")
Back to Journals » International Journal of Nanomedicine » Volume 11
Simvastatin increases the antineoplastic actions of paclitaxel carried in lipid nanoemulsions in melanoma-bearing mice
Authors Kretzer I, Maria D, Guido MC, Contente T, Maranhao R
Received 14 May 2015
Accepted for publication 17 October 2015
Published 7 March 2016 Volume 2016:11 Pages 885—904
DOI https://doi.org/10.2147/IJN.S88546
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Iara F Kretzer,1,2 Durvanei A Maria,3 Maria C Guido,1 Thaís C Contente,1 Raul C Maranhão1,2
1Laboratory of Metabolism and Lipids, Heart Institute of the Medical School Hospital, 2Department of Clinical Chemistry, Faculty of Pharmaceutical Sciences, University of São Paulo, 3Biochemistry and Biophysics Laboratories, Butantan Institute, São Paulo, Brazil
Purpose: Lipid nanoemulsions (LDEs) that bind to low-density lipoprotein (LDL) receptors used as carriers of paclitaxel (PTX) can decrease toxicity and increase PTX antitumoral action. The administration of simvastatin (Simva), which lowers LDL-cholesterol, was tested as an adjuvant to commercial PTX and to PTX associated with LDE (LDE-PTX).
Materials and methods: B16F10 melanoma-bearing mice were treated with saline solution or LDE (controls), Simva, PTX, PTX and Simva, LDE-PTX, and LDE-PTX and Simva: PTX dose 17.5 µmol/kg (three intraperitoneal injections, 3 alternate days): Simva 50 mg/kg/day by gavage, 9 consecutive days.
Results: Compared with saline controls, 95% tumor-growth inhibition was achieved by LDE-PTX and Simva, 61% by LDE-PTX, 44% by PTX and Simva, and 43% by PTX. Simva alone had no effect. Metastasis developed in only 37% of the LDE-PTX and Simva, 60% in LDE-PTX, and 90% in PTX and Simva groups. Survival rates were higher in LDE-PTX and Simva and in LDE-PTX groups. The LDE-PTX and Simva group presented tumors with reduced cellular density and increased collagen fibers I and III. Tumors from all groups showed reduction in immunohistochemical expression of ICAM, MCP-1, and MMP-9; LDE-PTX and Simva presented the lowest MMP-9 expression. Expression of p21 was increased in the Simva, LDE-PTX, and LDE-PTX and Simva groups. In the Simva and LDE-PTX and Simva groups, expression of cyclin D1, a proliferation and survival promoter of tumor cells, was decreased. Therapy with LDE-PTX and Simva showed negligible toxicity compared with PTX and Simva, which resulted in weight loss and myelosuppression.
Conclusion: Simva increased the antitumor activity of PTX carried in LDE but not of PTX commercial presentation, possibly because statins increase the expression of LDL receptors that internalize LDE-PTX.
Keywords: lipid nanoparticles, drug delivery, statins, cancer treatment, B16F10 melanoma
Introduction
In humans, low-density lipoprotein (LDL) is the lipoprotein fraction that contains most of the cholesterol carried in the plasma. LDL is removed from the circulation by receptors in the cell membrane.1 In cancer cells, overexpression of LDL receptors occurs, leading to increased uptake of the lipoprotein by those cells.2 This is probably caused by the increased need for input of cholesterol and other lipids to assist the membrane building required by the rapidly duplicating neoplastic cells. This phenomenon can be very intense: acute leukemia cells can take from three- to 100-fold more than normal cells of the corresponding lineage. In those leukemias and also in Hodgkin and non-Hodgkin lymphomas, LDL cholesterol levels may decrease, resulting from the increased removal of LDL from the circulation by the neoplastic tissues.3–6 In solid cancers, such as ovarian and breast carcinomas, the LDL uptake can be likewise severalfold higher than the corresponding normal tissue and may lead to decrease in LDL cholesterol.7–10
LDL-receptor overexpression in cancer tissues creates a major mechanism for drug targeting via the LDL receptor-mediated endocytic pathway, aiming to increase the uptake of the drugs by neoplastic tissues and to reduce influx to normal organs and tissues. This mechanism was initially explored by loading native LDL with anticancer drugs for injection in the bloodstream and subsequent concentration of the drug in the malignant tumors.6,11 However, the procedures to isolate native LDL from human serum and LDL drug loading are rather cumbersome and unlikely to be introduced in routine clinical practice.
Studies from our laboratory paved the way for the practical use of the LDL-receptor endocytic pathway as a drug-delivery strategy by substituting native LDL with artificially made lipid nanoemulsions (LDEs).3,4,9,10,12–14 Those nanoemulsions that resemble the structure of LDL can bind to the LDL receptors and concentrate in neoplastic cells and tissues with LDL-receptor overexpression. The nanoemulsions are made without protein, but in contact with plasma, they acquire several exchangeable apolipoproteins, such as ApoA, ApoC, and ApoE. ApoE is recognized by LDL receptors, so that the nanoemulsions can be taken up and internalized into the cells via the LDL receptor-mediated endocytic pathway.3,4 The nanoemulsions were termed LDEs to recall the resemblance with LDL and the ligand ApoE. The LDE system may be prone to industrial-scale manufacture, and thus was the first device with the ability to take full advantage of the LDL-receptor pathway to perform drug delivery.
Stable preparations of different chemotherapeutic agents associated with LDEs were obtained.13–15 To increase the yield of association with LDEs of those compounds and the stability of the preparations, an oleyl group was added to the molecular structure of the drugs, resulting in more lipophilic derivatives.16–19 A prominent aspect of the LDE–chemotherapeutic agent formulations is a remarkable reduction in drug toxicity. As tested in mice, several formulations developed in our laboratory, such as the association of LDE with carmustine, with etoposide, with paclitaxel (PTX), and with daunorubicin had severalfold less toxicity than the corresponding commercial preparations, as analyzed by classic pharmacological parameters, such as maximum tolerated dose or median lethal dose (LD50).16–21 In pilot clinical trials performed in patients with advanced cancers, LDE-carmustine, LDE-etoposide, and LDE-PTX showed no observable clinical and laboratorial toxicities at doses corresponding to those used in routine cancer treatment. Because it is prepared only with lipids present in the organism, LDE has no immunogenic potential and no safety issues related to materials in the nanoemulsion composition.5,14,22,23
Due to the aggressiveness of the malignant neoplastic processes, adjuvant strategies with the potential to increase the effectiveness of anticancer therapies should always be considered for testing. In this respect, statins, the most important cholesterol-lowering class of drugs, have been reported as inducers of apoptosis and antitumor activity of antineoplastic drugs, such as carmustine, cisplatin, 5-fluorouracil, doxorubicin, and PTX, and investigated as adjuvants in cancer treatment and prevention.24–31
Statins lower LDL cholesterol by inhibiting 3-hydroxy-3 methylglutaryl (HMG)-CoA reductase, the rate-limiting step enzyme of cholesterol synthesis.32,33 Depletion of intracellular cholesterol by statins leads to increased expression of the LDL receptors that play a key role in the drug-targeting properties of native LDL and the LDE-carrier system.34 In addition, statins may have pleiotropic actions that can intervene in processes related to cancer progression, such as cell growth, survival, and migration, as well as in angiogenesis.31,35–40
Therefore, this study aimed to test the effects of statin use on the treatment of tumor-bearing mice with PTX, a widely used anticancer chemotherapeutic agent belonging to the taxane class, as carried in LDE. To our knowledge, this is the first study to test the use of statins, the most frequently prescribed drug in the world, as an adjuvant to a chemotherapeutic treatment based on a drug-targeting system.
Materials and methods
Preparation of PTX formulations
To obtain a conventional PTX formulation, a derivative of castor oil Cremophor EL, purchased from Sigma-Aldrich (St Louis, MO, USA) was added to PTX (Calbiochem®; EMD Millipore, Billerica, MA, USA) and ethanol. Each 1 mL of the formulation solution consisted of 6 mg PTX, 527 mg Cremophor EL, and 49.7% v/v alcohol.
The PTX oleate synthesis was performed as described previously.17 LDE-PTX was prepared from a lipid mixture composed of 135 mg cholesteryl oleate and 6 mg cholesterol (Sigma-Aldrich), 333 mg egg phosphatidylcholine (Lipoid GmbH, Ludwigshafen, Germany), 132 mg Miglyol® 812 N (Sasol Ltd, Johannesburg, South Africa), and 6 mg of PTX oleate. The aqueous phase consisted of 100 mg polysorbate 80 (Tween 80) from EMD Millipore and 10 mL Tris-HCl buffer, pH 8.05. A pre-emulsion was obtained by ultrasonic radiation using a Sonifier 450 (Branson Ultrasonics, Danbury, CT, USA) until complete drug dissolution. Emulsification of all lipids, PTX oleate, and the aqueous phase was obtained by high-pressure homogenization using an EmulsiFlex-C5 homogenizer (Avestin Inc, Ottawa, Canada). After homogenization cycles, the formed emulsion was centrifuged and the nanoemulsion sterilized by passage through a 0.22 μm-pore polycarbonate filter (EMD Millipore) and kept at 4°C until it was used. The amount of drug associated with LDE was always measured by high-performance liquid chromatography (model LC-10AD; Shimadzu Corporation, Kyoto, Japan) before injection, and the yield of association was 90%. Particle diameter and polydispersity index, measured by laser-light scattering (ZetaPALS zeta potential analyzer, Brookhaven Instruments Corporation, Holtsville, NY, USA), were approximately 74 nm and 0.157, respectively. In this study, the formulation was used on the same day that it was prepared, but in previous assays in our laboratory, we had observed that LDE-PTX is stable at 4°C for at least 1 year.
Induction of B16 melanoma and mouse treatment groups
Specific pathogen-free (SPF) female C57BL/6J mice aged 8–12 weeks were purchased from the Central Animal Care Unit at the Butantan Institute (São Paulo, Brazil). The animals were housed in a temperature-controlled room at 25°C, with food and water provided ad libitum.
The murine B16F10 (H2b) melanoma cell line was obtained from the American Type Culture Collection (Manassas, VA, USA), and a variant of the B16 melanoma cell line originating from C57BL/6J mice was used in the experiments. Cells were cultured in Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 IU/mL of penicillin, and 100 μg/mL of streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). Cell suspensions were detached from plates with trypsin and 0.2% Versene. After trypsin inactivation with 10% fetal bovine serum, viable cells were counted by trypan blue dye exclusion. For tumor transfer, 5×104 cells suspended in 100 μL of phosphate-buffered saline (PBS) were injected subcutaneously into the flank region above the hind leg of C57BL/6J mice. At 10–14 days after inoculation, the tumors became macroscopically apparent.
Groups of mice with induced melanoma B16 tumors were randomly assigned to the following treatments:
- saline control: treated with 0.9% saline solution (n=23)
- LDE control: treated with LDE only (n=19)
- simvastatin (Simva): treated with 50 mg/kg body weight (n=19)
- PTX: treated with commercial formulation of PTX 17.5 μmol/kg (n=13)
- PTX and Simva: treated with commercial formulation of PTX 17.5 μmol/kg and with Simva 50 mg/kg (n=19)
- LDE-PTX: treated with PTX oleate 17.5 μmol/kg associated with LDE (n=19)
- LDE-PTX and Simva: treated with PTX oleate 17.5 μmol/kg associated with LDE and with Simva 50 mg/kg (n=19).
Saline, LDE, PTX, or LDE-PTX were administered by intraperitoneal route on days 11, 14, and 19, as counted from the initial tumor-inoculation day. The intraperitoneal route was adopted in accordance with the National Cancer Institute protocols for screening agents in mice implanted with B16 melanoma and also to avoid PTX-vesicant reactions that may lead to local severe necrosis.41 Simva (EMD Millipore) was macerated, suspended in water, and administered by daily oral gavage from day 11 to day 19, as counted from the initial tumor-inoculation day.
