YAP and β-Catenin Signaling in Gastric Cancer Cells
The Wnt pathway and Hippo pathway play critical roles in the carcinogenesis of gastric cancer.9,11 Therefore, we tested whether simvastatin treatment could regulate the core effectors of the Wnt pathway and Hippo pathway as well as their downstream targets.
As shown in Figure 2A, the expression levels of β-catenin, the core effector of the Wnt pathway, and its downstream targets c-Myc and Cyclin D1 were significant decreased with the increasing concentrations of simvastatin compared with control treatment.
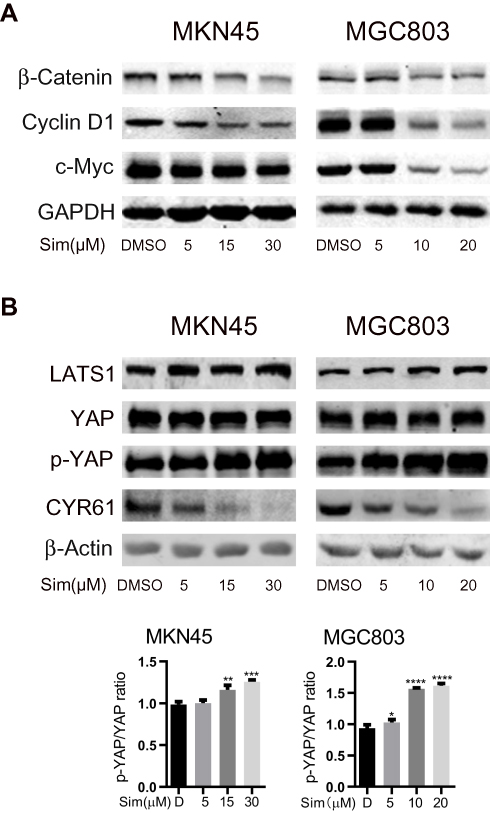 |
Figure 2 Simvastatin inhibits YAP and β-catenin signaling in gastric cancer cells. MKN45 and MGC803 cells were treated with simvastatin at increasing concentrations. Western blot analysis showed that simvastatin downregulated the β-catenin protein levels and the protein levels of the downstream molecules c-Myc and cyclin D1 (A). YAP activity was inhibited by simvastatin (B). Both effects were dose-dependent. The data are shown as the mean ± SD of three independent measures. A Student’s two-tailed t test was used for the statistical analysis (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). |
We also investigated the levels of YAP, the core effector of the Hippo pathway, Ser127-phosphorylated YAP and the YAP target CYR61. Phosphorylation of YAP at Ser127 induces translocation of YAP from the nucleus to the cytoplasm, and CYR61 is a target gene of the transcriptional YAP/TEAD complex. As shown in Figure 2B, compared with control treatment, simvastatin induced an increase in the LATS1 protein level and phosphorylation level of YAP at Ser127 and a decrease in the levels of CYR61 in MKN45 and MGC803 cells, while the total level of YAP protein remained almost unchanged. Together, these results indicated that simvastatin could inhibit the activity of YAP and β-catenin in gastric cancer cells.
YAP and β-Catenin Form a Positive Feedback Loop in Gastric Cancer Cells
Previous studies have revealed that YAP and β-catenin could form a positive feedback loop and play an important role in the carcinogenesis of several cancers.15,16 Here, we tried to determine whether positive feedback regulation also existed in gastric cancer. First, we investigated the effects of YAP expression knockdown in MKN45 and MGC803 cells. As expected, compared with normal YAP expression, the knockdown of YAP expression significantly reduced the expression of the downstream target CYR61 as well as the levels of β-catenin and its downstream targets c-Myc and Cyclin D1 (Figure 3A). Compared with normal β-catenin expression, the knockdown of β-catenin expression decreased the expression of its targets c-Myc and Cyclin D1, the activity of YAP and the protein level of the YAP downstream target CYR61 (Figure 3A). To investigate whether the overexpression of YAP could reverse the simvastatin treatment-induced downregulation of β-catenin and its downstream targets, the constitutively active YAP-5SA plasmid and the control plasmid were transfected into these two cell lines. The results showed that the forced expression of active YAP could partially counteract the effects of simvastatin treatment on the expression of CYR61, β-catenin, c-Myc and Cyclin D1 (Figure 3B). Moreover, we used two methods to enhance the Wnt/β-catenin pathway, overexpression of wild-type β-catenin or treatment with LiCl, which could induce the accumulation of β-catenin, enhancing the activation of Wnt/β-catenin signaling.17–19 As shown by Western blot we found that the upregulation of β-catenin could partially reverse the effects of simvastatin treatment on the expression of c-Myc, Cyclin D1, CYR61 and YAP (Figure 3C and D). In summary, simvastatin could inhibit the malignant behaviors of gastric cancer cells by simultaneously downregulating the activities of YAP and β-catenin, and YAP and β-catenin form a positive feedback loop in GC.
