")
Back to Journals » International Journal of Nanomedicine » Volume 12
Simultaneous delivery of anti-miR21 with doxorubicin prodrug by mimetic lipoprotein nanoparticles for synergistic effect against drug resistance in cancer cells
Authors Rui M , Qu Y, Gao T, Ge Y, Feng C, Xu X
Received 12 September 2016
Accepted for publication 1 December 2016
Published 30 December 2016 Volume 2017:12 Pages 217—237
DOI https://doi.org/10.2147/IJN.S122171
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Mengjie Rui, Yang Qu, Tong Gao, Yanru Ge, Chunlai Feng, Ximing Xu
Department of Pharmaceutics, School of Pharmacy, Jiangsu University, Zhenjiang, People’s Republic of China
Abstract: The development of drug resistance in cancer cells is one of the major obstacles to achieving effective chemotherapy. We hypothesized that the combination of a doxorubicin (Dox) prodrug and microRNA (miR)21 inhibitor might show synergistic antitumor effects on drug-resistant breast cancer cells. In this study, we aimed to develop new high-density lipoprotein-mimicking nanoparticles (HMNs) for coencapsulation and codelivery of this potential combination. Dox was coupled with a nuclear localization signal (NLS) peptide to construct a prodrug (NLS-Dox), thereby electrostatically condensing miR21 inhibitor (anti-miR21) to form cationic complexes. The HMNs were formulated by shielding these complexes with anionic lipids and Apo AI proteins. We have characterized that the coloaded HMNs had uniformly dispersed distribution, favorable negatively charged surface, and high coencapsulation efficiency. The HMN formulation effectively codelivered NLS-Dox and anti-miR21 into Dox-resistant breast cancer MCF7/ADR cells and wild-type MCF7 cells via a high-density-lipoprotein receptor-mediated pathway, which facilitated the escape of Pgp drug efflux. The coloaded HMNs consisting of NLS-Dox/anti-miR21 demonstrated greater cytotoxicity with enhanced intracellular accumulation in resistant MCF7/ADR cells compared with free Dox solution. The reversal of drug resistance by coloaded HMNs might be attributed to the suppression of miR21 expression and the related antiapoptosis network. Furthermore, the codelivery of anti-miR21 and NLS-Dox by HMNs showed synergistic antiproliferative effects in MCF7/ADR-bearing nude mice, and was more effective in tumor inhibition than other drug formulations. These data suggested that codelivery of anti-miR21 and chemotherapeutic agents by HMNs might be a promising strategy for antitumor therapy, and could restore the drug sensitivity of cancer cells, alter intracellular drug distribution, and ultimately enhance chemotherapeutic effects.
Keywords: drug resistance, microRNA21 inhibitor, doxorubicin prodrug, nuclear localization signal peptide, lipoprotein-mimicking nanoparticles, breast cancer therapy
Introduction
Cancer is one of the leading causes of morbidity and mortality worldwide, and its incidence has gradually increased over the past few decades.1,2 Despite chemotherapy bringing significant progress to a variety of cancers, the efficacy of cancer treatment has been largely limited by the development of drug resistance. The emergence of drug resistance mainly arises from the overexpression of membrane proteins that activate the efflux pump, thereby exporting drugs before they reach their intracellular sites of action.3–5 Pgp, a typical drug-efflux transporter, has received increasing attention, due to the fact that it plays a vital role in drug resistance.6 Accordingly, considerable efforts have been focused on inhibiting the function of Pgp and ultimately increasing intracellular accumulation of drugs.7 Previous studies have reported various inhibitors that were designed to block Pgp activity.8–10 Nevertheless, the applications of these inhibitors were more or less likely to interfere with the chemotherapeutic agents, leading to impaired antitumor efficacy and undesirable side reactions. Therefore, an alternative approach is to circumvent the pump-out effect of Pgp, rather than to inhibit its function directly, by developing anticancer agents that are not substrates for the Pgp transporter.
The anthracycline doxorubicin (Dox) is one of the most efficacious anticancer chemotherapeutics for the treatment of a broad range of neoplasms, such as breast cancer, acute leukemia, and malignant lymphoma.11 In clinical trials, the emerging drawback of Dox has been acquired drug resistance of cancer cells that led to reduced intracellular concentration of drugs. Previous studies have demonstrated that Dox cannot efficiently enter the nucleus of drug-resistant MCF7/ADR breast cancer cells, whereas Dox accumulates highly in the nuclei of parental MCF7 cells.6,12 This discrepancy stems from the pump-out effect of overexpressed Pgp proteins, which transport Dox away from its target. To address the drug-resistance issue, researchers have synthesized a large number of Dox prodrugs, which have demonstrated great potential in anticancer treatment.13–15 One of these attempts is to escape the recognition of the Pgp transporter and efficiently deliver Dox into nuclei inside cancer cells. Recently, the nuclear localization signal (NLS) peptides have demonstrated the inspiring ability to facilitate active transportation toward nuclei, potentially bypassing the export of drugs by Pgp proteins. According to previous studies, the most widely used NLS peptide is the SV40 large T antigen (Pro-Lys-Lys-Lys-Arg-Lys-Val), which enabled effective and nucleus-targeting delivery of nanocrystals,16 gold nanoparticles,17 and DNA18 to cancer cells. In the light of the promising property of NLS peptide, we supposed that the coupling of NLS peptide to Dox could provide a strategy to deliver Dox efficiently to nuclei and eventually improve the therapeutic effect in Dox-resistant cancer cells.
Besides activated drug-efflux pumps, the occurrence of drug resistance involves various factors, such as inhibited apoptosis, altered cell-cycle regulation, and intrinsic compensation pathways. Therefore, it is apparent that in order to suppress resistance to chemotherapy effectively, a rational therapeutic approach is to combine multiple agents with diverse molecular mechanisms for synergistic treatment, such as employing the combination of chemical drugs and microRNA (miRNA) therapy. miRNAs, a class of small noncoding RNAs, posttranscriptionally regulate the translation of mRNAs and can influence a variety of biological processes. Therefore, dysregulation of miRNAs can partially contribute to tumorigenesis and cancer progression, supported by the distinctive miRNA-expression profile in tumors compared with that in healthy tissues. Unlike small-interfering RNA, an individual miRNA molecule can target hundreds of mRNAs on the basis of partial sequence complementarity and simultaneously regulate diverse signaling pathways in cancer cells. This broad regulatory activity of miRNA confers it the potential ability to coordinate the complex cancer-related signaling networks, resulting in improved outcomes in tumor treatment. miRNA21 (miR21) is one of the most overexpressed miRNAs in various cancers, such as breast cancer, hepatocellular carcinoma, and hematologic malignancies. As a result, the increased cell proliferation and declining levels of apoptosis have been triggered in cancer cells by repressing miR21’s downstream targets, including PTEN, tropomyosin 1, and PDCD4.19,20 Previous reports have demonstrated that inhibition of miR21 can restore the suppressive functions of endogenous miRNAs, induce cell apoptosis, and enhance the antiproliferative effects of anticancer drugs.21,22 In addition, current reports have revealed that overexpression of miR21 is significantly associated with Dox-induced resistance in breast cancer through downregulation of its target, PTEN proteins,23 indicating a promising therapeutic target for reversing drug resistance. As such, the combination of anticancer prodrugs with miR21-targeted therapy is a more practical strategy for synergistic cancer therapy against drug-resistant tumors compared with monochemotherapy.