This study was approved by the Ethics Committee for Animal Use of the Faculty of Pharmaceutical Sciences of the University of São Paulo (protocol 246) and by the Ethical Committee for Animal Research of the Butantan Institute of São Paulo (protocol 638/09). The study experiments were performed in agreement with the Ethical Principals in Animal Research adopted by the Brazilian College of Animal Experimentation.
Tumor growth
Tumor sizes were measured three times a week using a caliper-like instrument during the experiment. The size measurement was converted to tumor volume by the equation:
|
|
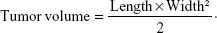
Observation of tumor volume was ended on day 31, but survival was monitored over 60 days.
Necropsies were performed by an investigator who was blinded to the treatment groups, and the presence of macroscopic metastasis was verified in the following locations: brain, lungs, heart, liver, spleen, kidneys, intestine, stomach, and lymph nodes (cervical, mediastinal, axillary, lateral axillary, mesenteric, renal, para-aortic, iliac, superficial inguinal, popliteal, and sacral). Micrometastases were not evaluated. Primary tumors were then resected and processed for histological examination.
Tumor histology
Tumor fragments were excised from the tumor-bearing mice as described earlier, fixed in 10% buffered formalin solution, and then embedded in paraffin according to standard histological procedures. Paraffin-embedded tissues were sectioned at 5 μm thickness, deparaffinized, and then stained with hematoxylin–eosin or picrosirius red, in which the collagen stained red.
Photomicrographs of 18 fields from six tissue sections per group (three fields for each tissue section) were captured at 200× magnification using a Nikon 80i microscope (Nikon Corporation, Tokyo, Japan) coupled with a digital camera and analyzed with NIS-Elements 3.2 Advanced Research image-analysis software (Nikon). The mean percentage of the stained area on all measured fields for each group was calculated.
Tumor immunohistochemistry
Additional tumor sections were prepared as described earlier, and then processed for immunohistochemistry analysis using anti-ICAM/CD54 antibody (R&D Systems Inc, Minneapolis, MN, USA), anti-MMP-9 antibody (Abcam PLC, Cambridge, UK), anti-MCP-1 antibody (R&D Systems), and anti-LDL-receptor antibody (Abcam). For immunostaining, antigen retrieval was performed by heating tumor-tissue sections with 10 mM citrate buffer, pH 6.0, for 30 minutes and cooling for 5 minutes at room temperature. Endogenous peroxidase activity was blocked by incubation in 3% hydrogen peroxide, and nonspecific reaction was blocked by incubation in fetal bovine serum for 1 hour at 42°C. The sections were then incubated overnight at 4°C with one of the following antibodies: anti-ICAM/CD54 (1:100 dilution), anti-MMP-9 (1:100 dilution), anti-MCP-1 (1:10 dilution), anti-LDL receptor (1:100 dilution). Next, the sections were incubated for 30 minutes at room temperature with the SuperPicture polymer-detection system (Thermo Fisher Scientific). The sections were then incubated with a 3,3′-diamino-benzidine (DAB) chromogen system (Agilent Technologies, Santa Clara, CA, USA) for 2 minutes at room temperature and counterstained with hematoxylin. Between incubations, the slides were rinsed three times for 5 minutes with PBS. In the negative-control experiment, primary antibodies were replaced with 1% bovine serum albumin.
Photomicrographs were taken and analyzed as described earlier. For quantitative image analysis of immunostaining for ICAM/CD54, MMP-9, MCP-1, and LDL receptors, the color-detection threshold was chosen for the DAB chromogen in tissue sections. The settings for chromogen staining were optimized so that all positively labeled cells were scored by the image-analysis software.
Flow-cytometry analyses
Three mice from each group were killed by cervical dislocation 48 hours after the end of the treatments, and tumor samples were collected and processed for evaluation of cell mitochondrial membrane potential by rhodamine 123 staining and for the expression of proteins p21, p27, and cyclin D1.
Briefly, macerated tumors were prepared in PBS. The cell suspension was filtered, added to an alcohol ribonuclease solution, and then centrifuged. The supernatant was discarded, the cell pellet was added to a FACSFLow™ buffer (BD Biosciences, San Jose, CA, USA), and each cell suspension was divided into four samples. One cell-suspension sample was used for rhodamine 123 staining, in which 10 μL of rhodamine 123 (5 mg/mL; Sigma-Aldrich) was added for each 100 μL of cell suspension containing 106 cells. After cell staining, the sample was analyzed by flow cytometry.
In the three remaining samples of the cell suspension, 10 μL of Triton X-100 0.1% was added for cell permeabilization. For each 100 μL of cell suspension with 106 cells, 1 μg of one of the following antibodies was added: anti-p21, anti-p27, and anti-cyclin D1 (Santa Cruz Biotechnology Inc, Dallas, TX, USA). After overnight incubation at a temperature of 4°C, each 100 μL of cell suspension containing 106 cells received 1 μg of anti-IgG secondary antibody (Santa Cruz Biotechnology). The cells were then washed, and a paraformaldehyde solution was added prior to flow-cytometry analysis. All flow-cytometry analyses were performed on a FACScalibur FACScan flow-cytometry system (BD Biosciences, San Jose, CA, USA), and data were analyzed with WinMDI 2.8 software (Scripps Research Institute, San Diego, CA, USA).
Drug toxicity in mice without tumors
SPF female BALB/c mice were randomly allocated to six groups of eleven animals, namely LDE-PTX, Simva, LDE-PTX and Simva, PTX and Simva, LDE, or saline. On days 1, 4, and 9, animals were treated with intraperitoneal injections of LDE-PTX, PTX, LDE, or saline. Simva was administered by daily oral gavage from day 1 to day 9. Drug doses were as referred to earlier. Survival, weight changes, blood cell counts, and clinical chemistry parameters were monitored over a 24-day period.
Hematological profile
Peripheral blood samples were collected from mouse submandibular veins by incision with a sterile metal tip, and blood cell counts were determined using the veterinary hematology analyzer ABX ABC Vet (Horiba Ltd, Kyoto, Japan). The hematological profile was determined in BALB/c mice without tumors and also in C57BL/6J melanoma-bearing mice.
Blood clinical chemistry
Plasma from BALB/c mice without tumors was obtained from blood samples collected as described earlier, and used for the determination of creatinine concentration and of the activities of alanine transaminase (ALT), aspartate transaminase (AST), and creatine kinase (CK). The analyses were performed using the automated analyzer Modular Analytics P800 (Hoffman-La Roche Ltd, Basel, Switzerland) at the Division of Central Laboratory in the Clinics Hospital of the University of São Paulo Medical School (São Paulo, Brazil).
Statistical analysis
Data with two or more independent variables were tested by two-way analysis of variance and tested with Bonferroni post hoc test. Fisher’s exact test was used to compare the number of animals with metastatic nodes in the studied groups. Survival comparison between groups was made with Kaplan–Meier survival curves. All values are expressed as means ± standard error of mean. In all analyses, P<0.05 was considered statistically significant. Statistical analyses were carried out using the GraphPad Prism version 4.02 statistical software (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Comparison of the control groups: saline and LDE
When comparing the two control groups of animals – one treated with saline solution and the other with blank LDE – no significant differences were observed regarding the results of tumor-growth inhibition, survival rates, percentage of animals with metastatic sites, tumor histological examination, mitochondrial membrane potential, expression of proteins p21, p27, and cyclin D1, or hematological profile of melanoma-bearing mice. Furthermore, there were no differences either when those groups were compared regarding the evaluation of drug toxicity in mice, such as analysis of animal survival, body weight, blood cell counts, and clinical chemistry parameters (data not shown).
Assuming that LDE itself had no influence on the results of the several measured parameters, the saline group was designated the single control group for the ensuing results of all the experiments with the different animal treatments.
Antitumor activity
Figure 1 depicts three major aspects of tumor-development evaluation under the different treatments, ie, tumor growth, animal survival, and tumor metastasis. Figure 1A shows the 31-day follow-up of tumor growth observed in the different treatment groups. Based on the data obtained on the last day of observation (day 31) and in comparison with the control data (saline group), treatment with Simva alone did not significantly inhibit tumor growth. Compared to the saline group, all treatments with PTX (PTX alone, PTX and Simva, LDE-PTX, and LDE-PTX and Simva groups) resulted in significant inhibition of tumor growth. The addition of Simva to PTX treatment did not result in enhancement of tumor-growth inhibition, but when Simva was added to the treatment with LDE-PTX, tumor growth was indeed further decreased compared to LDE-PTX without Simva. If tumor growth in the control saline-treated animals is taken to be 100%, the average tumor-growth inhibition was 95% in the group treated with LDE-PTX and Simva, in contrast to 61% in the group treated with LDE-PTX, 44% in the group treated with PTX and Simva, and 42% in the group treated with PTX alone.
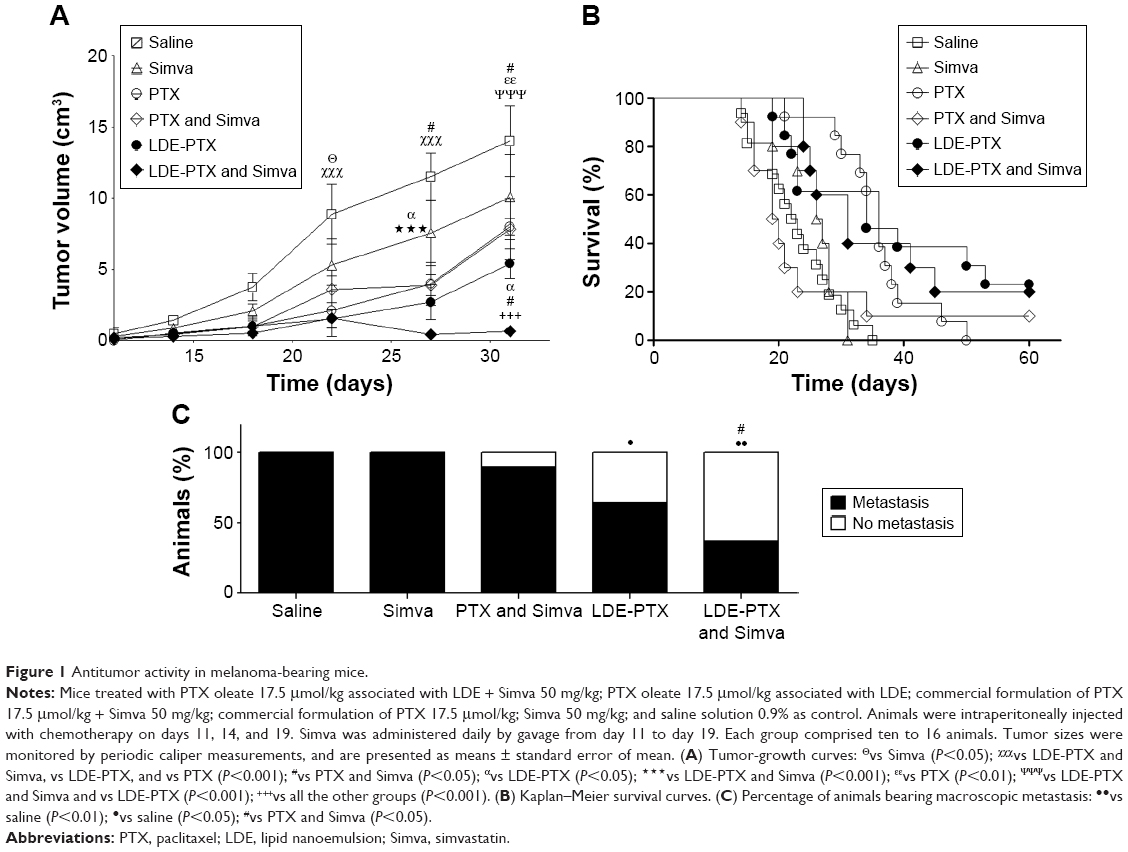 | Figure 1 Antitumor activity in melanoma-bearing mice. |
Figure 1B shows the Kaplan–Meier survival curves of tumor-bearing mice undergoing the different treatments. The groups of animals treated with PTX associated with LDE (LDE-PTX and Simva and LDE-PTX) or with PTX alone presented increased survival rates in comparison with the control group (P<0.01), but in the PTX and Simva group, survival was not significantly affected. As observed at day 35, when all the animals from the control and Simva groups had died, the survival rates were 46% for the groups treated with LDE-PTX or PTX alone and 40% for the group treated with LDE-PTX and Simva.