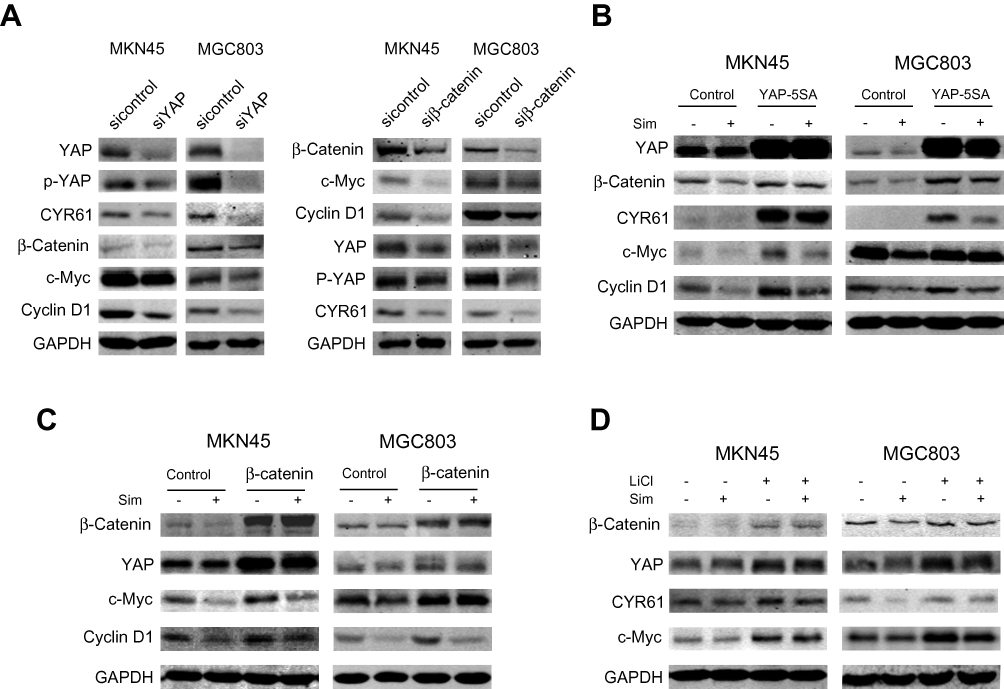 |
Figure 3 The crosstalk between YAP and β-catenin in gastric cancer cells. (A) MKN45 and MGC803 cells were transfected with control, siYAP or siβ-catenin. MKN45 and MGC803 cells were transfected with control, YAP-5SA (B) or β-catenin plasmid (C). LiCl-treated MKN45 and MGC803 cells were assessed by Western blotting (D). |
RhoA Mediated the Simvastatin-Induced Inhibition of YAP and β-Catenin Signaling in Gastric Cancer Cells
From the microscopy studies, we found that simvastatin treatment could change the morphology of gastric cancer cells. Further immunofluorescence experiments showed that simvastatin treatment induced F-actin cytoskeleton fragmentation (Figure 4A). Filamentous actin is the main component of the cytoskeleton, and Rho GTPases are well-known organizers of the F-actin cytoskeleton.20 Thus, we detected the activity of RhoA and found that the level of total RhoA increased with increasing concentrations of simvastatin, while active RhoA levels were found to be decreased by GST pulldown assay (Figure 4B). Several previous studies have shown that the mevalonate pathway regulates YAP activity through the activation of Rho GTPases.21,22 Next, we investigated whether simvastatin was able to affect YAP and β-catenin activity through RhoA in gastric cancer cells. As shown by Western blotting, the knockdown of RhoA expression decreased the levels of YAP and β-catenin in MKN45 and MGC803 cells (Figure 4C). Moreover, the overexpression of constitutively active RhoA-V14 was sufficient to counteract the effects of simvastatin on YAP and β-catenin in the HEK293 cell line (Figure 4D). In conclusion, simvastatin changes the morphology of gastric cancer cells by suppressing the activity of Rho GTPases, which in turn inhibits YAP and β-catenin signaling.