With regard to the weak effect of NLS peptides on cellular uptake,24,25 the simultaneous transportation of chemical prodrugs and miRNA inhibitors is probably a challenging task. In terms of tumor-targeted delivery, a suitable delivery system is required that helps this combination escape from the recognition of macrophage systems, penetrate into tumor tissues, and accumulate in their intracellular sites of action. In recent years, significant progress in combination therapy has been made with cationic nanoparticles formed with liposomes,26,27 polymers,28,29 micelles,30,31 and dendrimers.29 Among these, endogenous lipoproteins have attracted growing attention, due to their advantages, such as ease of formulation, excellent biocompatibility, and clinical potential. High-density lipoprotein (HDL), which is composed of anionic Apo AI and phospholipids, has shown great potential in biomedical applications, including controlled release, target-specific delivery, and effective gene transfection.32–35 Furthermore, HDL plays a crucial role in the reverse cholesterol-transport process, acting as a vehicle that transports cholesterols to a variety of organs for metabolism.36–38 It is worth mentioning that cholesterol is an essential element to form cell membranes. Since a large number of cholesterols are required for maintaining the high rate of cancer-cell proliferation,39,40 HDL nanoparticles as cholesterol carriers are naturally endowed with tumor-targeting function. This specific property of HDL was validated by the fact that HDL receptors were found to be highly expressed on the surfaces of a variety of tumor cells,41,42 including MCF7 breast cancer cells and the drug-resistant MCF7/ADR cell line. On the other hand, decoration with Apo AI proteins in HDL nanoparticles could reduce undesirable interaction with blood proteins, and what is more, prevent opsonization by the mononuclear phagocytic system, which has always troubled cationic nanoparticles applied in gene delivery. In this regard, we speculated that a drug formulation mimicking natural HDLs would be an ideal carrier, and facilitate the combination of diverse therapeutic approaches for synergistic therapy.
In the present study, HDL-mimicking nanoparticles (HMNs) were designed as a drug and gene cocarrier for the treatment of drug-resistant breast cancers, as shown in Figure 1. Initially, an original NLS peptide (CPKKKRKV-NH2) was conjugated with Dox through an amide linkage to construct the prodrug NLS-Dox, following condensation of anti-miR21 to form cationic complexes. Subsequently, HMNs were prepared in which the NLS-Dox/anti-miR21 complexes were shielded by anionic lipids and Apo AI proteins. The encapsulation efficiency of the NLS-Dox conjugate and anti-miR21, and physiochemical properties, including particle size, surface ζ-potential, were characterized. Meanwhile, the cytotoxicity of HMNs and inhibitory efficiency of HMNs on miR21 expression were evaluated in the in vitro setting. Furthermore, the synergistic effect and the antitumor mechanism of coloaded HMNs were investigated through in vitro cellular assays, as well as in vivo nude mouse models, in the treatment of drug-resistant breast cancer.
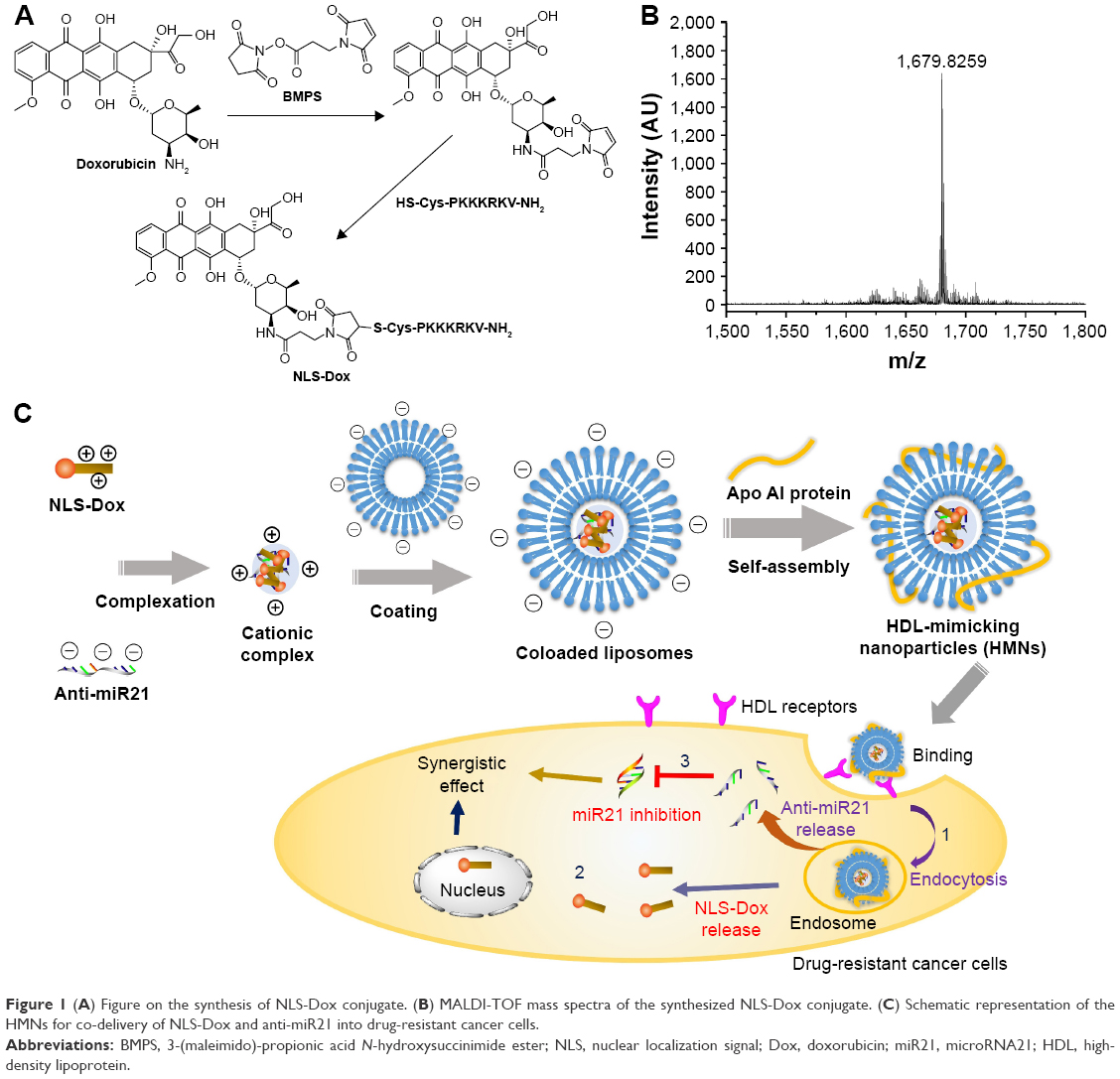 | Figure 1 (A) Figure on the synthesis of NLS-Dox conjugate. (B) MALDI-TOF mass spectra of the synthesized NLS-Dox conjugate. (C) Schematic representation of the HMNs for co-delivery of NLS-Dox and anti-miR21 into drug-resistant cancer cells. |
Materials and methods
Materials
1,2-Ditetradecanoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DMPG) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). 3-(Maleimido)-propionic acid N-hydroxysuccinimide ester (BMPS) (≥98.5%) was obtained from Sigma-Aldrich (St Louis, MO, USA). Dox HCl was purchased from Meilun Biotech Co Ltd (Dalian, China). The NLS peptides (CPKKKRKV-NH2) used in this work were synthesized by China Peptides (Shanghai, China). GelRed dye was purchased from Biotium Inc (Hayward, CA, USA). Nucleopore track-etched membranes were obtained from GE Healthcare (Little Chalfont, UK). The human breast cancer cell lines MCF7 (Dox-sensitive) and MCF7/ADR (Dox-resistant) were purchased from Kaiji Biology Co Ltd (Nanjing, China). Dulbecco’s Modified Eagle’s Medium, fetal bovine serum, and penicillin–streptomycin were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Trypsin was provided by Sango Co Ltd (Shanghai, China). Other materials were purchased from various suppliers and were of reagent grade or better. Apo AI recombinant proteins were expressed in Escherichia coli-bearing pNFXex plasmids,43 and subsequently purified using HisTrap affinity chromatography, as described by Ryan et al.44 For fluorescence observation, fluorescein isothiocyanate (FITC) was labeled with Apo AI proteins according to the manufacturer’s instructions.