Figure 1C shows the effects of each treatment on the appearance of metastasis, as observed macroscopically. All the animals from the control and Simva groups developed metastatic nodes. The LDE-PTX and Simva (P<0.01) and the single LDE-PTX (P<0.05) treatments significantly inhibited the appearance of metastatic sites, while PTX and Simva was ineffective.
Tumor histology
Figure 2 shows photomicrographs of tumor sections stained with hematoxylin–eosin and with picrosirius red, which stains types I and III collagen. The photomicrographs belonging to the control group typically show a great number of neoplastic cells (Figure 2A) and decreased fibrillar collagen-network architecture (Figure 2F). The inhibition of tumor growth by treatment with LDE-PTX and Simva was evidenced by small-cell density, the presence of extensive areas of necrosis and fibrosis, and the intense deposition of collagen fibers, as illustrated in Figure 2E and J, respectively. In fact, treatment with LDE-PTX and Simva dramatically reduced the tumor-cell population (Figure 2K) and increased the presence of collagen fibers (Figure 2L) in tumor tissue in comparison to all the other treatments, namely LDE-PTX, PTX and Simva, Simva, and saline. Treatment of the tumor-bearing mice with Simva (Figure 2B), PTX and Simva (Figure 2C), or LDE-PTX (Figure 2D) also led to a reduced tumor-cell population compared to the control group (Figure 2K). However, the presence of collagen fibers in the tumors of those groups (Figure 2G–I) was not significantly different from the tumors pertaining to the saline-treated controls (Figure 2L).
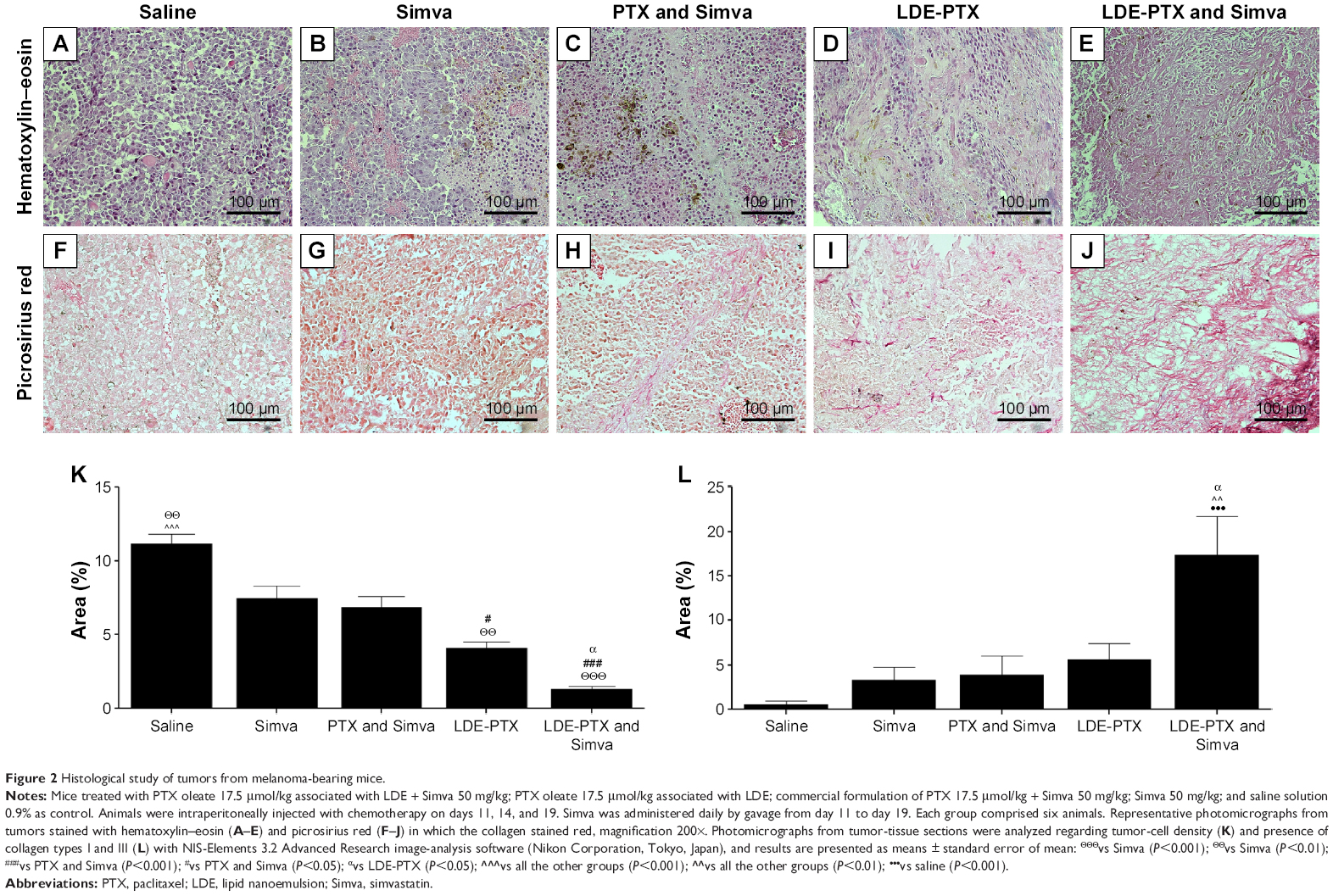 | Figure 2 Histological study of tumors from melanoma-bearing mice. |
Tumor immunohistochemistry
Figure 3 shows the photomicrographs of tumor sections processed for immunostaining for ICAM/CD54 (Figure 3A–E), MMP-9 (Figure 3F–J), MCP-1 (Figure 3K–O), and LDL receptors (Figure 3P–T) and the percentages of stained areas for each one of the antibodies (Figure 3U–X). In comparison to the control group, all treatment groups, namely LDE-PTX and Simva, LDE-PTX, PTX and Simva, and Simva, strongly reduced the expression of ICAM/CD54, MMP-9, and MCP-1 in tumor tissues. If the stained area in the control saline-treated animals is taken to be 100%, ICAM/CD54 expression was reduced by 96% in the group treated with LDE-PTX and Simva (P<0.001), 86% in the LDE-PTX-treated group (P<0.001), 69% in the PTX and Simva-treated group (P<0.001), and 46% in the Simva-treated group (P<0.05) (Figure 3U).
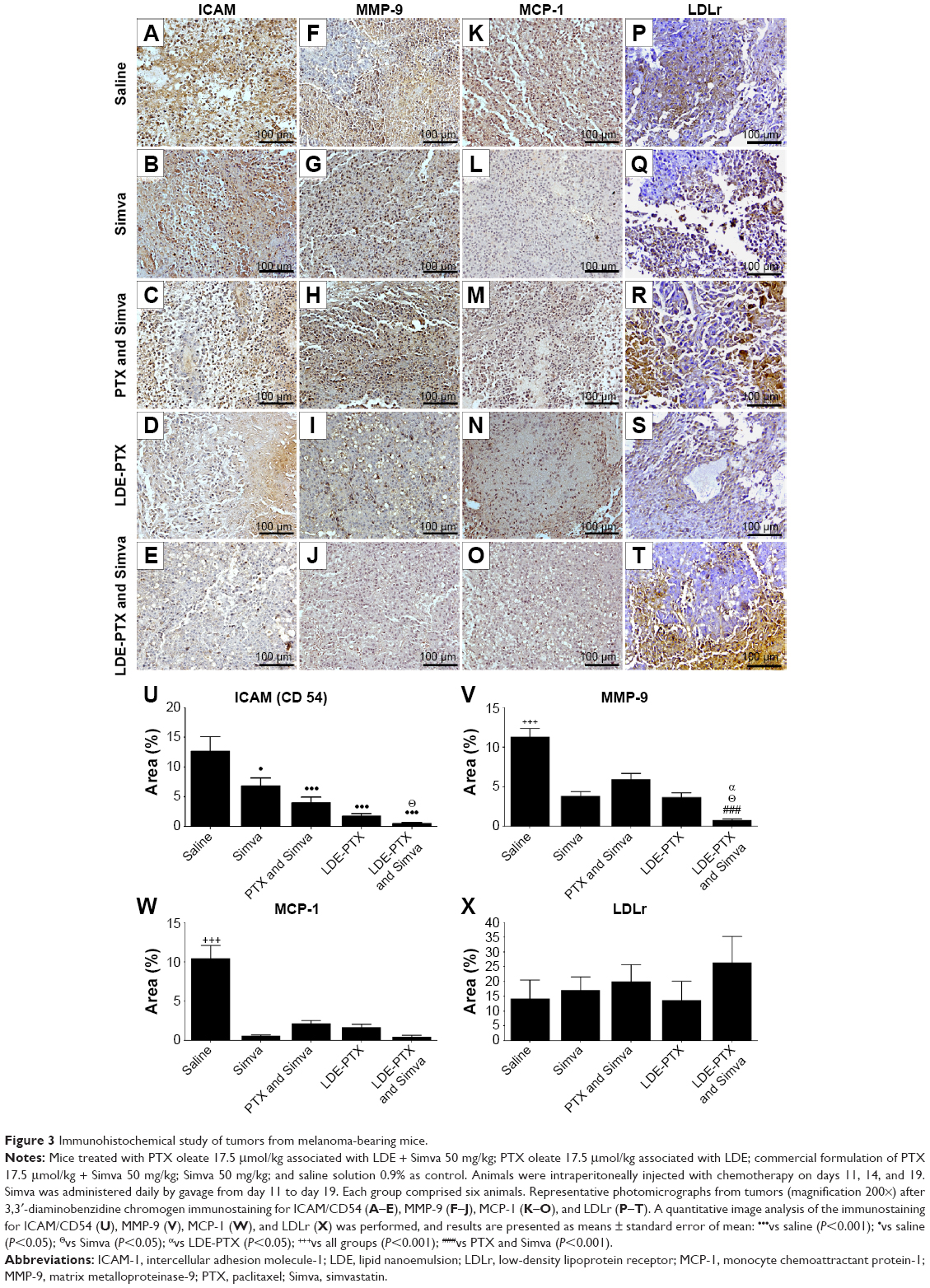 | Figure 3 Immunohistochemical study of tumors from melanoma-bearing mice. |
With regard to MMP-9 (Figure 3V), the reduction in the metalloproteinase expression was 93% compared to the control group for LDE-PTX and Simva (P<0.001), 64% for both LDE-PTX and Simva (P<0.001), and 45% for PTX and Simva (P<0.001). In respect of MCP-1 (Figure 3W), when compared to the control group, the expression of the protein was reduced by approximately 95% with the LDE-PTX and Simva and Simva treatments (P<0.001), 83% with LDE-PTX treatment (P<0.001), and 80% with the PTX and Simva treatment (P<0.001).
Figure 3X shows that in all the Simva-treatment groups, namely Simva, PTX and Simva, and LDE-PTX and Simva, there was an apparent increase in the expression of the LDL receptors within the tumor tissue in comparison with the control group. The increase in the percentage of stained areas for LDL receptors was approximately 88% for LDE-PTX and Simva, 43% for PTX and Simva, and 21% for Simva, but this difference did not attain statistical significance. In tumors of animals treated with LDE-PTX alone, the expression of LDL receptors appeared not to be affected by the treatment.