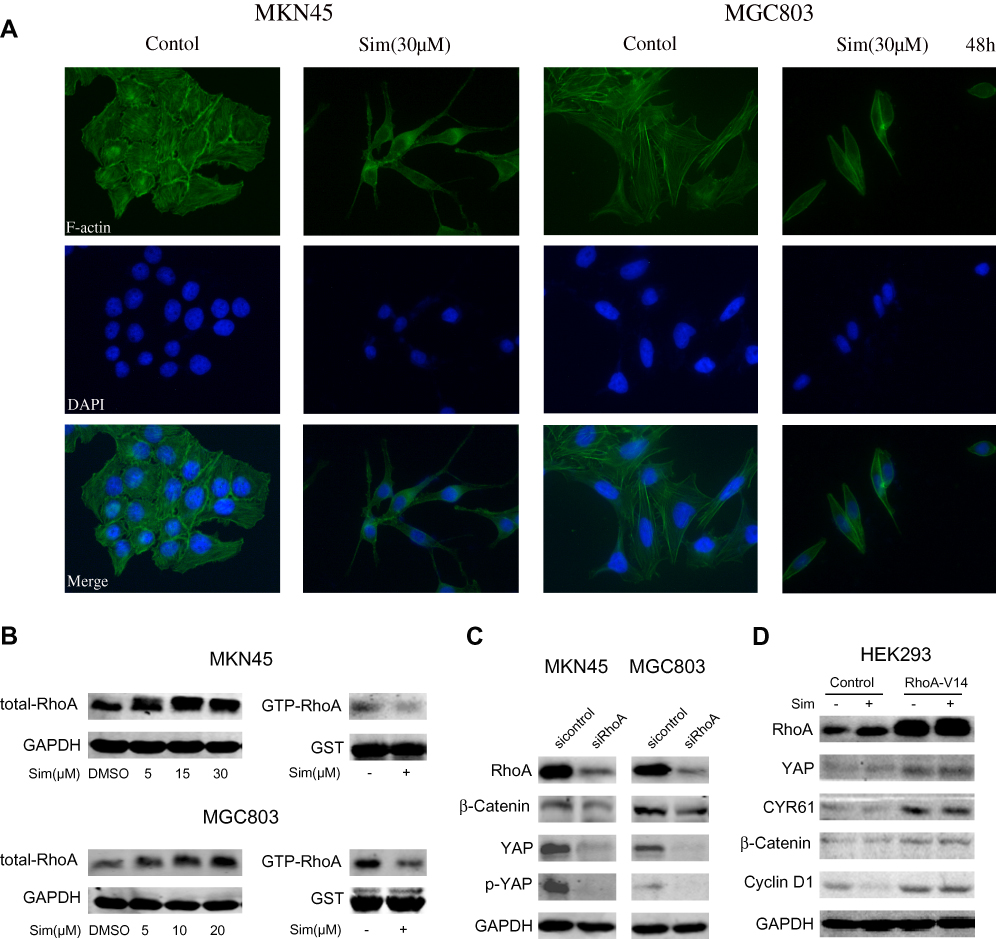 |
Figure 4 RhoA-mediated regulation of the cytoskeleton mediated the simvastatin-induced inhibition of YAP and β-catenin signaling in gastric cancer cells. (A) Simvastatin changed the cell morphology and induced F-actin cytoskeleton fragmentation in MKN45 and MGC803 cells. (B) The levels of total RhoA and GTP-RhoA in MKN45 and MGC803 cells after treatment with simvastatin are shown. (C) MKN45 and MGC803 cells were transfected with control or RhoA siRNAs. (D) HEK293 cells were transfected with control or RhoA-V14 plasmid. |
Geranylgeranyl Pyrophosphate Prevents Simvastatin-Induced Effects on Gastric Cancer Cells
Since the mevalonate-derived prenyl groups, which are intermediates in the HMG-CoA reductase pathway, are essential lipid anchors for active small GTPase proteins in cells, we tried to determine whether farnesyl pyrophosphate (FPP) or geranylgeranyl pyrophosphate (GGPP) could reverse the effects of simvastatin on the two gastric cancer lines or whether they were the upstream regulators that mediated the simvastatin-induced downregulation of YAP activity and β-catenin expression in the gastric cancer cell lines. As shown by a Cell Counting Kit-8 assay, the simvastatin-induced inhibition of cell viability was reversed by supplementation with GGPP (Figure 5A), while FPP supplementation could not reverse this effect (Figure 5B). Western blot results also showed that GGPP, but not FPP, reversed the effects of simvastatin on the YAP, CYR61, β-Catenin, Cyclin D1, and c-Myc protein levels (Figure 5C and D). GGPP, but not FPP, almost completely prevented the statin-induced inhibition of cell viability seen in this study. Together, the above data indicated that GGPP mainly mediated the simvastatin-induced inhibition of YAP and β-catenin.
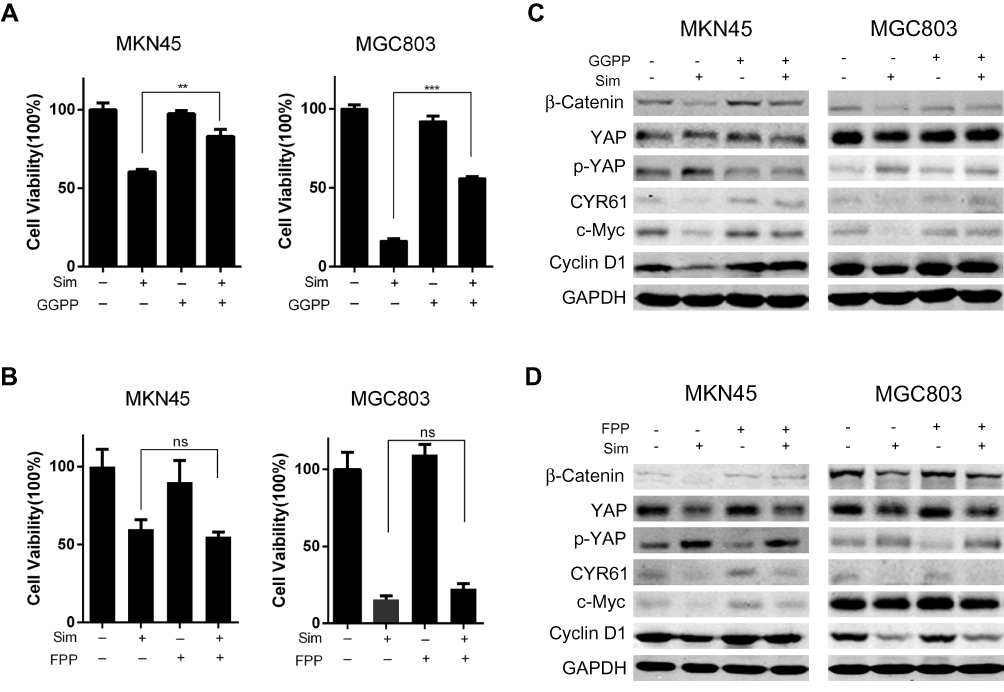 |
Figure 5 Geranylgeranyl pyrophosphate reverses the simvastatin-induced effects on gastric cells. GGPP reversed the effects of simvastatin on the proliferation (A) and protein levels (C) of MKN45 and MGC803 cells. FPP could not change the effects of simvastatin (B and D). The data are shown as the mean ± SD of three independent measures. A Student’s two-tailed t test was used for statistical analysis (**P < 0.01 and ***P < 0.001). |