Design and synthesis of miRNA inhibitors
The inhibitor anti-miR21 was designed according to the principle of sequences complementary to mature hsa-5p-miR21, while the scramble control contained no complementarity to the miR21 seed region. The sequences used in this study were as follows: anti-miR21, 5′-UCAACAUCAGUCUGAUAAGCUA-3′; scramble control, 5′-CAGUACUUUUGUGUAGUACAA-3′. The anti-miR21, scramble control, and fluorescein-labeled anti-miR21 (FAM) compounds were synthesized chemically by GenePharma (Shanghai, China), and were composed of 2′-O-methoxyethyl-modified nucleotides to make the RNA fragments resistant to RNase degradation.
Synthesis of NLS-Dox conjugate
The conjugate NLS-Dox was synthesized by coupling Dox with NLS peptide through a linker of BMPS. The synthesis procedure of NLS-Dox is shown in Figure 1. Dox (43.5 mg, 0.075 mmol) and BMPS (22 mg, 0.082 mmol) were dissolved in 2 mL of anhydrous dimethyl sulfoxide (DMSO). N, N-diisopropylethylamine (DIPEA; 28 μL, 0.15 mmol) was added, then the reaction mixture was maintained under stirring in the dark at ambient temperature overnight. The synthetic process was monitored by thin-layer chromatography (chloroform:methanol:NH3 70:30:3). The mixture was precipitated by cold anhydrous diethyl ether (4°C), and the resulting red solid separated by centrifugation at 8,000 ×g for 5 minutes. The precipitation was repeated three times, and red Dox-SMP was dried under vacuum (43 mg, 82.7%).
Then, Dox-BMPS (3.9 mg, 5.6 μmol) was dissolved in 0.25 mL of anhydrous DMSO. Meanwhile, the NLS peptide (5.5 mg, 5.6 μmol) was dissolved in another 0.25 mL of anhydrous DMSO. Then, the NLS-peptide solution and DIPEA (13 μL, 0.07 mmol) were added into Dox-BMPS and stirred for 24 hours in dark to form the NLS-Dox. The reaction mixture was precipitated by the aforementioned method, and the final product of NLS-Dox with a 76% yield was obtained under vacuum drying. The red solid was verified by a matrix-assisted laser desorption/ionization time-of-flight mass spectrometry.
Preparation of NLS-Dox/anti-miR21 complex
The complex was prepared by the rapid mixing of anti-miR21 with varying amounts of NLS-Dox. Briefly, the NLS peptides provided five positive charges at neutral pH. The desired charge ratio was calculated as nitrogen of positively charged amino acids in peptide (positive charge) to anti-miR21 phosphate (negative charge) ratios. Then, according to a variety of N:P molar ratios, 0.5 μg of anti-miR21 and varying amounts of NLS-Dox were separately diluted in complexation buffer (10 mM HEPES, 5% glucose, pH 7.4) to a final volume of 25 μL. Then, the Dox-NLS solution was transferred to the anti-miR solution by rapid addition and vortexed for 1 minute. The resultant complexes were incubated at room temperature for 20 minutes, and aliquots of 20 μL (40% of the total mixture volume) were removed from each complex and loaded directly onto 2% agarose gels for electrophoresis. The free anti-miR21 was visualized by staining with GelRed.
Preparation of HDL-mimicking nanoparticles
The NLS-Dox/anti-miR21 complexes were packaged into the HMNs through several steps. Firstly, the positively charged complexes were wrapped by anionic liposomes. Anionic liposomes composed of DMPG and DOPE in a molar ratio of 1:1 were prepared by thin-film hydration. Briefly, appropriate amounts of DMPG and DOPE were dissolved in chloroform and dried by a rotary evaporator to obtain a lipid film. The dry lipid film was hydrated with complexation buffer (10 mM HEPES, 5% glucose, pH 7.4) by subjection to a vigorous vortex for 30 minutes to obtain a lipid suspension. After hydration, the liposomes were extruded eleven times through polycarbonate track-etched membranes (pore sizes 0.4, 0.2, and 0.1 μm, respectively) in a miniextruder (Avanti Polar Lipids). Secondly, the liposomes and NLS-Dox/anti-miR21 complex were mixed at various ratios to coat the complexes with a lipid bilayer. The prepared NLS-Dox/anti-miR21 complex solution was diluted with complexation buffer, then slowly added into an equal volume of liposomes while stirring the mixture continuously. The nanoparticles encapsulating NLS-Dox/anti-miR21 complexes were prepared upon incubation for 30 minutes in ambient temperature. Alternatively, the liposome solutions were added into an equal volume of complex solution to form wrapped nanoparticles using a similar method. Eventually, the nanoparticles encapsulating NLS-Dox/anti-miR21 complexes were assembled with Apo AI protein at a lipid: Apo AI weight ratio of 10:1 for 6 hours at 4°C.
The HMNs were further purified by size-exclusion chromatography (CL-4B column). The column was prewashed three times with HEPES buffer (10 mM HEPES, 5% glucose, pH 7.4). A total of 22 fractions of 1 mL each were collected for each sample. The purified samples were characterized with respect to complex formulation, size distribution, ζ-potential, and encapsulation efficiency.
Agarose gel retardation assays
Gel-electrophoresis studies were performed using 2% w/v agarose gel stained with GelRed. A variety of samples were mixed with 6× loading dye before gel electrophoresis. The anti-miR21 alone was used as control. After samples were loaded onto the gel, the 2% gel was run in Tris–borate–ethylenediaminetetraacetic acid (EDTA) buffer at 110 V for 20 minutes. The gel was visualized using a standard 300 nm ultraviolet transilluminator.
Characterization of HMNs
Particle-size and ζ-potential measurements
The HMNs were diluted in complexation buffer (10 mM HEPES, 5% glucose, pH 7.4) to obtain an optimal scattering intensity. The hydrodynamic diameter, size distribution, and ζ-potential were measured using a ZetaPlus particle analyzer (Brookhaven Instruments Corporation, Santa Barbara, CA, USA). Scattered light was detected at 25°C at an angle of 90°.
Transmission electron microscopy
The morphology of the HMNs was observed using transmission electron microscopy (TEM; Philips Tecnai 12) operated at 120 kV. According to the process described by Forte and Nordhausen45 with some modifications, samples were diluted with ammonium acetate buffer (0.125 M ammonium acetate, 2.6 mM ammonium carbonate, 0.26 mM tetrasodium EDTA, pH 7.4). Then, a drop of the diluted samples at a final protein concentration of 0.5 mg/mL was applied to carbon-coated 100-mesh copper grids and adsorbed for approximately 5 minutes, followed by negative staining with a drop of phosphotungstic acid (2% v/v). Excess solutions were blotted by filter papers, and the grids were air-dried at room temperature before observation under TEM.
Determination of encapsulation efficiency
To determine the efficiency of NLS-Dox and anti-miR21 encapsulation in the HMNs, 10 μL of 20% Triton X-100 (v/v) and 20% heparin (w/v) were added to 100 μL of fractionated samples and incubated for 30 minutes at ambient temperature. The incubation completely released the NLS-Dox and FAM from the lipid-coated nanoparticles or HMNs. Then, the solution was diluted with complexation buffer, producing appropriate dilutions for fluorescence determination. The fluorescence intensities of NLS-Dox and FAM were measured using spectrofluorometry (RF-5301PC; Shimadzu, Kyoto, Japan) at 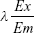 of 470/590 nm and 490/518 nm, respectively. Concentration was calculated by interpolation using a standard curve. Encapsulation efficiency was calculated as the percentage of the NLS-Dox incorporated into the nanoparticles or HMNs relative to the initial total amount of the NLS-Dox, as follows:
of 470/590 nm and 490/518 nm, respectively. Concentration was calculated by interpolation using a standard curve. Encapsulation efficiency was calculated as the percentage of the NLS-Dox incorporated into the nanoparticles or HMNs relative to the initial total amount of the NLS-Dox, as follows:
|
|
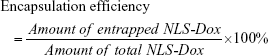
and anti-miR21 encapsulation efficiency was also determined by this equation.