Rhodamine 123 staining of viable cells
Mitochondrial membrane potential was measured by rhodamine 123 staining to distinguish viable from nonviable cells within tumor tissue, as illustrated in Figure 4. In tumors from control saline-treated animals, the rhodamine fluorescence emission (Figure 4A) indicated an amount of 87% of living cells with active mitochondria (Figure 4B). In contrast, all single and combined treatments, namely LDE-PTX and Simva, PTX and Simva, LDE-PTX, and Simva, had the ability to reduce strongly the mitochondrial fluorescence of rhodamine. The proportion of viable cells was 30% in group Simva, 23% in group PTX and Simva, 20% in group LDE-PTX, and 14% in group LDE-PTX and Simva (Figure 4B).
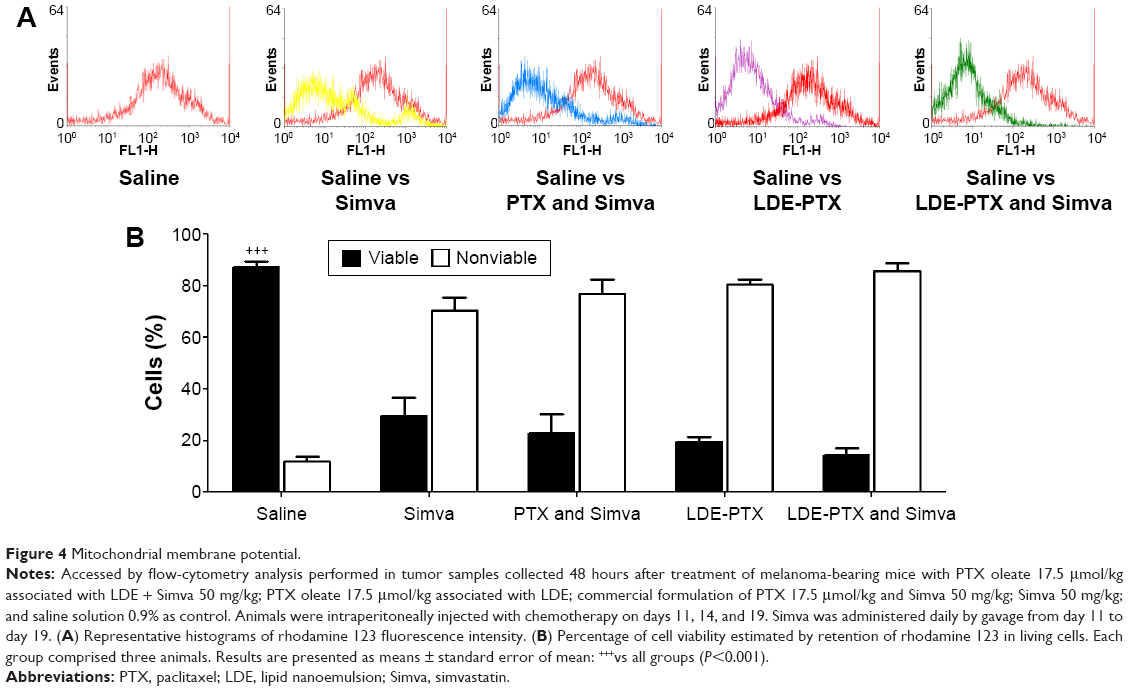 | Figure 4 Mitochondrial membrane potential. |
Expression of proteins cyclin D1, p21, and p27
Figure 5 illustrates the expression of the proteins cyclin D1, p21, and p27 accessed by flow-cytometry analysis of tumor samples of melanoma-bearing mice. It is shown that in comparison to control, Simva (P<0.05) or LDE-PTX and Simva (P<0.01) were the only treatments in which a decrease in the percentage of cells expressing cyclin D1 was observed (Figure 5A and B). This percentage was 28% in tumors from the control group, 17% for the Simva-treated group, and 16% for the LDE-PTX and Simva-treated group (Figure 5A).
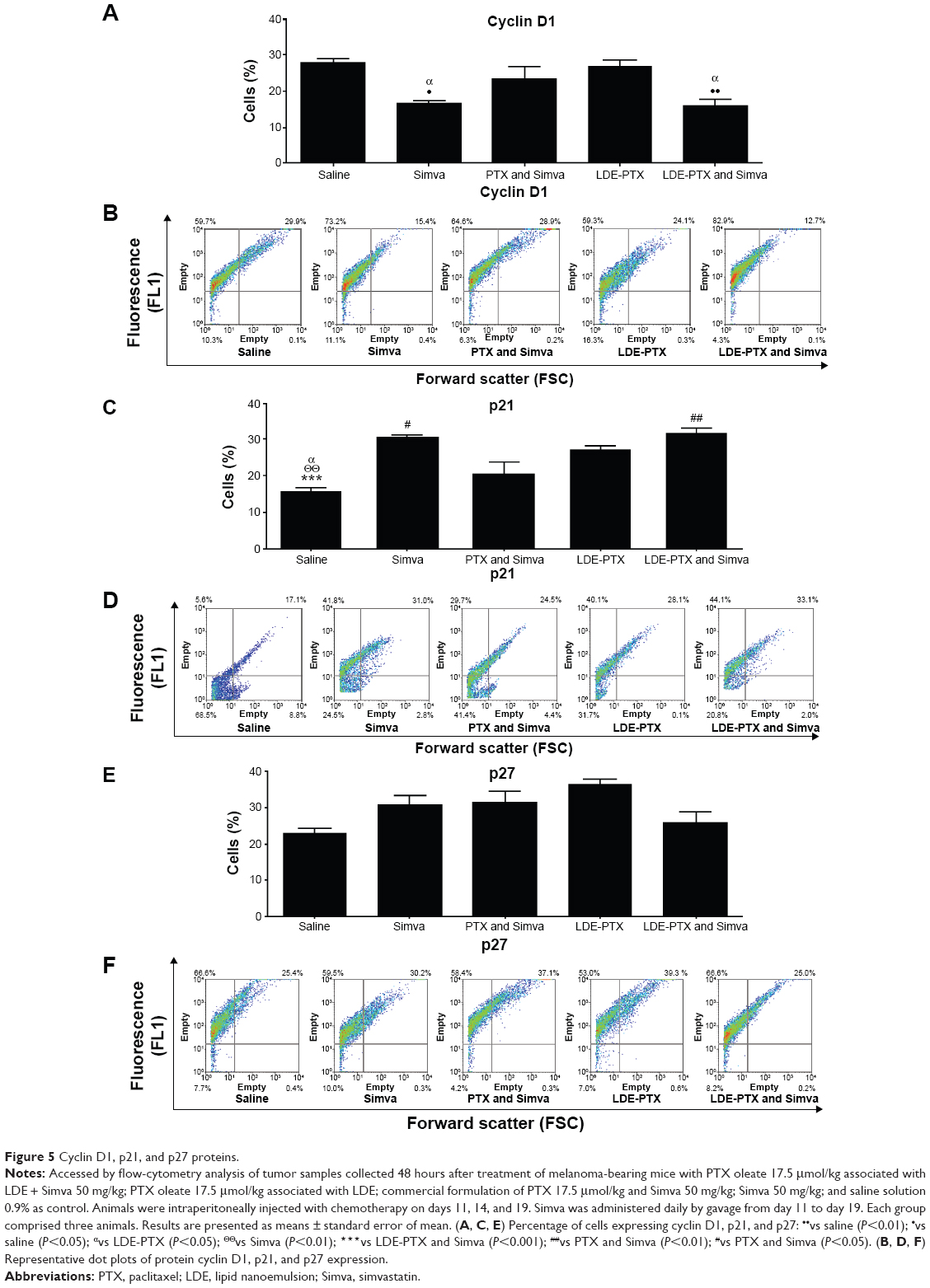 | Figure 5 Cyclin D1, p21, and p27 proteins. |
In respect of p21 expression in tumor tissue (Figure 5C and D), treatment with LDE-PTX and Simva (P<0.001), Simva (P<0.01), and LDE-PTX (P<0.05) resulted in increased percentages of cells expressing p21 (32%, 30%, and 27%, respectively) in comparison to the control group (16%). This latter effect was not achieved with PTX and Simva treatment (20%).
With regard to the expression of p27 within tumor tissue (Figure 5E and F), analysis of the results presented in Figure 5E might suggest that treatment with Simva, PTX and Simva, or LDE-PTX resulted in increased percentages of cells expressing the protein in comparison to the control group. However, this was not statistically significant.
Hematological profile of the treated mice
Figure 6 shows the hematological profile of tumor-bearing mice under treatment with LDE-PTX and Simva, PTX and Simva, LDE-PTX, Simva, and saline, and of healthy untreated mice before tumor-cell inoculation, termed the basal group. When the control group of saline-treated melanoma-bearing animals were compared with non-tumor-bearing mice (basal), the control group had anemia (Figure 6C–E) and thrombocytosis (Figure 6B).
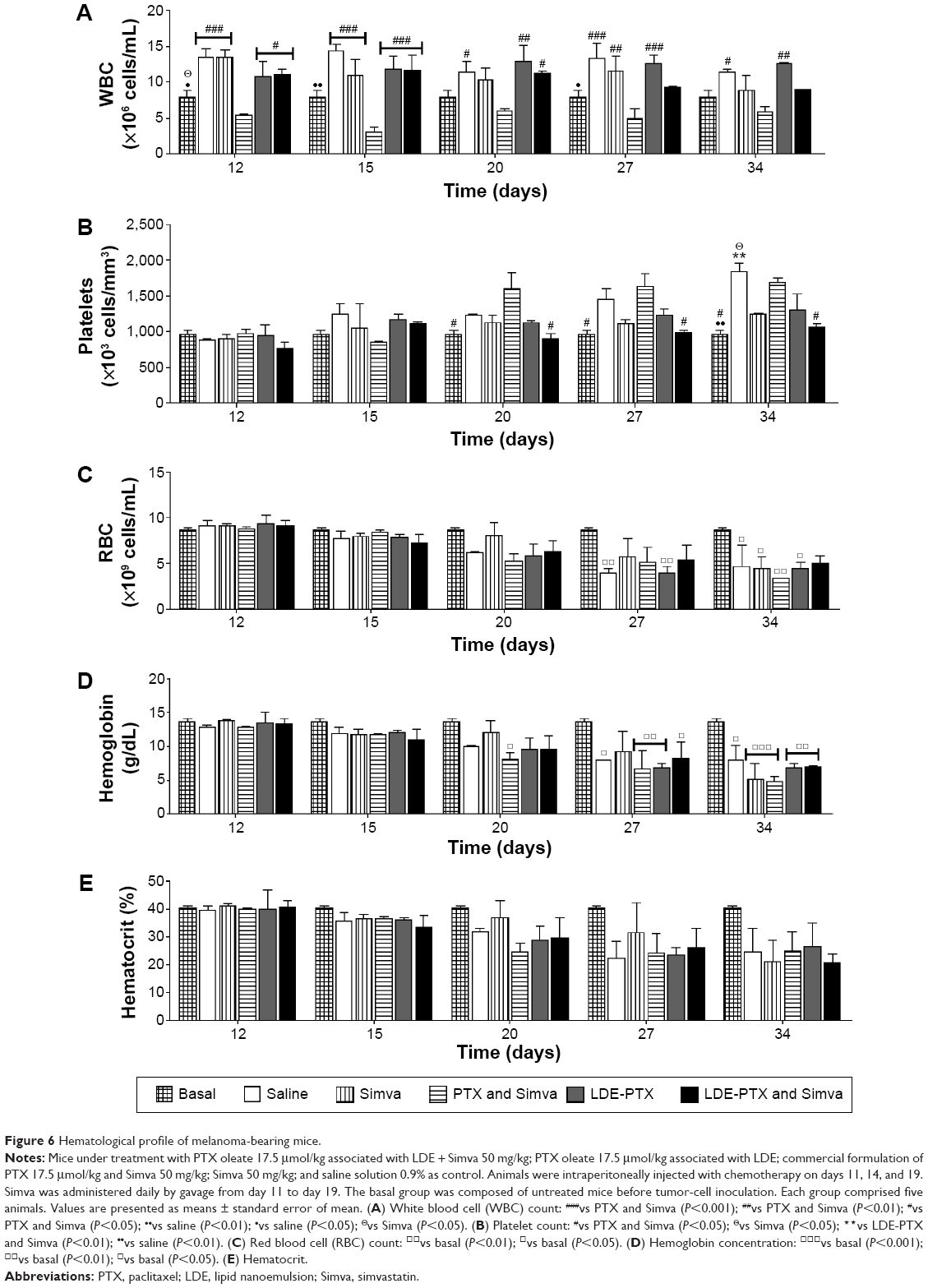 | Figure 6 Hematological profile of melanoma-bearing mice. |
In Figure 6A, it is shown that leukocyte counts were unchanged during treatments with combined LDE-PTX and Simva, single LDE-PTX, or single Simva. In contrast, combined treatment with PTX and Simva was myelotoxic, as it resulted in a strong reduction of leukocyte counts in comparison to the control group. The tumor-related thrombocytosis (Figure 6B) observed in the controls was improved by treatment with LDE-PTX and Simva, LDE-PTX, and Simva. However, the combined treatment with PTX and Simva was ineffective in this aspect, since platelet counts increased as much as in the control group.