Discussion
Recently, some clinical studies have shown that statin use may be associated with a lower risk of GC,23–25 whereas other studies have shown no beneficial effect.26,27 The reasons for the negative clinical trial results might be that the sample sizes of the randomized clinical trials were too small, the concentrations of statins in these trials were lower, or the treatment durations were shorter than those of the in vitro experiments. In studies about the effects and mechanisms of statins on cancer cells, like our study, the concentration of statins that can inhibit cancer cells is more than 5 μM (2.09ug/mL). However, the IC50 of simvastatin inhibits sterol synthesis in vitro is less than 20nM (8.37ng/mL).28 And for humans, taking simvastatin as a lipid-regulating agent, the Cmax of simvastatin is less than 120nM (50.19ng/mL).29 Correlation between in vitro and in vivo concentration-effect relationships can be so complicated that we should not simply compare these concentrations.30 But we may be able to assume that the dose of simvastatin needed to treat cancer is much higher than the one used for treating cardiovascular diseases. Therefore, the differences between placebo and statins as well as the antitumor effects of statins still need further exploration.
YAP, the core effector of the Hippo pathway, has been defined as the candidate oncogene in several cancers.31 Studies in vivo have shown that YAP inhibition dramatically suppressed tumor formation driven by other Hippo pathway alterations. Dysregulation of the Hippo pathway promotes the proliferation and metastasis of gastric cancer, whereas the inhibition of the Hippo pathway effector proteins YAP and TAZ shows great promise in many preclinical models. The Wnt/β-catenin pathway plays a key role in gastric cancer pathogenesis, and its complex functions are highly dependent on the transcriptional output of β-catenin.32 β-catenin activation is associated with carcinogenesis and metastasis in gastric carcinoma. Consequently, patients with gastric cancer may benefit from drugs targeting both pathways. In our study, we found a positive feedback loop between YAP and β-catenin, which contrasts with previous studies that showed that YAP/TAZ inhibit Wnt/β-catenin signaling