Cell culture
The human breast cancer cell lines MCF7 (Dox-sensitive) and MCF7/ADR (Dox- or adriamycin-resistant) cells were cultivated in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin solution (HyClone; GE Healthcare) in a humidified atmosphere containing 5% CO2 at 37°C. For maintenance of the Dox-resistant phenotype, 1 μg/mL of Dox was added to the culture medium and removed 1 week before experiments. Cells were passaged with 0.5% trypsin and 0.2 mg/mL EDTA when they reached 80% confluence. The culture medium was replaced every 3 days. Before experiments, cells were detached, washed in phosphate-buffered saline (PBS), and counted. Defined numbers of cells were then seeded and left overnight to adhere.
In vitro cellular uptake and intracellular trafficking of HMNs
HMNs were prepared with NLS-Dox and anti-miR21 for fluorescence observation. MCF7 and MCF7/ADR cells were seeded into 35 mm glass-bottom culture dishes (2×104 cells/well) for confocal microscopy imaging, and into 12-well cell culture plates (105 cells/well) for flow cytometry. After 24 hours, the medium was replaced with fresh cell medium containing HMNs at an NLS-Dox concentration of approximately 12 μg/mL. The equivalent dose of free Dox solution was used as a control.
For uptake studies, cells were incubated with different formulations for 1, 4, and 12 hours, respectively. Then, cells were washed twice with cold PBS (0.1 M, pH 7.4), detached with 0.25% trypsin, and washed with cold PBS. Finally, the cells were resuspended in 0.5 mL of PBS and subjected to flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA, USA). The autofluorescence of the cells was used as a control. Results are expressed as mean and standard deviation obtained from three samples.
To investigate the uptake mechanism, cells were treated with a variety of preparations for 6 hours using similar methods. To achieve lysosome staining, cells were incubated with 100 nM LysoTracker green (Thermo Fisher Scientific) for 30 minutes at 37°C prior to fixation. Double-stained HMNs were formulated with FAM or FITC-labeled Apo AI proteins. After further incubation for 6 hours, the cells were washed twice with cold PBS (0.1 M, pH 7.4) and fixed with 4% formaldehyde for 15 minutes at room temperature. Nuclei were stained by 4′,6-diamidino-2-phenylindole according to the standard protocol provided by the supplier. The confocal images of cells were observed using laser confocal laser-scanning microscopy (CLSM; TCS SP5 II; Leica Microsystems, Wetzlar, Germany).
For the competitive binding assay, a 10- and 50-molar excess of natural HDL solution was coincubated with the HMNs while treating MCF7/ADR cells for 6 hours at 37°C, respectively. After incubation, the uptake levels of NLS-Dox were examined using flow cytometry. Results are expressed as mean and standard deviation obtained from three experiments.
In vitro cytotoxicity and apoptosis assay
Cytotoxicity was evaluated by a Cell Counting Kit (CCK)-8 assay (Dojindo, Kumamoto, Japan). Cells were seeded at a density of 0.5×104 cells per well in 100 μL of growth medium in 96-well plates and grown overnight. Then, several HMNs, including either NLS-Dox/anti-miR21, NLS-Dox–scramble control, or NLS peptide–anti-miR21, were added into cells over a range of concentrations and incubated for 48 and 96 hours. Meanwhile, cells treated with free Dox solution, free NLS-Dox solution, and HMNs consisting of NLS peptide–scramble control were used as control. According to the manufacturer’s instructions, 10 μL of CCK-8 solution was added to each well, followed by incubation for 1 hour at 37°C. Absorbance at 450 nm was determined by a multiplate reader (BioTek, Winooski, VT, USA). Cell viability was calculated as follows:
|
|
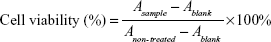
and data are presented as average ± standard deviation (n=5).
To evaluate apoptosis, MCF7/ADR cells were seeded into 12-well plates at a density of 105 cells/well. After incubation for 24 hours, the cells were treated with the free Dox solution (20 μM) or various HMNs (Dox dosage of 20 μM and/or anti-miR21 dosage of 213.3 nM) for another 48 hours. Then, the cells were washed with PBS three times, detached with trypsin, and collected by centrifugation. After being washed with PBS three times, the cells were resuspended in 0.5 mL of binding buffer. Subsequently, 10 μL of propidium iodide and 5 μL of annexin V–FITC were added into the solution and incubated for 15 minutes at room temperature in the dark. Then, the cells were subjected to flow cytometry (FACSCalibur). Cells without treatment were used as blank controls. The experiments were repeated three times.
In vitro miR21-expression assay
To examine the efficiency of anti-miR21, MCF7 and MCF1/ADR (105 cells/well) cells were seeded in 12-well culture plates and incubated for 24 hours. The cells were then treated with HMNs containing anti-miR21 or scramble control at a final concentration of 213.3 nM for 48 hours.
Total RNA was extracted from cells with Trizol reagent (Thermo Fisher Scientific), and reverse-transcribed to complementary DNA using a TaqMan miRNA reverse-transcription kit (Takara, Kyoto, Japan). Then, real-time polymerase chain reaction (PCR) was performed on an Applied Biosystems 7500 (Thermo Fisher Scientific) using the TaqMan kit according to the standardized protocol. Both reverse transcription and PCR primers were purchased from GenePharma. miR21 expression was normalized using the 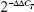 method relative to human U6 small nuclear RNA. All reactions were performed in triplicate. The change in miR21 expression was calculated as the fold variation relative to the untreated control.
method relative to human U6 small nuclear RNA. All reactions were performed in triplicate. The change in miR21 expression was calculated as the fold variation relative to the untreated control.
Western blot assay
After the same treatments, the cells were lysed in lysis buffer (20 mM Tris [pH 7.4], 150 mM NaCl, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 1% NP40, 100 μg/mL phenylmethylsulfonyl fluoride, 2 μg/mL aprotinin, 1 μg/mL pepstatin, and 10 μg/mL leupeptin) for 30 minutes on ice. The supernatant was collected after centrifugation at 12,000 ×g for 15 minutes at 4°C. Equal amounts of lysates were resolved on a 10% sodium dodecyl sulfate polyacrylamide-gel electrophoresis, transferred onto polyvinylidene difluoride membranes, and blocked with 5% nonfat dry milk in TBS-T (10 mM Tris–HCl pH 7.5, 100 mM NaCl, and 0.05% Tween 20), followed by incubation with an antibody specific for PTEN (1:1,000) overnight at 4°C. Blots were washed and incubated with horseradish peroxidase-conjugated secondary antibody. The blots were developed using an enhanced chemiluminescence kit. The density of the blots was determined by ImageJ analysis software.
In vivo antitumor efficacy
Female BALB/c nude mice (5–7 weeks old) were purchased from Shanghai Laboratory Animal Center (Shanghai, China). The mice were housed in specific pathogen-free conditions following guidelines for laboratory animals and the ethics committee of Jiangsu University. The animal care and use committee of Jiangsu University approved the use of animals in this study. To establish xenograft models of MCF7/ADR, mice were subcutaneously injected in the right flank with 107 cells suspended in 200 μL of PBS. When the tumors reached approximately 100 mm3, the mice were randomized into five groups (n=6): PBS, free Dox solution (7.6 mg/kg), HMNs (NLS peptide–anti-miR21, dose of 1 mg/kg anti-miR21), HMNs (NLS-Dox–scramble inhibitor, 23.6 mg/kg +1 mg/kg), and HMNs (NLS-Dox/anti-miR21, 23.6 mg/kg +1 mg/kg). Mice were dosed via the tail vein every 3 days five times. Animal weight and tumor volume were measured and calculated once every 3 days for 3 weeks. Tumor volume was calculated using the formula:
|
|
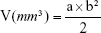
where a and b represent the major and minor axes of the tumor, respectively.
Statistical analysis
Data represent mean and standard deviation in each experiment, which was repeated at least three times. All data are shown as mean ± standard deviation unless particularly outlined. Student’s t-test or two-way analysis of variance was performed in statistical evaluation, with the Student–Newman–Keuls test employed as a post hoc test. Results were considered statistically significant at P<0.05 and highly significant at P<0.01.