Erythrogram results are represented in Figure 6C–E, comprising red blood cell count, hemoglobin concentration, and hematocrit. All melanoma-bearing mice developed anemia by days 27–34, as pointed out by the comparison with their basal values. Anemia was tumor-induced rather than resultant from toxicity, because anemia also appeared in the saline-treated group (Figure 6C and D).
Drug toxicity
Figure 7A contains the Kaplan–Meier survival curves of mice without tumors treated with PTX and Simva and with the other treatments, namely LDE-PTX and Simva, LDE-PTX, Simva, or saline. Whereas 100% of animals from the saline-treated control group and from groups treated with Simva, LDE-PTX, and LDE-PTX and Simva survived during the 25-day observation period, the survival of animals treated with PTX and Simva was only 36%.
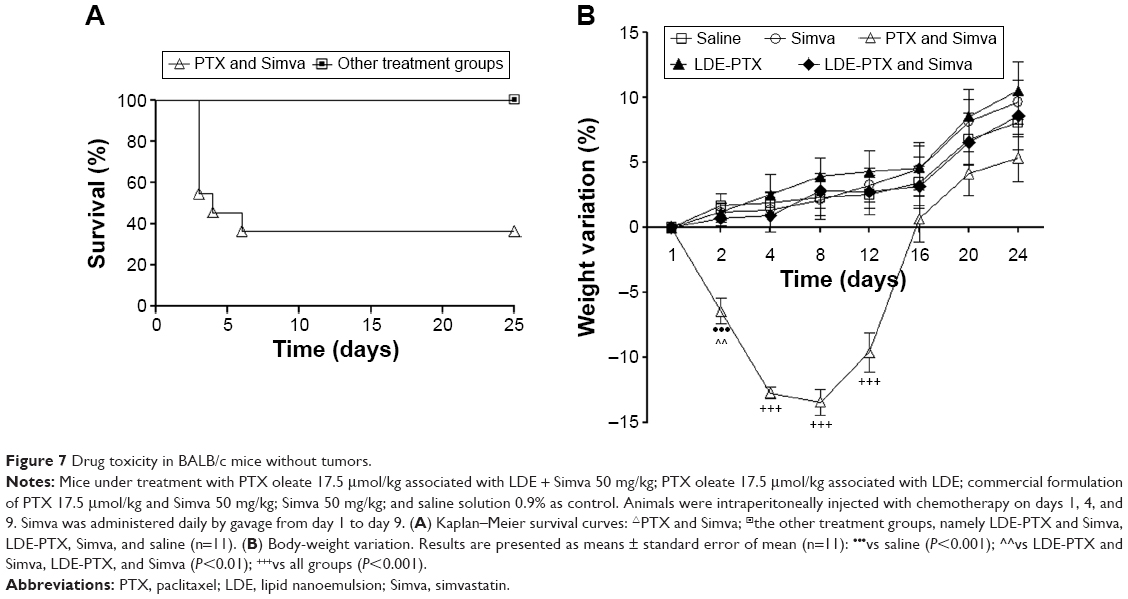 | Figure 7 Drug toxicity in BALB/c mice without tumors. |
Figure 7B shows the variation in body weight of the animals during the 25 days ensuing from the beginning of the treatments. It is shown that treatment of mice with saline, Simva, or LDE-PTX did not inhibit body-weight gain during the observation period. Combined treatment with LDE-PTX and Simva had also no effect on weight gain. In contrast, combined treatment with PTX and Simva resulted in a sharp loss of weight, so that the pretreatment weight was recovered only 2 weeks after the beginning of the treatment, when the animals started to gain weight.
Figure 8 shows the hematological profile of the mice submitted to the different treatments. It is shown that in comparison to the control group, both leukocyte (Figure 8A) and platelet (Figure 8B) counts were unchanged after treatment with Simva and LDE-PTX and combined treatment with LDE-PTX and Simva. On the other hand, combined treatment with commercial PTX and Simva strongly reduced those cells counts. With regard to mouse erythrograms, no treatments had any effect on red blood cell count (Figure 8C), hemoglobin concentration (Figure 8D), or hematocrit (Figure 8E).
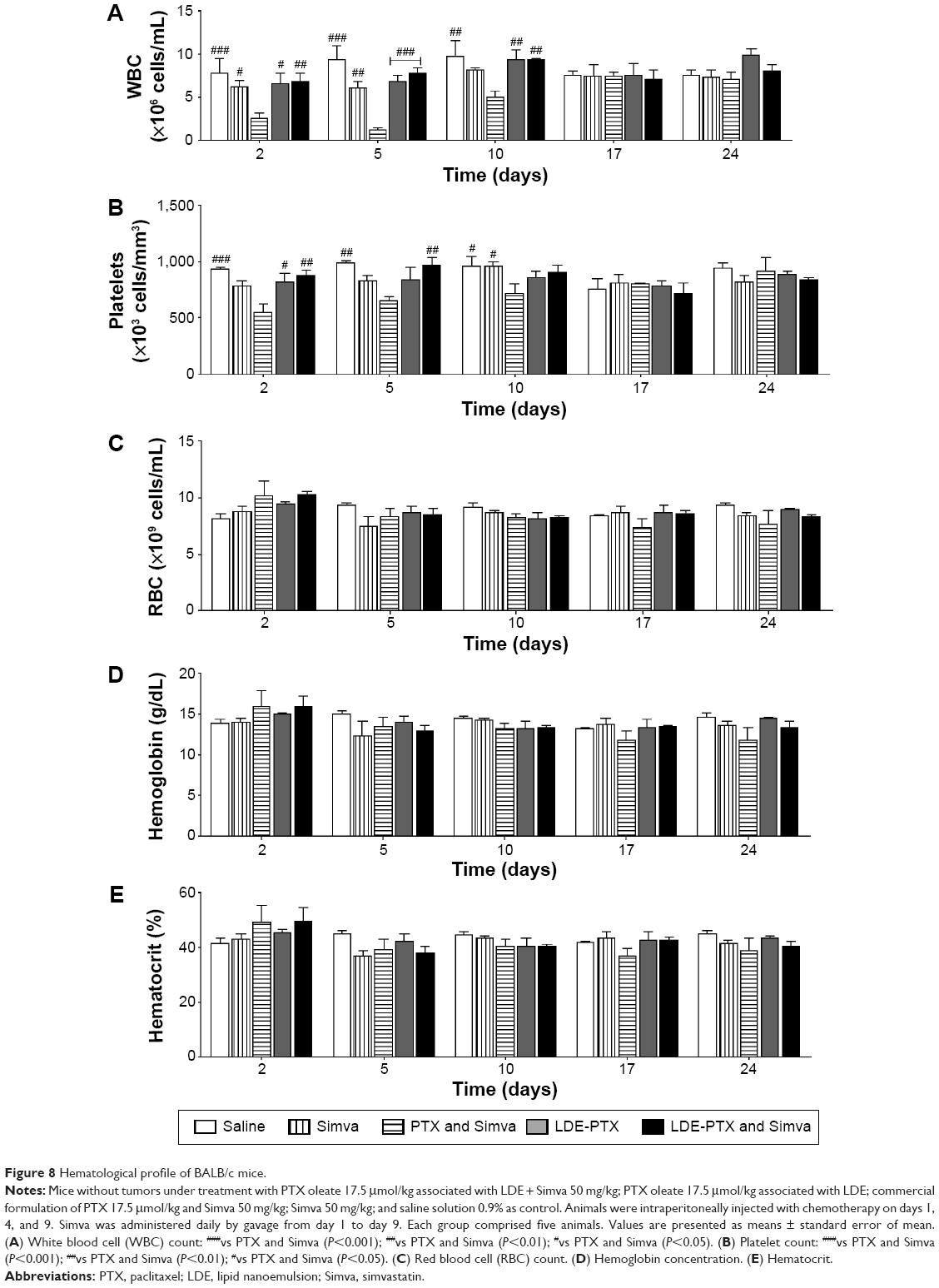 | Figure 8 Hematological profile of BALB/c mice. |
Analysis of the clinical chemistry parameters of mice under treatment with LDE-PTX and Simva, PTX and Simva, LDE-PTX, Simva, or saline is shown in Figure 9. One week after the end of treatment, all the groups that received chemotherapy, namely LDE-PTX and Simva, PTX and Simva, and LDE-PTX, presented a slight increase in plasmatic activities of ALT (Figure 9A) and AST (Figure 9B) in comparison to the control group (P<0.05). These latter effects were reversed in the second week after the end of chemotherapy. On the other hand, none of the single or combined treatments affected mice CK plasma activity (Figure 9C) or creatinine concentration (Figure 9D).