in vivo and in vitro.33,34 The alternative Wnt signaling pathway activates YAP/TAZ, which antagonize Wnt/β-catenin signaling via TEAD-mediated gene transcription during adipocyte differentiation.15 However, we found that the reciprocal activation of YAP and β-catenin was an important mechanism for the effect of simvastatin on gastric cancer cells. We still lack a unified model regarding the mechanism of feedback, and more research should be conducted to further our investigation.
GGPP could almost completely prevent the statin-induced inhibition of cell viability seen in this study, but FPP could not. This result was in agreement with previous studies that reported that the addition of GGPP but not FPP overcame statin-induced apoptosis or growth inhibition in tumor cells.35–37 The lack of an effect from the addition of FPP might be explained by the lack of isopentenyl-PP, which is required to convert FPP to GGPP.38 Interesting, in Figure 5, we could see that although FPP could not reverse the effect of simvastatin on YAP, FPP did have some effects on the β-Catenin, Cyclin D1, and c-Myc protein levels; the mechanism needs further study. RhoA, a geranylgeranylated protein that can be modified by GGPP, can regulate the activation of YAP. In our study, we found that RhoA could regulate β-catenin, but whether it was an immediate action or an effect that occurred through the regulation of YAP needs further investigation.
Conclusions
The potential role of statins as anticancer agents in various tumor types has been revealed systematically. In this study, we demonstrated that simvastatin could promote apoptosis and inhibit proliferation and metastasis in gastric cancer cells. The mechanism might be that the statin inhibits HMGCR, downregulating geranylgeranyl pyrophosphate (GGPP), which is an essential substrate for the posttranslational modification of RhoA. Then, the decrease in the active RhoA level leads to the inhibition of YAP and β-catenin activity, while the decreases in the YAP and β-catenin activity levels could mutually promote the decrease in the activity of the other protein (Figure 6). Together, statins may be promising drugs for the treatment of gastric cancer, and further investigations should be carried out in clinical trials.