Results
Synthesis of NLS-Dox conjugate
The synthesis of NLS-Dox was designed and conducted according to the scheme shown in Figure 1A. This conjugate was synthesized through the linker of BMPS, and the whole process was completed following two steps. Initially, an NLS peptide modified with cysteine (CPKKKRKV-NH2) was synthesized by standard fluorenylmethoxycarbonyl solid-peptide synthesis with 99.35% purity. Meanwhile, hydrophilic Dox HCl was neutralized by the removal of hydrochloride salt using DIPEA. Therefore, the 3′-amino group of daunosamine sugar of Dox was favorable for cross-linking with the active ester of BMPS, yielding Dox-BMPS. Secondly, NLS-Dox was obtained by covalently conjugating the cysteine of NLS peptide to the maleimide of Dox-BMPS through a Michael addition. Successful synthesis of NLS-Dox was confirmed by the molecular shifts in the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis: 1,680 (calculated), 1,679.83 (found), as depicted in Figure 1B.
Preparation of liposomes incorporating NLS-Dox/anti-miR21 complexes
HMNs encapsulating NLS-Dox/anti-miR21 complex were prepared by a previously established procedure,35 with some modifications. As illustrated in Figure 1C, anti-miR21 initially complexed with appropriate amounts of NLS-Dox and was condensed into a cationic core. HMNs were subsequently prepared by coating the preformed complexes with anionic lipid bilayers, in which the Apo AI proteins were embedded. The inserted Apo AI proteins not only stabilized the nanoparticles but provided similar functions to natural HDL nanoparticles.
As for the preparation of complexes, NLS-Dox, which carried five positively charged amino acids (lysines and arginines), was used to condense constant amounts of anti-miR21 at varying N:P molar ratios. Complexation efficiency was determined by electrophoresis on 2% agarose gels, since the formed complexes were retained in wells and only free anti-miR21 was able to migrate on gels. As shown in Figure 2A, we observed a gradual rise in the retarded proportion of anti-miR21 as the N:P ratio increased, and anti-miR21 was completely condensed at ratios higher than 10. With respect to the stability and surface potential of complexes, an N:P ratio of 20, at which overall cationic complexes were prepared, was chosen in the following experiments.
 | Figure 2 Agarose gel electrophoresis analysis. (A) Gel retardation assay of complexation efficiency of anti-miR21 by NLS-Dox. Lane 1: free anti-miR21 control, lane 2-9: complexation at various N/P molar ratios. (B) Gel retardation assay of the incorporation efficiency of complexes into anionic DMPG liposomes composed, lane 1-8: encapsulation at varying DMPG:anti-miR21 weight ratios. (C) Gel retardation assay of the incorporation efficiency of complexes into anionic liposomes composed of DMPG–DOPE (1:1, m/m ). (D) Gel retardation assay of the incorporation efficiency after the preparation of the HMNs followed by incubation of Apo AI with preformed DMPG–DOPE liposomes at varying DMPG:anti-miR21 weight ratios. |
To envelop the NLS-Dox/anti-miR21 complexes, different anionic liposomes composed of either DMPG or DMPG–DOPE (1:1, mol/mol) were prepared by thin-lipid rehydration. Followed by extrusion using 100 nm polycarbonate membranes, homogeneous liposomes were obtained with an average diameter of 117.7 nm and a polydispersity-index value of 0.18 (data not shown). As a result of the incubation of liposomes with NLS-Dox/anti-miR21 complexes, the spontaneous coating of complexes with lipid bilayers was most likely to be driven by electrostatic interaction between negative DMPG and cationic complexes.
Moreover, other parameters, such as the lipid composition of liposomes and DMPG:anti-miR21 ratios, were critical for the stability and encapsulation efficiency of complexes. As shown in Figure 2B, the free anti-miR21 on gels indicated that the increased amounts of DMPG liposomes triggered the dissociation of complexes. This undesirable release of anti-miR21 implied that if intact complexes were not quickly incorporated into liposomes, they would be disrupted under the strong competition of anionic DMPG lipids for NLS-Dox. To tackle this problem, DOPE, a neutral fusogenic lipid, was added to liposome composition with the aim of accelerating the coating process and improving encapsulation efficiency. Optimal coating of complexes, represented in Figure 2C, was observed with DMPG–DOPE liposomes, leading to the almost-complete retention of anti-miR21 in wells. Furthermore, the ratio of DMPG:anti-miR21 had an impact on the average diameter and ζ-potentials of formed nanoparticles. As shown in Figure 3A, large and heterogeneous aggregates were observed at the DMPG:anti-miR21 weight ratio of 16, presumably as a result of the formation of isoelectric particles that were prone to aggregation. At either higher or lower ratios, much smaller nanoparticles were prepared. Consequently, nanoparticles were unimodal in size with a mean diameter of 74.5±6.9 nm as the ratio of DMPG:anti-miR21 reached 40. On the other hand, the ζ-potential of nanoparticles decreased gradually while the DMPG:anti-miR21 ratio rose (Figure 3B). We observed that the net surface charge of nanoparticles was 0.45 mV at a ratio of 16 and decreased to −16.1 mV at a ratio of 40, confirming the complete coating of cationic complexes with negatively charged DMPG lipids.
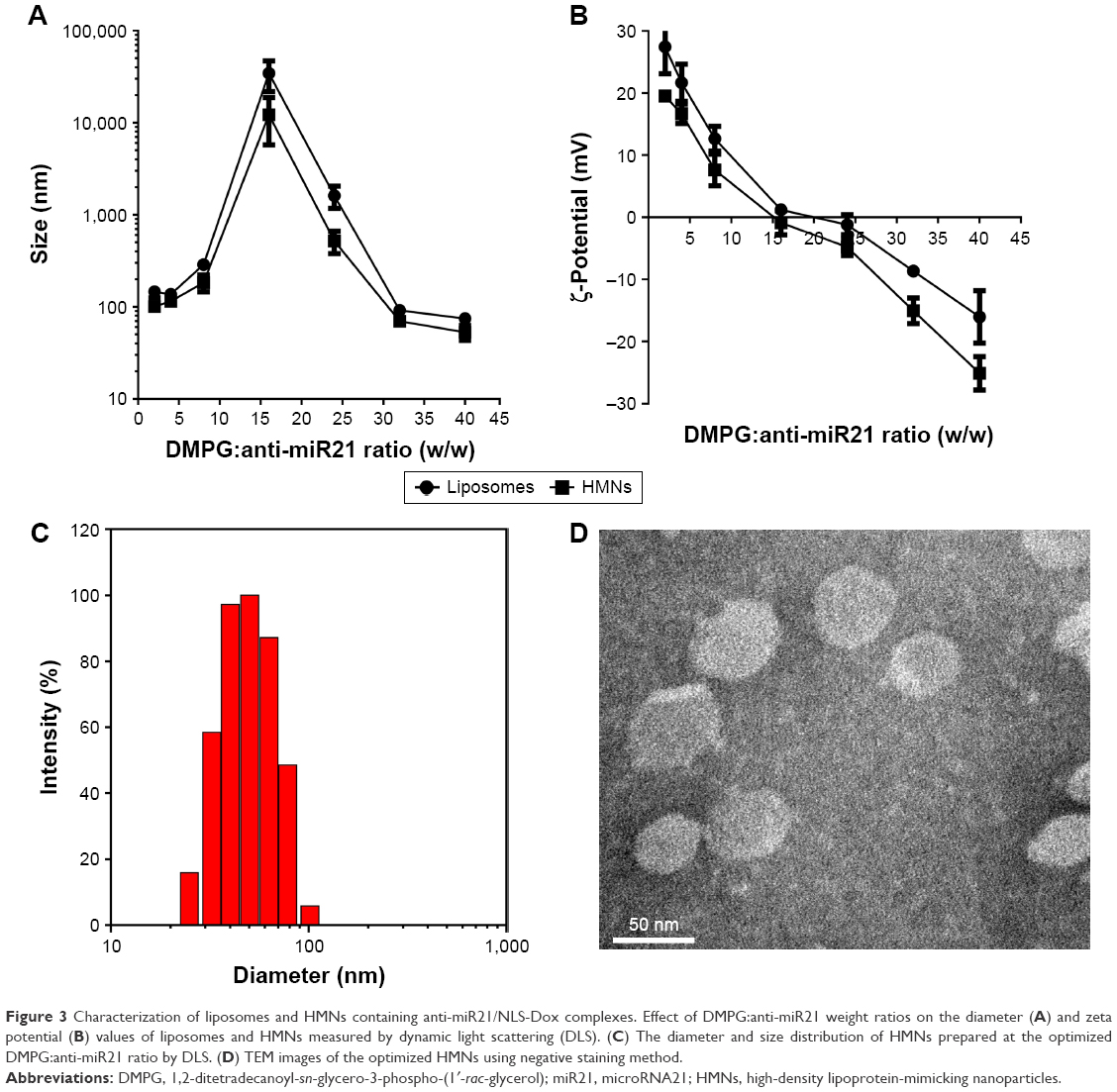 | Figure 3 Characterization of liposomes and HMNs containing anti-miR21/NLS-Dox complexes. Effect of DMPG:anti-miR21 weight ratios on the diameter (A) and zeta potential (B) values of liposomes and HMNs measured by dynamic light scattering (DLS). (C) The diameter and size distribution of HMNs prepared at the optimized DMPG:anti-miR21 ratio by DLS. (D) TEM images of the optimized HMNs using negative staining method. |
Size-exclusion chromatography showed that more than 90% of NLS-Dox/anti-miR21 complexes was eluted at void volume, reflecting the encapsulated complexes in liposomes (Figure S1A). According to the elution profile, the encapsulation efficiencies of NLS-Dox and anti-miR21 were 91.3%±4.6% and 92.7%±1.8%, respectively, at a DMPG:anti-miR21 ratio of 40. The complex ratio of NLS-Dox and anti-miR21 in purified HMNs was calculated as approximately 20.3±1.8, which accorded with the initial ratios. Collectively, the optimized DMPG–DOPE liposomes encapsulating NLS-Dox/anti-miR21 complexes were prepared at a DMPG:anti-miR21 weight ratio of 40.