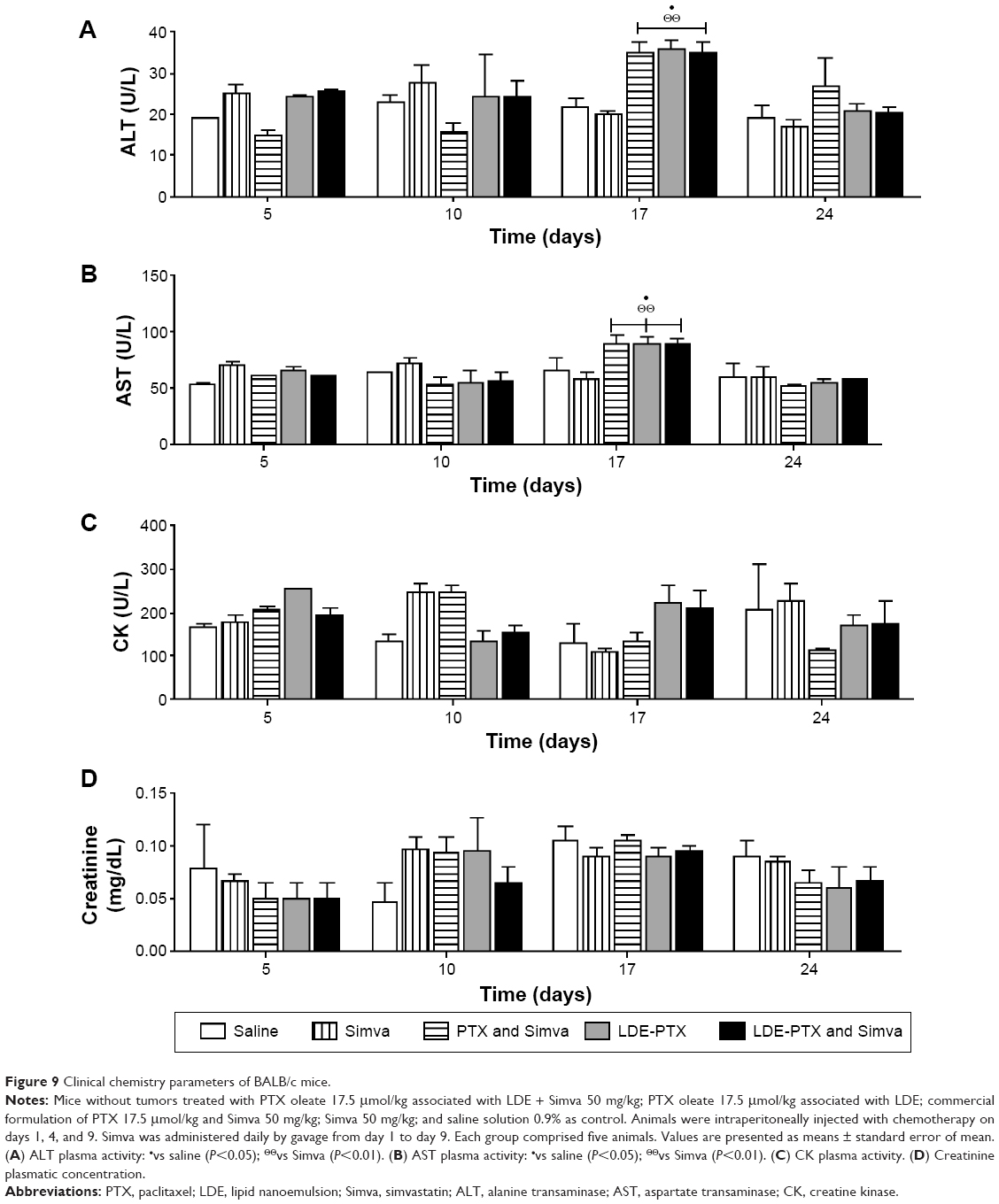 | Figure 9 Clinical chemistry parameters of BALB/c mice. |
Discussion
This study confirms our previous observations that treatment with PTX carried by LDE is superior to PTX dissolved in Cremophor EL, the commercial formulation, in inhibiting the growth of melanoma B16 implanted in mice.17 The main novel finding was that Simva had the ability to increase the antitumor activity of PTX associated with LDE. In contrast, Simva had no effect on the tumor inhibition obtained with treatment with PTX in the commercially used formulation. Statin use with LDE-PTX also resulted in a smaller percentage of animals with macroscopic metastasis, but this effect was not achieved in animals treated with the commercial PTX formulation. While the association with LDE increased the survival of the animals treated with PTX, the accretion of Simva to the treatment with the commercial formulation of PTX seemed to be ineffective. It is noteworthy to point out that B16 melanoma is a highly aggressive tumor that can severely damage the host, so that death may occur even in animals with reduced tumor volume. In this regard, the aggressiveness of the melanoma can be illustrated by the fact that tumor-induced anemia was not completely abolished by any of the drug treatments. Therefore, the increase in survival rates observed in the LDE-PTX and Simva group compared to the Simva and PTX groups is relevant and can be ascribed to increased antitumor effectiveness of this treatment.
The melanoma B16 mice were chosen for the current experiments because this had been previously characterized as an adequate model for evaluation of LDL receptor-based drug delivery.42 By the immunoperoxidase technique, our group had previously shown that the presence of LDL receptors in B16 melanoma cells is intense, and that after injection in melanoma-bearing mice, LDE concentrates in the tumor.17 In those mice, the tumor was second only to the liver regarding the LDE-uptake rate. The tumor uptake of PTX carried in LDE was approximately 60% of that of the liver, and at least three- to fourfold that of the surrounding normal dermal tissue. Finally, we had shown that this tumor model is responsive to LDE-PTX treatment.17
The potential anticancer actions of statins can be ascribed either to their effects on the lipid metabolism or to the so-called pleiotropic effects ascribed to these drugs. Statins are competitive inhibitors of HMG-CoA reductase, the rate-limiting step enzyme of cholesterol synthesis.32,33 Downstream of this pathway, the inhibition of HMG-CoA reductase also reduces the synthesis of isoprenoids, which are intermediates in the cholesterol-synthesis chain. Isoprenoids are responsible for membrane localization and function of Ras and Rho GTPase.43,44 Since the inhibition of the normal function of those proteins affects the signaling pathways of important events regarding apoptosis and angiogenesis, as well as cell growth and migration, the potential interference of statins in events related to neoplasia development could be hypothesized.31,35–40
Different statins were reportedly tested in conjunction with such drugs as carmustine, cisplatin, 5-fluorouracil, doxorubicin, and PTX in treatments of neoplastic cell-cultured systems or in experiments with animal oncologic models, and have also been evaluated in clinical trials with effects that were either beneficial or absent.24–30,45 In fact, the effects of statins on tumor growth under conventional anticancer chemotherapy are not homogenous and depend on the malignant cell lineage, the time of statin-treatment commencement, the statin type and dose, and the antineoplastic agent, among other factors.
Results are lacking in the literature from tumor-bearing animal models or clinical trials regarding treatments with the PTX and Simva combination, the current study being the first to report antineoplastic effects in vivo. With respect to studies in cells, Ahn et al reported that in myeloid leukemia cells, Simva enhanced PTX cytotoxic effects while increasing the apoptosis of the malignant cells.30 Those authors observed that the combination of PTX and Simva suppressed TNFα-induced NFκB activation, which could account for the superimposed effects of the statin. In cultured ovarian carcinoma cells, Robinson et al found that Simva associated with PTX treatment had effects on the autophagy pathway, which might contribute to the cytotoxic activity of the chemotherapeutic agent.46
Because PTX in the commercial formulation was not affected by Simva use, while LDE-PTX antitumor action was further increased by the statin, it can be hypothesized whether the superimposed effect of Simva was related to the status of receptor activity or by the changed intracellular cholesterol dynamics created by the statin treatment. As HMG-CoA reductase inhibitors lead to an increase in LDL receptors, it is also possible that Simva could have further increased LDL-receptor overexpression, allowing the internalization of larger amounts of PTX associated with LDE into the melanoma tumor cells.34 This would enhance the tumor-growth inhibition effect of LDE-PTX, but conceivably not that of the commercial PTX formulation using Cremophor EL as the vehicle.
Enrichment of the plasma membrane and intracellular cholesterol pool is an essential metabolic requirement for the increase in mitosis rates.47 Increase in LDL-receptor expression is the chief mechanism to supply the rapidly dividing cancer cell cholesterol requirements. Other mechanisms can also come into play, including the increase in the endogenous synthesis of cholesterol and fatty acids regulated by sterol regulatory element-binding proteins (SREBPs), but statins can impair this mechanism by inhibiting HMG-CoA reductase.48 This could further increase the need for LDL-receptor pathway as lipid suppliers for the neoplastic cells, and consequently increase the drug-targeting effect of LDE. In prostate cancer, it was also shown that reduction of the cholesterol efflux through ATP-binding cassette (ABC) class A transporters such as ABCA1 may also occur, but it is difficult to figure the possible effects of statins on this mechanism.49 Another factor that may have influenced the results refers to PCSK9, an enzyme that catabolizes the LDL receptors. It has been reported that PCSK9 activity in human serum is increased under statin treatment,50 but PCSK9 status in cancer is largely unexplored, although it has been found increased in some tumor tissues. It is of note that although the values were not statistically significant for each group, in all the groups treated with Simva, there was an apparent increase in LDL receptors. This is an important issue, in that statin treatment may have the ability to increase the LDL receptors of tumors, and might explain, at least in part, the superior pharmacological action of LDE-PTX and Simva.
Solid tumors, such as melanoma, are composed of cancer cells that are surrounded by a complex microenvironment consisting of blood vessels, collagen fibers, fibroblasts, immune cells, and soluble molecules that play an important role in tumor progression.51,52 In tumors excised from the LDE-PTX and Simva group, the effect of the treatment in reducing the number of neoplastic cells was markedly greater than in those excised from the commercial PTX and Simva group. Furthermore, LDE-PTX and Simva was the only treatment that increased the presence of collagen fibers in the tumor stroma. This is an important finding, given that the inhibition of type I collagen synthesis in melanoma advanced stages can contribute to further progression of the tumor and its degradation by metalloproteinases is associated with tumor invasiveness.52,53 These assumptions may support a link between the results of the histological analysis and our findings regarding the metastasis evaluation in which the LDE-PTX and Simva treatment resulted in marked reduction of animals with metastasis, while PTX and Simva was ineffective. In fact, monotherapy with LDE-PTX also showed the ability to reduce metastasis development, although to a lower degree than the combined therapy with LDE-PTX and Simva. Therefore, it is likely that Simva contributed to the superior ability of LDE-PTX in impairing tumor dissemination. In fact, the properties of Simva in promoting the reduction of tumor invasiveness have been previously described.54,55 Furthermore, taking into account that LDEs target cancer cells through LDL receptors and that statins are known to increase their expression, the coadministration of Simva with LDE-PTX may also have favored LDE-drug uptake wherever neoplastic cells were located.4,34
Another interesting finding of this study regards the effect of the treatments on other components of the melanoma microenvironment, namely MCP-1, ICAM-1, and MMP-9.51,56–62 Studies regarding the anti-inflammatory and immunomodulatory effects of statins in such diseases as atherosclerosis have shown that the inhibition of the formation of isoprenoid derivatives promoted by Simva resulted in inhibition of ICAM-1 expression and also in decreased release of MMP-9 and MCP-1 by endothelial cells and macrophages.63–65 It is important to point out that in cancer, these proteins are associated with tumor progression and metastasis.51,56–62 Of note, in this study all treatments reduced the expression of those proteins in the tumor tissues, but the effect on ICAM-1 and MMP-9 was stronger under LDE-PTX and Simva than under the other treatments. The suggestion is then left that the greater reduction of metastasis development observed in this group could be the result of the combination of the changes in tumor microenvironment elicited by both Simva and LDE-PTX. These findings may account for the superior antitumor activity of the combined treatment with Simva and PTX, when PTX is carried by a lipid drug-targeting system.
In this study, we employed flow-cytometry analysis of tumor-tissue cells using rhodamine 123 as a marker to assess the mitochondrial bioenergetics in living cells. A loss of mitochondrial inner-membrane potential was observed after the administration of all the different treatments, demonstrating their induction of cell death, especially in the case of LDE-PTX and Simva, which promoted a remarkable reduction in cell viability. Taking into account that tumors from the LDE-PTX and Simva-treated group presented the smallest tumor sizes, we may assume that by the end of the drug-administration period, this treatment may have resulted in a lower proportion of viable residual cells than with the other treatments. This may have favored the increased antitumor activity of LDE-PTX and Simva.