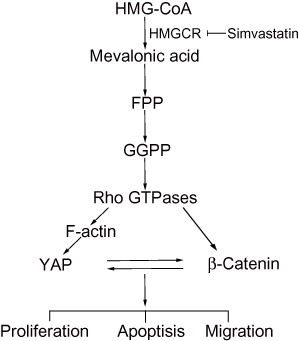 |
Figure 6 A proposed model for the regulation of YAP and β-catenin signaling by simvastatin in gastric cancer. |
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers 81502655, 81572731, and 81602124).
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors have no conflicts of interest to declare in this work.
References
1. Beranard W, Stewart CPW. World Cancer Report 2014. World Health Organization press; 2014: 544–559.
2. Haghighat P, Bekaii-saab T. An update on biochemotherapy of advanced gastric and gastroesophageal adenocarcinoma. J Natl Compr Cancer Net. 2008;6(9):895–900. doi:10.6004/jnccn.2008.0068
3. Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425–430. doi:10.1038/343425a0
4. Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer. 2005;5(12):930–942. doi:10.1038/nrc1751
5. Chushi L, Wei W, Kangkang X, Yongzeng F, Ning X, Xiaolei C. HMGCR is up-regulated in gastric cancer and promotes the growth and migration of the cancer cells. Gene. 2016;587(1):42–47. doi:10.1016/j.gene.2016.04.029
6. Gazzerro P, Proto MC, Gangemi G, et al. Pharmacological actions of statins: a critical appraisal in the management of cancer. Pharmacol Rev. 2012;64(1):102–146. doi:10.1124/pr.111.004994
7. Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13(4):246–257. doi:10.1038/nrc3458
8. Zhou G-X, Li X-Y, Zhang Q, et al. Effects of the hippo signaling pathway in human gastric cancer. Asian Pac J Cancer Prev. 2013;14(9):5199–5205. doi:10.7314/APJCP.2013.14.9.5199
9. Choi W, Kim J, Park J, et al. YAP/TAZ initiates gastric tumorigenesis via upregulation of MYC. Cancer Res. 2018;78(12):3306–3320. doi:10.1158/0008-5472.CAN-17-3487
10. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi:10.1016/j.cell.2012.05.012
11. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127(3):469–480. doi:10.1016/j.cell.2006.10.018
12. Cristescu R, Lee J, Nebozhyn M, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21(5):449–456. doi:10.1038/nm.3850
13. Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–209. doi:10.1038/nature13480
14. Flanagan DJ, Vincan E, Phesse TJ. Winding back Wnt signalling: potential therapeutic targets for treating gastric cancers. Br J Pharmacol. 2017;174(24):4666–4683. doi:10.1111/bph.v174.24
15. Park HW, Kim YC, Yu B, et al. Alternative Wnt signaling activates YAP/TAZ. Cell. 2015;162(4):780–794. doi:10.1016/j.cell.2015.07.013
16. Pan JX, Xiong L, Zhao K, et al. YAP promotes osteogenesis and suppresses adipogenic differentiation by regulating beta-catenin signaling. Bone Res. 2018;6:18. doi:10.1038/s41413-018-0018-7
17. Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93(16):8455–8459. doi:10.1073/pnas.93.16.8455
18. Hedgepeth CM, Conrad LJ, Zhang J, Huang H-C, Lee VM, Klein PS. Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev Biol. 1997;185(1):82–91. doi:10.1006/dbio.1997.8552
19. Hao HP, Wen LB, Li JR, et al. LiCl inhibits PRRSV infection by enhancing Wnt/beta-catenin pathway and suppressing inflammatory responses. Antiviral Res. 2015;117:99–109. doi:10.1016/j.antiviral.2015.02.010
20. Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11(5):353–365. doi:10.1038/nrm2890
21. Sorrentino G, Ruggeri N, Specchia V, et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol. 2014;16(4):357–366. doi:10.1038/ncb2936