Preparation and characterization of HMNs
We prepared HMNs coloaded with NLS-Dox and anti-miR21 after the self-assembly of Apo AI proteins with corresponding liposomes, as illustrated in Figure 1C. The formation of HMNs had negligible influence on the rigidity of complexes, due to no migration of free anti-miR21 on gels (Figure 2D). After purification by size-exclusion chromatography, the incorporated efficiencies of NLS-Dox and anti-miR21 in HMNs were 87.4%±3.7% and 89.1%±4.5%, respectively (Figure S1B). More important, the assembly of Apo AI effectively decreased the diameter of the formed HMNs compared with that of corresponding liposomes across a wide range of ratios, showing that HMNs had a mean diameter of 52.9±9.6 nm at the optimal ratio (Figure 3C). Because of the negative Apo AI proteins, the ζ-potential of HMNs fell to −25.11 mV, lower than the −16.1 mV of liposomes. In addition, TEM images of HMNs, as presented in Figure 3D, demonstrated well-dispersed, spherical morphology with slightly smaller diameter than dynamic light-scattering results.
Cellular uptake and intracellular trafficking of HMNs in MCF7/ADR cells
To elucidate whether NLS-Dox could efficiently accumulate in drug-resistant cancer cells, the uptake of HMNs coloaded with NLS-Dox/anti-miR21 was investigated in Dox-resistant MCF7/ADR cells and parental MCF7 cells by flow cytometry. As shown in Figure 4A, we observed that free Dox rapidly accumulated in MCF7 cells after 1 hour’s incubation, followed by a slight increase with prolonged incubation time. Not surprisingly, the uptake of free Dox by MCF7/ADR cells was significantly subdued, showing obvious resistance to Dox. On the other hand, NLS-Dox in HMNs was taken up gradually by both cell lines, and demonstrated similar uptake efficiency (Figure 4B). This uptake efficiency of NLS-Dox implied that Pgp-mediated drug efflux was partially counteracted by the nucleus-targeting ability of conjugated NLS peptides. To illustrate the codelivery capacity of HMNs, anti-miR21 was labeled with fluorescein, a typical green fluorescent dye, for further uptake investigation. As represented in Figure 4C, the steady increase in intracellular concentration of anti-miR21 was also observed in two cell lines as the incubation time was extended, verifying the simultaneous uptake of HMNs by cancer cells. With respect to uptake efficiency, the mean fluorescence values of different drugs were detected (Figure 4D). NLS-Dox had similar intracellular concentration in MCF7 cells as free Dox. Nevertheless, NLS-Dox showed a 2.43-fold increase in intracellular concentration in MCF7/ADR cells in comparison with free Dox after 12 hours of incubation. The mean fluorescence intensity of FAM showed a similar trend in cellular accumulation to that of NLS-Dox, confirming the effective codelivery pattern of the HMNs.
 | Figure 4 Flow cytometry analysis of in vitro uptake of free Dox (A), the HMNs containing NLS-Dox (B) and FAM labeled anti-miR21 (C) by Dox-sensitive MCF7 cells and Dox-resistant MCF7/ADR cells after incubation for various hours, respectively. (D) The mean fluorescent intensity of different formulations in MCF7 cells and MCF7/ADR cells were measured at various time points, respectively. |
Labeled by multiple fluorescence dyes, intracellular trafficking mechanisms of HMNs were illustrated in more detail using CLSM. Based on the aforementioned results, the acid organelles, including late endosomes and lysosomes, were stained with LysoTracker green. As shown in Figure 5A, a part of red fluorescent NLS-Dox was colocated in green fluorescent endosomes/lysosomes, which indicated that the HMNs might have been endocytosed into drug-resistant cells. Some red fluorescent Dox did not overlap with LysoTracker signals, implying the escape of HMNs from lysosomes and subsequent release of cargos in cytoplasm. Moreover, we prepared HMNs including FAM. After endocytosis by MCF7/ADR cells, the green fluorescent signal of FAM was mainly localized in cytosol, where the anti-miR21 could efficiently inhibit the expression and function of its target – miR21 (Figure 5B).
 | Figure 5 The confocal microscopy images of MCF7/ADR cells after incubation with varying HMNs. The acid organelles in cells were stained by LysoTracker green (A). The HMNs containing FAM-labeled anti-miR21 (B) and FITC-labeled Apo AI proteins (C) were observed, respectively. Then the nucleus were stained with DAPI (blue) after fixation. Competitive inhibitory effect of natural HDL amounts on the accumulation levels of NLS-Dox in MCF7/ADR cells was determined by FACS analysis (D). Data are shown as mean ± SD (n = 3). **P<0.01. |
To investigate whether HDL receptor-mediated mechanism was indeed involved in the uptake of HMNs, Apo AI was cross-linked with FITC to monitor its internalization. Green Apo AI signals were overlain with a portion of red NLS-Dox signals, indicating that the HMNs were taken up by holoparticle endocytosis (Figure 5C). Furthermore, a competition assay was conducted in which HMNs were coincubated with natural HDL when treating MCF7/ADR cells. As determined by flow-cytometry analysis, coincubation of native HDL significantly influenced cellular uptake of NLS-Dox and exhibited an inhibitory effect on uptake as a function of competing HDL amounts. The mean fluorescence intensity of NLS-Dox was reduced by 38% and 95.5% after treatment with tenfold and 50-fold excess HDL, respectively, compared to the control group (Figure 5D). These results validated our assumption that the cellular internalization of HMNs was probably mediated by particular HDL receptors on the surface of MCF7/ADR cells.