In order to understand further the antitumor effects of the combined use of Simva with PTX chemotherapy, the expression of cyclin D1 was also evaluated. This protein is involved in the transition from G1 to S phase of the cell cycle, and its overexpression is related to breast tumors, lymphomas, melanomas, and colorectal cancer.66,67 Besides being a proliferation promoter, cyclin D1 can also act as a survival factor for tumor cells.68–70 Here, we have shown a reduction in the percentage of cells positive for cyclin D1 in tumors from the Simva and LDE-PTX and Simva groups. In addition, in tumors from the Simva, LDE-PTX, and LDE-PTX and Simva groups, there was an increase in positive cells for the cyclin-kinase inhibitor p21. The link between cyclin D1 reduction, p21 increase, and the antiproliferative effects of statins have been previously described as being related to the cell-cycle block in the G1 phase promoted by these drugs.36,71–74 It is noteworthy that Simva alone did not decrease melanoma growth. However, its antitumor effects may have been added to the effects of PTX associated with LDE, thereby enhancing the effectiveness of chemotherapy. On the other hand, when Simva was used in combination with commercial PTX, neither p21 increase nor cyclin D1 reduction were observed. A previous study has shown that treatment of ovarian cancer cells with Simva combined with carboplatin or PTX could result in mild antagonistic interactions between the drugs.46 Therefore, it is possible that the combined use of Simva and the commercial preparation of PTX may have resulted in drug interactions that could have interfered with the mechanisms of cell-death induction related to those drugs.
Confirming our previous studies with mice, rabbits, and also with patients with advanced cancers, the toxicity of PTX was pronouncedly reduced upon association of this drug with LDE.17,23,75 As estimated by animal survival, body-weight variation, and blood cell count after the different treatments of mice without tumors, a remarkable enhancement in the tolerability of the therapy was achieved by PTX association with LDE. The severe weight loss, myelosuppression, and survival reduction observed in the animals under commercial PTX and Simva treatment were absent in the animals treated with LDE-PTX preparations alone or in combination with Simva. In fact, these results are consistent with our previous findings that association of PTX with LDE resulted in remarkable toxicity reduction, probably by the new biodistribution of the drug created by the association with LDE, and by the replacement of the commercial PTX toxic vehicle with LDE, which is virtually nontoxic.17 It is noteworthy that reduction of chemotherapy toxicity is extremely valuable when considering its concomitant administration with other drugs and consequently the possibility of accumulation of drug toxicities. With regard to the analysis of the clinical chemistry parameters of mice under all the different treatments, no alterations indicative of muscle or kidney toxicity were found, as none of the single or combined treatments affected mice CK plasma activity or creatinine concentration. In respect of liver enzymes, only a minor alteration on AST and ALT was observed 1 week after the end of chemotherapy. It is important to point out that despite the statistical significance observed, the values of AST and ALT activities reported in this study are within the normal values for these enzymes described in the literature, and probably do not reflect an important liver injury.76
Another important set of data is related to the analysis of the hematological profile of melanoma-bearing mice. The picture observed in the leukocyte-count data was quite similar to that observed in the toxicity tests performed in mice without tumors. The leukopenia observed in animals under commercial PTX and Simva treatment was absent when LDE was used as a carrier of PTX, which confirms the diminution of the toxicity promoted by the LDE-targeting system.
With regard to platelet counts, thrombocytosis occurred as a paraneoplastic symptom. The increase in platelet count is indeed a common event in cancer, and is often associated with tumor progression, metastasis development, and poorer prognosis.77–81 Therefore, the circumvention of tumor-induced thrombocytosis seems to be an additional advantage of the combined treatment with LDE-PTX and Simva in comparison to commercial PTX and Simva, which was clearly unsuccessful in this respect.
The results of the toxicity parameters obtained from LDE-PTX and Simva are particularly relevant in view of the fact that statins are substrates of the CYP3A4 and CYP2C9 enzymes. The combinations of statins with drugs that are also metabolized by the cytochrome P450 enzyme system are eventually prone to drug interactions that may increase the occurrence of statin-related adverse effects, such as myopathy, rhabdomyolysis, and hepatotoxicity.82,83 The lack of noticeable toxicity in the LDE-PTX and Simva group is reassuring, in that statins did not change the remarkable ability of LDE to decrease the toxicity of PTX.
High toxicity and low pharmacological index are major drawbacks that present serious hazards to patients, and limit the effectiveness, duration, and indications of cancer chemotherapy. As shown here and elsewhere in tumor-bearing animals, the use of an LDE system had the ability to increase the effectiveness of carried chemotherapeutic agents, such as carmustine, etoposide, daunorubicin, and PTX, while drastically reducing their toxicity.17,19,21,84 In pilot clinical trials performed in patients with advanced cancers, LDE-carmustine, LDE-etoposide, and LDE-PTX showed no observable clinical or laboratorial toxicities in doses corresponding to those used in routine cancer treatment.5,14,23 Therefore, the LDE system and eventually others that explore the LDL-receptor endocytic pathways as drug-delivery gateway are creditable candidates for introduction in clinical oncology practice. In this setting, the use of statins can further enhance the anticancer activity of drugs carried in this type of drug-delivery system.
Conclusion
Although Simva had no relevant effect on the inhibition of tumor growth obtained with the commercial formulation of PTX, it significantly increased the antitumor action of PTX carried in LDE. Therefore, Simva can be a potential adjuvant to chemotherapy using drug-carrier systems internalized into the neoplastic cells via the LDL receptor-mediated endocytic pathway.
Acknowledgments
This study was supported by the National Council for Scientific and Technological Development (CNPq, Brasilia, Brazil). RCM has a Research Career Award, and IFK received a scholarship from CNPq. The authors are grateful to Dr Ricardo Ambrósio Fock, Ms Débora F Deus, and Dr Aleksandra Tiemi for their help with the experiments.
Disclosure
The authors report no conflicts of interest in this work.
References
Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29(4):431–438. | ||
Ho YK, Smith RG, Brown MS, Goldstein JL. Low-density lipoprotein (LDL) receptor activity in human acute myelogenous leukemia cells. Blood. 1978;52(6):1099–1114. | ||
Maranhão RC, Garicochea B, Silva EL, Llacer PD, Pileggi FJ, Chamone DA. Increased plasma removal of microemulsions resembling the lipid phase of low-density lipoproteins (LDL) in patients with acute myeloid leukemia: a possible new strategy for the treatment of the disease. Braz J Med Biol Res. 1992;25(10):1003–1007. | ||
Maranhão RC, Garicochea B, Silva EL, et al. Plasma kinetics and biodistribution of a lipid emulsion resembling low-density lipoprotein in patients with acute leukemia. Cancer Res. 1994;54(17):4660–4666. | ||
Pinheiro KV, Hungria VT, Ficker ES, Valduga CJ, Mesquita CH, Maranhão RC. Plasma kinetics of a cholesterol-rich microemulsion (LDE) in patients with Hodgkin’s and non-Hodgkin’s lymphoma and a preliminary study on the toxicity of etoposide associated with LDE. Cancer Chemother Pharmacol. 2006;57(5):624–630. | ||
Vitols S, Angelin B, Ericsson S, et al. Uptake of low density lipoproteins by human leukemic cells in vivo: relation to plasma lipoprotein levels and possible relevance for selective chemotherapy. Proc Natl Acad Sci U S A. 1990;87(7):2598–2602. | ||
Rudling MJ, Ståhle L, Peterson CO, Skoog L. Content of low density lipoprotein receptors in breast cancer tissue related to survival of patients. Br Med J (Clin Res Ed). 1986;292(6520):580–582. | ||
Avall-Lundqvist EH, Peterson CO. Serum cholesterol and apolipoprotein B levels may reflect disease activity in ovarian cancer patients. Acta Oncol. 1996;35(8):1007–1010. | ||
Ades A, Carvalho JP, Graziani SR, et al. Uptake of a cholesterol-rich emulsion by neoplastic ovarian tissues. Gynecol Oncol. 2001;82(1):84–87. | ||
Graziani SR, Igreja FA, Hegg R, et al. Uptake of a cholesterol-rich emulsion by breast cancer. Gynecol Oncol. 2002;85(3):493–497. | ||
Lundberg B. Preparation of drug-low density lipoprotein complexes for delivery of antitumoral drugs via the low density lipoprotein pathway. Cancer Res. 1987;47(15):4105–4108. | ||
Maranhão RC, Cesar TB, Pedroso-Mariani SR, Hirata MH, Mesquita CH. Metabolic behavior in rats of a nonprotein microemulsion resembling low-density lipoprotein. Lipids. 1993;28(8):691–696. | ||
Dorlhiac-Llacer PE, Marquezini MV, Toffoletto O, Carneiro RC, Maranhão RC, Chamone DA. In vitro cytotoxicity of the LDE: daunorubicin complex in acute myelogenous leukemia blast cells. Braz J Med Biol Res. 2001;34(10):1257–1263. | ||
Maranhão RC, Graziani SR, Yamaguchi N, et al. Association of carmustine with a lipid emulsion: in vitro, in vivo and preliminary studies in cancer patients. Cancer Chemother Pharmacol. 2002;49(6):487–498. | ||
Rodrigues DG, Covolan CC, Coradi ST, Barboza R, Maranhão RC. Use of a cholesterol-rich emulsion that binds to low-density lipoprotein receptors as a vehicle for paclitaxel. J Pharm Pharmacol. 2002;54(6):765–772. | ||
Valduga CJ, Fernandes DC, Lo Prete AC, Azevedo CH, Rodrigues DG, Maranhão RC. Use of a cholesterol-rich microemulsion that binds to low-density lipoprotein receptors as vehicle for etoposide. J Pharm Pharmacol. 2003;55(12):1615–1622. | ||
Rodrigues DG, Maria DA, Fernandes DC, et al. Improvement of paclitaxel therapeutic index by derivatization and association to a cholesterol-rich microemulsion: in vitro and in vivo studies. Cancer Chemother Pharmacol. 2005;55(6):565–576. | ||
Teixeira RS, Valduga CJ, Benvenutti LA, Schreier S, Maranhão RC. Delivery of daunorubicin to cancer cells with decreased toxicity by association with a lipidic nanoemulsion that binds to LDL receptors. J Pharm Pharmacol. 2008;60(10):1287–1295. | ||
Contente TC, Kretzer IF, Filippin-Monteiro FB, Maria DA, Maranhão RC. Association of daunorubicin to a lipid nanoemulsion that binds to low-density lipoprotein receptors enhances the antitumour action and decreases the toxicity of the drug in melanoma-bearing mice. J Pharm Pharmacol. 2014;66(12):1698–1709. | ||
Teixeira RS, Curi R, Maranhão RC. Effects on Walker 256 tumour of carmustine associated with a cholesterol-rich microemulsion (LDE). J Pharm Pharmacol. 2004;56(7):909–914. | ||
Lo Prete AC, Maria DA, Rodrigues DG, Valduga CJ, Ibañez OC, Maranhão RC. Evaluation in melanoma-bearing mice of an etoposide derivative associated to a cholesterol-rich nano-emulsion. J Pharm Pharmacol. 2006;58(6):801–808. | ||