22. Wang Z, Wu Y, Wang H, et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci U S A. 2014;111(1):E89–98. doi:10.1073/pnas.1319190110
23. Singh PP, Singh S. Statins are associated with reduced risk of gastric cancer: a systematic review and meta-analysis. Ann Oncol. 2013;24(7):1721–1730. doi:10.1093/annonc/mdt150
24. Chiu HF, Ho SC, Chang CC, Wu TN, Yang CY. Statins are associated with a reduced risk of gastric cancer: a population-based case-control study. Am J Gastroenterol. 2011;106(12):2098–2103. doi:10.1038/ajg.2011.277
25. Lee J, Lee SH, Hur KY, Woo SY, Kim SW, Kang WK. Statins and the risk of gastric cancer in diabetes patients. BMC Cancer. 2012;12:596. doi:10.1186/1471-2407-12-596
26. Kim WS, Kim MM, Choi HJ, et al. Phase II study of high-dose lovastatin in patients with advanced gastric adenocarcinoma. Invest New Drugs. 2001;19(1):81–83. doi:10.1023/A:1006481423298
27. Kim ST, Kang JH, Lee J, et al. Simvastatin plus capecitabine-cisplatin versus placebo plus capecitabine-cisplatin in patients with previously untreated advanced gastric cancer: a double-blind randomised Phase 3 study. Eur J Cancer. 2014;50(16):2822–2830. doi:10.1016/j.ejca.2014.08.005
28. Slater EE, MacDonald JS. Mechanism of action and biological profile of HMG CoA reductase inhibitors. A New Therapeutic Alternative. Drugs. 1988;36(Suppl 3):72–82.
29. Srinivas NR. Limited sampling strategy for the prediction of Area Under the Curve (AUC) of statins: reliability of a single time point for AUC prediction for pravastatin and simvastatin. Drug Res (Stuttg). 2016;66(2):82–93. doi:10.1055/s-0035-1549983
30. Huntjens DR, Spalding DJ, Danhof M, Della Pasqua OE. Correlation between in vitro and in vivo concentration-effect relationships of naproxen in rats and healthy volunteers. Br J Pharmacol. 2006;148(4):396–404. doi:10.1038/sj.bjp.0706737
31. Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15(2):73–79. doi:10.1038/nrc3876
32. Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13(12):767–779. doi:10.1038/nrm3470
33. Imajo M, Miyatake K, Iimura A, Miyamoto A, Nishida E. A molecular mechanism that links Hippo signalling to the inhibition of Wnt/beta-catenin signalling. EMBO J. 2012;31(5):1109–1122. doi:10.1038/emboj.2011.487
34. Barry ER, Morikawa T, Butler BL, et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature. 2013;493(7430):106–110. doi:10.1038/nature11693
35. Xia Z, Tan MM, Wong WW, Dimitroulakos J, Minden MD, Penn LZ. Blocking protein geranylgeranylation is essential for lovastatin-induced apoptosis of human acute myeloid leukemia cells. Leukemia. 2001;15(9):1398–1407. doi:10.1038/sj.leu.2402196
36. Agarwal B, Rao CV, Bhendwal S, et al. Lovastatin augments sulindac-induced apoptosis in colon cancer cells and potentiates chemopreventive effects of sulindac. Gastroenterology. 1999;117(4):838–847. doi:10.1016/S0016-5085(99)70342-2
37. van de Donk NW, Lokhorst HM, Nijhuis EH, Kamphuis MM, Bloem AC. Geranylgeranylated proteins are involved in the regulation of myeloma cell growth. Clin Cancer Res. 2005;11(2 Pt 1):429–439.
38. Danesi R, McLellan CA, Myers CE. Specific labeling of isoprenylated proteins: application to study inhibitors of the post-translational farnesylation and geranylgeranylation. Biochem Biophys Res Commun. 1995;206(2):637–643. doi:10.1006/bbrc.1995.1090
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.