In vitro cytotoxicity of HMNs
Once it was confirmed that the targeted delivery of HMNs led to the remarkable accumulation of NLS-Dox and anti-miR21 in MCF7/ADR cells, we proceeded to evaluate the potential antitumor effect of HMNs combining chemotherapeutics and miR21 downregulation. The viability of MCF7/ADR cells and parental MCF7 cells was determined after treatment with HMN coencapsulation of NLS-Dox/anti-miR21 compared with a variety of drug formulations. As expected, the cytotoxicity of free Dox was remarkably reduced in MCF7/ADR cells in comparison with MCF7 cells (Figure 6), verifying the high Dox resistance of MCF7/ADR. Meanwhile, control HMNs (NLS peptide/scramble inhibitor) showed negligible toxicities toward two cell lines across all examined concentrations during a 96-hour exposure period, demonstrating the excellent biocompatibility of the HMNs. The anti-miR21 alone encapsulated in HMNs induced a moderate decrease by approximately 40% in two cell lines when its concentration reached 160 nM, and antitumor efficacy was close to the maximum platform whether or not it was incubated for a further 96 hours. As for the formulations consisting of NLS-Dox alone or NLS-Dox/anti-miR21, similar cytotoxicities were shown as a function of drug concentration in MCF7 cells when compared to free DOX solution. However, HMNs composed of NLS-Dox/anti-miR21 demonstrated the most effective anticancer activity in MCF7/ADR cells.
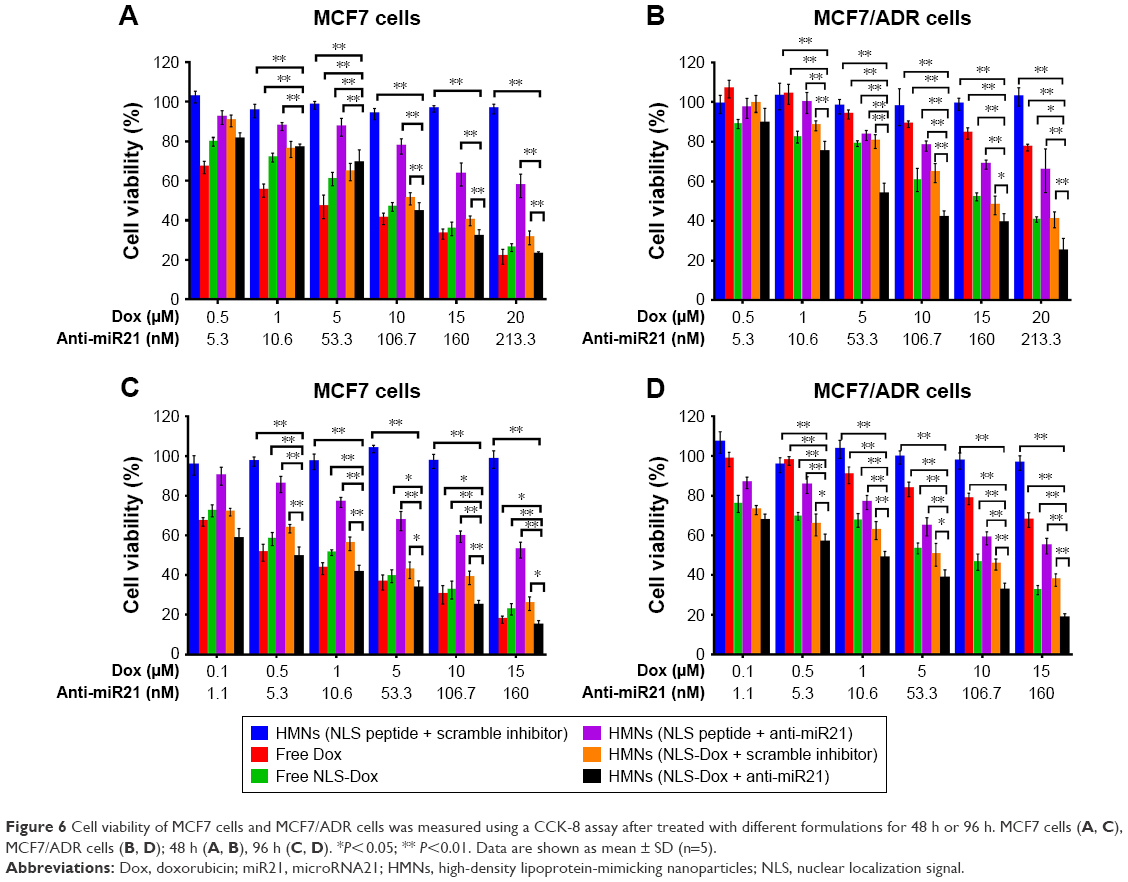 | Figure 6 Cell viability of MCF7 cells and MCF7/ADR cells was measured using a CCK-8 assay after treated with different formulations for 48 h or 96 h. MCF7 cells (A, C), MCF7/ADR cells (B, D); 48 h (A, B), 96 h (C, D). *P<0.05; ** P<0.01. Data are shown as mean ± SD (n=5). |
As summarized in Table 1, the half-maximal inhibitory concentration (IC50) of free Dox solution in MCF7/ADR cells (49.39 μM) was over 20-fold higher than that in parent MCF7 cells (2.43 μM) after incubation for 48 hours. A similar increase in IC50 values was also observed when cells were exposed to free Dox for a further 96 hours, from 0.61 μM in wild-type MCF7 cells to 29.96 μM in resistant MCF7/ADR cells. Also, we found that NLS-Dox itself showed lower IC50 values in MCF7/ADR cells when it was used in either free drug solution or encapsulated in HMNs, resulting from the fact that NLS-Dox was able to escape the drug efflux mediated by Pgp pumps. Among all treatments, dual-drug-loaded HMNs were the best at killing resistant MCF7/ADR cells, with lowest IC50 values of 6.06 μM at 48 hours of incubation and 1.02 μM at 96 hours of incubation, respectively. These results were associated with enhanced accumulation of NLS-Dox in drug-resistant cells, and demonstrated that codelivery of NLS-Dox and anti-miR21 caused a remarkable decline in drug resistance and provided good synergistic antitumor effects in vitro.
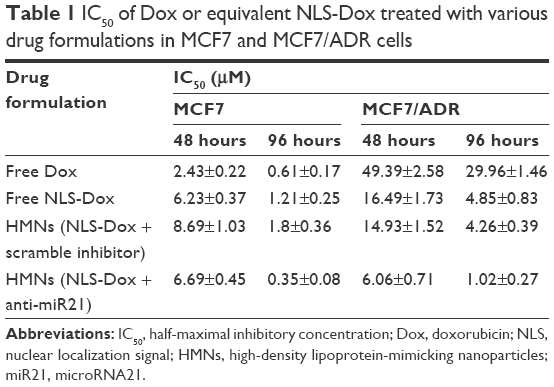 | Table 1 IC50 of Dox or equivalent NLS-Dox treated with various drug formulations in MCF7 and MCF7/ADR cells |
We thereafter investigated whether the cytotoxic effects of NLS-Dox/anti-miR21 were regulated by enhanced cellular apoptosis. Cell apoptosis was subsequently examined by flow cytometry (Figure 7). MCF7/ADR cells did not show visible apoptosis after 48 hours of incubation with control HMNs. Likewise, only 10.75% of cells were detected to be apoptotic when treated with free DOX solutions, indicating pump-out effects of Pgp proteins. The anti-miR21 itself in HMNs showed a slight therapeutic effect on MCF7/ADR cells, which was consistent with the results of the CCK-8 assay. Moreover, a significant increase in cell apoptosis was found in cells after treatment of HMNs, including NLS-Dox alone, suggesting that the nuclear delivery of NLS-Dox could avoid drug efflux and subsequently trigger cell apoptosis. The highest percentage of cell apoptosis was obtained in cells treated with HMNs coloaded with NLS-Dox/anti-miR21, which indicated that this combination therapy assisted by HMNs could significantly improve cell apoptosis and in turn increase chemotherapeutic efficacy.
 | Figure 7 Induction of cell apoptosis in MCF7/ADR cell treated with different formulations for 48 h. The apoptotic cells and necrotic cells were analyzed by flow cytometry. |
In vitro inhibition of miR21 expression in MCF7/ADR cells
The antitumor activity of HMNs coloaded with NLS-Dox/anti-miR21 presumably accounted for the efficient inhibition of miR21 expression, which led to the resensitization of MCF7/ADR cells to Dox. We determined the expression of miR21 by quantitative reverse-transcription PCR and the downstream targets by Western blot assay after 48 hours of incubation. The scramble inhibitor was used as a negative control. As shown in Figure 8A, the relative expression level of miR21 was reduced the most, approximately 60.2%, when cells were treated with NLS-Dox/anti-miR21. Compared with the coloaded formulation, the other two HMNs containing the NLS peptide–anti-miR21 or NLS-Dox/negative control reduced only 46.6% and 16.2% of miR21 expression, respectively, which suggested that NLS-Dox itself slightly decreased the expression of miR21. By contrast, the delivery of NLS-peptide/negative control by HMNs had negligible effect on the levels of miR21 in cells, confirming that the remarkable inhibition of miR21 resulted mainly from the effect of the anti-miR21, and of NLS-Dox to some extent.