Dias ML, Carvalho JP, Rodrigues DG, Graziani SR, Maranhão RC. Pharmacokinetics and tumor uptake of a derivatized form of paclitaxel associated to a cholesterol-rich nanoemulsion (LDE) in patients with gynecologic cancers. Cancer Chemother Pharmacol. 2007;59(1):105–111. | ||
Pires LA, Hegg R, Valduga CJ, Graziani SR, Rodrigues DG, Maranhão RC. Use of cholesterol-rich nanoparticles that bind to lipoprotein receptors as a vehicle to paclitaxel in the treatment of breast cancer: pharmacokinetics, tumor uptake and a pilot clinical study. Cancer Chemother Pharmacol. 2009;63(2):281–287. | ||
Soma MR, Baetta R, De Renzis MR, et al. In vivo enhanced antitumor activity of carmustine [N,N’-bis(2-chloroethyl)-N-nitrosourea] by simvastatin. Cancer Res. 1995;55(3):597–602. | ||
Feleszko W, Zagozdzon R, Gołab J, Jakóbisiak M. Potentiated antitumour effects of cisplatin and lovastatin against MmB16 melanoma in mice. Eur J Cancer. 1998;34(3):406–411. | ||
Agarwal B, Bhendwal S, Halmos B, Moss SF, Ramey WG, Holt PR. Lovastatin augments apoptosis induced by chemotherapeutic agents in colon cancer cells. Clin Cancer Res. 1999;5(8):2223–2229. | ||
Holstein SA, Hohl RJ. Synergistic interaction of lovastatin and paclitaxel in human cancer cells. Mol Cancer Ther. 2001;1(2):141–149. | ||
Werner M, Sacher J, Hohenegger M. Mutual amplification of apoptosis by statin-induced mitochondrial stress and doxorubicin toxicity in human rhabdomyosarcoma cells. Br J Pharmacol. 2004;143(6):715–724. | ||
Kozar K, Kaminski R, Legat M, et al. Cerivastatin demonstrates enhanced antitumor activity against human breast cancer cell lines when used in combination with doxorubicin or cisplatin. Int J Oncol. 2004;24(5):1149–1157. | ||
Ahn KS, Sethi G, Aggarwal BB. Reversal of chemoresistance and enhancement of apoptosis by statins through down-regulation of the NF-κB pathway. Biochem Pharmacol. 2008;75(4):907–913. | ||
Martirosyan A, Clendening JW, Goard CA, Penn LZ. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: potential therapeutic relevance. BMC Cancer. 2010;10:103. | ||
Mol MJ, Erkelens DW, Leuven JA, Schouten JA, Stalenhoef AF. Effects of synvinolin (MK-733) on plasma lipids in familial hypercholesterolaemia. Lancet. 1986;2(8513):936–939. | ||
Alberts AW. Lovastatin and simvastatin – inhibitors of HMG CoA reductase and cholesterol biosynthesis. Cardiology. 1990;77 Suppl 4:14–21. | ||
Tobert JA. New developments in lipid-lowering therapy: the role of inhibitors of hydroxymethylglutaryl-coenzyme A reductase. Circulation. 1987;76(3):534–538. | ||
Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation. 2002;105(6):739–745. | ||
Collisson EA, Kleer C, Wu M, et al. Atorvastatin prevents RhoC isoprenylation, invasion, and metastasis in human melanoma cells. Mol Cancer Ther. 2003;2(10):941–948. | ||
Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer. 2005;5(12):930–942. | ||
Hoque A, Chen H, Xu XC. Statin induces apoptosis and cell growth arrest in prostate cancer cells. Cancer Epidemiol Biomarkers Prev. 2008;17(1):88–94. | ||
Favero GM, Otuki MF, Oliveira KA, et al. Simvastatin impairs murine melanoma growth. Lipids Health Dis. 2010;9:142. | ||
Relja B, Meder F, Wilhelm K, Henrich D, Marzi I, Lehnert M. Simvastatin inhibits cell growth and induces apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int J Mol Med. 2010;26(5): 735–741. | ||
Geran RI, Greenberg NH, MacDonald MM, Schumacher AM, Abbott BJ. Protocols for Screening Chemical Agents and Natural Products Against Animal Tumors and Other Biological Systems. 3rd ed. Bethesda (MD): National Institutes of Health; 1972. | ||
Versluis AJ, van Geel PJ, Oppelaar H, van Berkel TJ, Bijsterbosch MK. Receptor-mediated uptake of low-density lipoprotein by B16 melanoma cells in vitro and in vivo in mice. Br J Cancer. 1996;74(4):525–532. | ||
Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425–430. | ||
Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118(Pt 5):843–846. | ||
Lee J, Jung KH, Park YS, et al. Simvastatin plus irinotecan, 5-fluorouracil, and leucovorin (FOLFIRI) as first-line chemotherapy in metastatic colorectal patients: a multicenter phase II study. Cancer Chemother Pharmacol. 2009;64(4):657–663. | ||
Robinson E, Nandi M, Wilkinson LL, Arrowsmith DM, Curtis AD, Richardson A. Preclinical evaluation of statins as a treatment for ovarian cancer. Gynecol Oncol. 2013;129(2):417–424. | ||
Fernández C, Lobo M, Gómez-Coronado D, Lasunción MA. Cholesterol is essential for mitosis progression and its deficiency induces polyploid cell formation. Exp Cell Res. 2004;300(1):109–120. | ||
Griffiths B, Lewis CA, Bensaad K, et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013;1(1):3. | ||
Lee BH, Taylor MG, Robinet P, et al. Dysregulation of cholesterol homeostasis in human prostate cancer through loss of ABCA1. Cancer Res. 2013;73(3):1211–1218. | ||
Careskey HE, Davis RA, Alborn WE, Troutt JS, Cao G, Konrad RJ. Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res. 2008;49(2):394–398. | ||
Villanueva J, Herlyn M. Melanoma and the tumor microenvironment. Curr Oncol Rep. 2008;10(5):439–446. | ||
Labrousse AL, Ntayi C, Hornebeck W, Bernard P. Stromal reaction in cutaneous melanoma. Crit Rev Oncol Hematol. 2004;49(3):269–275. | ||
Wandel E, Raschke A, Hildebrandt G, et al. Fibroblasts enhance the invasive capacity of melanoma cells in vitro. Arch Dermatol Res. 2002;293(12):601–608. | ||
Glynn SA, O’Sullivan D, Eustace AJ, Clynes M, O’Donovan N. The 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors simvastatin, lovastatin and mevastatin inhibit proliferation and invasion of melanoma cells. BMC Cancer. 2008;8:9. | ||
Relja B, Meder F, Wang M, et al. Simvastatin modulates the adhesion and growth of hepatocellular carcinoma cells via decrease of integrin expression and ROCK. Int J Oncol. 2011;38(3):879–885. | ||
Kuroda T, Kitadai Y, Tanaka S, et al. Monocyte chemoattractant protein-1 transfection induces angiogenesis and tumorigenesis of gastric carcinoma in nude mice via macrophage recruitment. Clin Cancer Res. 2005;11(21):7629–7636. | ||
Gazzaniga S, Bravo AI, Guglielmotti A, et al. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J Invest Dermatol. 2007;127(8):2031–2041. | ||
Koga M, Kai H, Egami K, et al. Mutant MCP-1 therapy inhibits tumor angiogenesis and growth of malignant melanoma in mice. Biochem Biophys Res Commun. 2008;365(2):279–284. | ||
Johnson JP, Stade BG, Holzmann B, Schwäble W, Riethmüller G. De novo expression of intercellular-adhesion molecule 1 in melanoma correlates with increased risk of metastasis. Proc Natl Acad Sci U S A. 1989;86(2):641–644. | ||
Slattery MJ, Dong C. Neutrophils influence melanoma adhesion and migration under flow conditions. Int J Cancer. 2003;106(5):713–722. | ||
Ghislin S, Obino D, Middendorp S, Boggetto N, Alcaide-Loridan C, Deshayes F. LFA-1 and ICAM-1 expression induced during melanoma-endothelial cell co-culture favors the transendothelial migration of melanoma cell lines in vitro. BMC Cancer. 2012;12:455. | ||
Johnsen M, Lund LR, Rømer J, Almholt K, Danø K. Cancer invasion and tissue remodeling: common themes in proteolytic matrix degradation. Curr Opin Cell Biol. 1998;10(5):667–671. | ||
Han KH, Ryu J, Hong KH, et al. HMG-CoA reductase inhibition reduces monocyte CC chemokine receptor 2 expression and monocyte chemoattractant protein-1-mediated monocyte recruitment in vivo. Circulation. 2005;111(11):1439–1447. | ||
Veillard NR, Braunersreuther V, Arnaud C, et al. Simvastatin modulates chemokine and chemokine receptor expression by geranylgeranyl isoprenoid pathway in human endothelial cells and macrophages. Atherosclerosis. 2006;188(1):51–58. | ||
Montecucco F, Burger F, Pelli G, et al. Statins inhibit C-reactive protein-induced chemokine secretion, ICAM-1 upregulation and chemotaxis in adherent human monocytes. Rheumatology (Oxford). 2009;48(3):233–242. | ||
Donnellan R, Chetty R. Cyclin D1 and human neoplasia. Mol Pathol. 1998;51(1):1–7. | ||
Coupland SE, Bechrakis N, Schüler A, et al. Expression patterns of cyclin D1 and related proteins regulating G1-S phase transition in uveal melanoma and retinoblastoma. Br J Ophthalmol. 1998;82(8):961–970. | ||
Inohara S, Kitagawa K, Kitano Y. Expression of cyclin D1 and p53 protein in various malignant skin tumors. Dermatology. 1996;192(2):94–98. | ||
Driscoll B, Buckley S, Barsky L, Weinberg K, Anderson KD, Warburton D. Abrogation of cyclin D1 expression predisposes lung cancer cells to serum deprivation-induced apoptosis. Am J Physiol. 1999;276(4 Pt 1):L679–L687. | ||
Sauter ER, Yeo UC, von Stemm A, et al. Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res. 2002;62(11):3200–3206. | ||
Ukomadu C, Dutta A. P21-dependent inhibition of colon cancer cell growth by mevastatin is independent of inhibition of G1 cyclin-dependent kinases. J Biol Chem. 2003;278(44):43586–43594. | ||
Denoyelle C, Albanese P, Uzan G, et al. Molecular mechanism of the anti-cancer activity of cerivastatin, an inhibitor of HMG-CoA reductase, on aggressive human breast cancer cells. Cell Signal. 2003;15(3):327–338. | ||
Takeda I, Maruya S, Shirasaki T, et al. Simvastatin inactivates β1-integrin and extracellular signal-related kinase signaling and inhibits cell proliferation in head and neck squamous cell carcinoma cells. Cancer Sci. 2007;98(6):890–899. | ||
Saito A, Saito N, Mol W, et al. Simvastatin inhibits growth via apoptosis and the induction of cell cycle arrest in human melanoma cells. Melanoma Res. 2008;18(2):85–94. | ||
Lourenço-Filho DD, Maranhão RC, Méndez-Contreras CA, Tavares ER, Freitas FR, Stolf NA. An artificial nanoemulsion carrying paclitaxel decreases the transplant heart vascular disease: a study in a rabbit graft model. J Thorac Cardiovasc Surg. 2011;141(6):1522–1528. | ||
Quimby FW, Luong RH. Clinical chemistry of the laboratory mouse. In: Fox JG, Davisson MT, Quimby FW, Barthold SW, Newcomer CE, Smith AL, editors. The Mouse in Biomedical Research. 2nd ed. Burlington (MA): Academic Press; 2007:171–216. | ||
Levin J, Conley CL. Thrombocytosis associated with malignant disease. Arch Intern Med. 1964;114(4):497–500. | ||
Menczer J, Schejter E, Geva D, Ginath S, Zakut H. Ovarian carcinoma associated thrombocytosis. Correlation with prognostic factors and with survival. Eur J Gynaecol Oncol. 1998;19(1):82–84. | ||
Ikeda M, Furukawa H, Imamura H, et al. Poor prognosis associated with thrombocytosis in patients with gastric cancer. Ann Surg Oncol. 2002;9(3):287–291. | ||
Nash GF, Turner LF, Scully MF, Kakkar AK. Platelets and cancer. Lancet Oncol. 2002;3(7):425–430. | ||
Nolte I, Przibylla H, Bostel T, Groden C, Brockmann MA. Tumor-platelet interactions: glioblastoma growth is accompanied by increasing platelet counts. Clin Neurol Neurosurg. 2008;110(4):339–342. | ||
Clark LT. Treating dyslipidemia with statins: the risk-benefit profile. Am Heart J. 2003;145(3):387–396. | ||
Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther. 2006;80(6):565–581. | ||
Kretzer IF, Maria DA, Maranhão RC. Drug-targeting in combined cancer chemotherapy: tumor growth inhibition in mice by association of paclitaxel and etoposide with a cholesterol-rich nanoemulsion. Cell Oncol (Dordr). 2012;35(6):451–460. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.