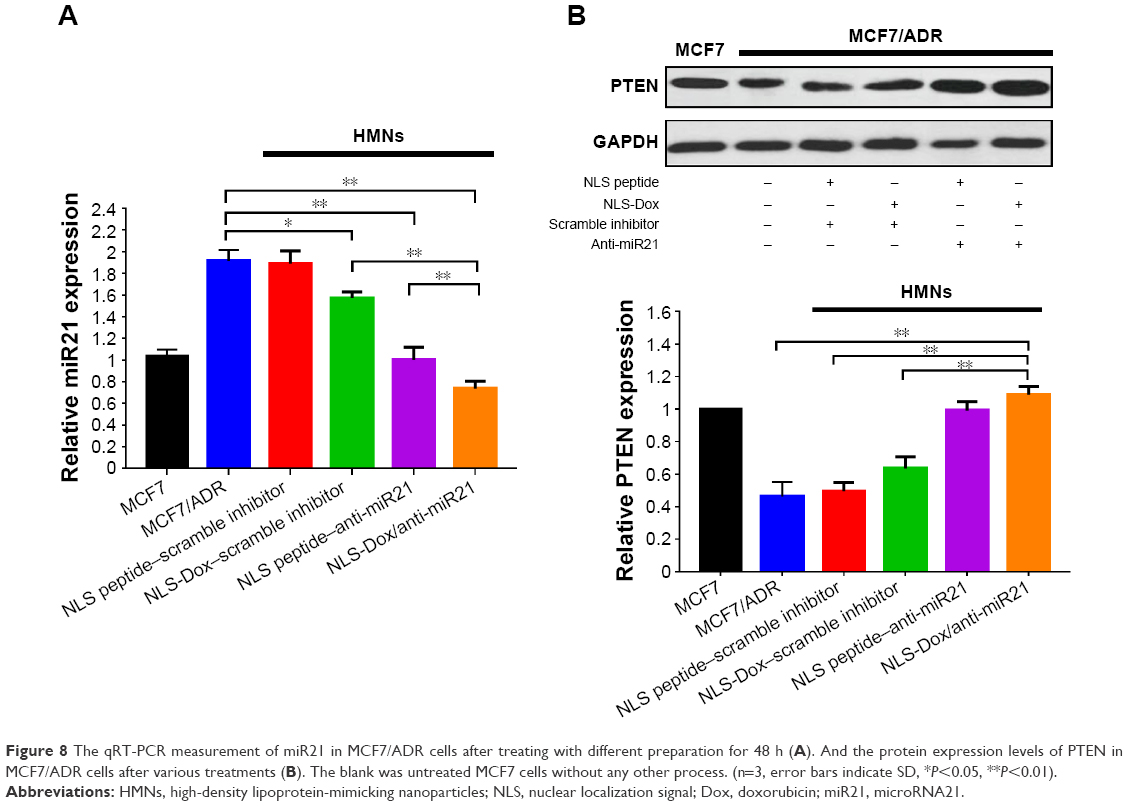 | Figure 8 The qRT-PCR measurement of miR21 in MCF7/ADR cells after treating with different preparation for 48 h (A). And the protein expression levels of PTEN in MCF7/ADR cells after various treatments (B). The blank was untreated MCF7 cells without any other process. (n=3, error bars indicate SD, *P<0.05, **P<0.01). |
Furthermore, we evaluated the expression levels of the downstream target of miR21. Based on previous reports, PTEN was chosen, due to its vital role in tumor-suppressive signaling networks.46,47 As shown in Figure 8B, relative levels of PTEN expression in MCF7/ADR cells were significantly lower than in wild-type MCF7 cells. Treatment with NLS peptide–anti-miR21 and NLS-Dox/anti-miR21 resulted in a remarkable increase in PTEN expression in MCF7/ADR cells, leading to a 4.17- and 4.75-fold upregulation compared to untreated MCF7/ADR, respectively. Meanwhile, a slight increase in expression levels of PTEN was observed after treatment with NLS-Dox/control inhibitor. All these results validated the PTEN being regulated by miR21 in MCF7/ADR cells and its expression being efficiently upregulated by the combination of NLS-Dox and anti-miR21.
In vivo antitumor efficacy
MCF7/ADR tumor-bearing mice were further used to investigate the antitumor efficacy of various HMNs or free DOX in vivo. As shown in Figure 9A, the free Dox solution had a slight inhibitory effect on the growth of MCF7/ADR xenografts. The delivery of either NLS-Dox or anti-miR21 alone using HMNs displayed moderate suppression of tumor growth. By contrast, the combination of NLS-Dox and anti-miR21 codelivered by HMNs had the best antitumor efficacy, showing a tumor-inhibition ratio of 68.5% compared with the PBS control group and prolonged antitumor effect.
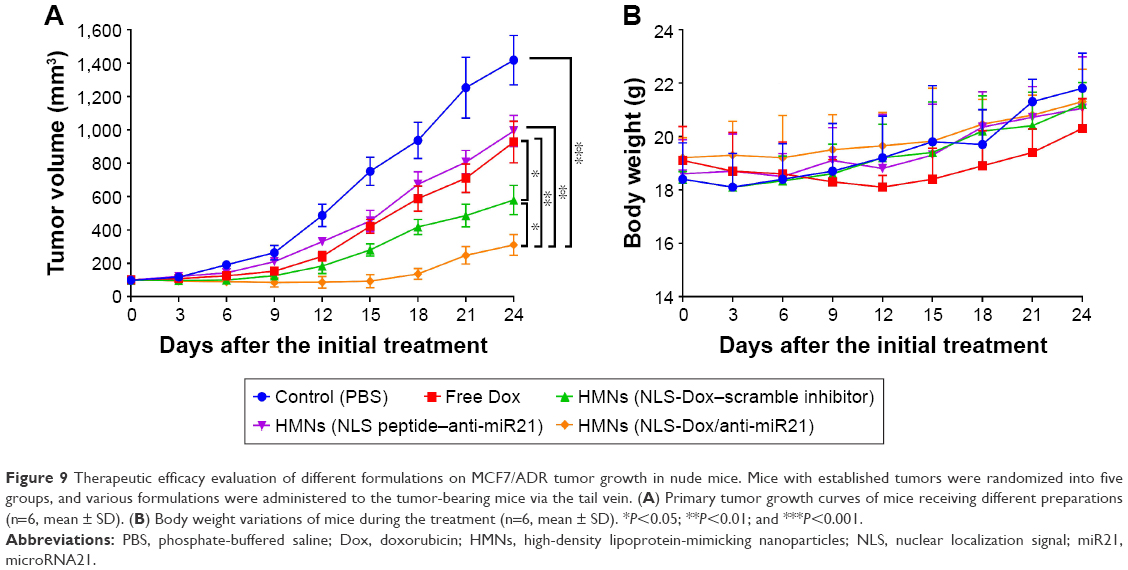 | Figure 9 Therapeutic efficacy evaluation of different formulations on MCF7/ADR tumor growth in nude mice. Mice with established tumors were randomized into five groups, and various formulations were administered to the tumor-bearing mice via the tail vein. (A) Primary tumor growth curves of mice receiving different preparations (n=6, mean ± SD). (B) Body weight variations of mice during the treatment (n=6, mean ± SD). *P<0.05; **P<0.01; and ***P<0.001. |
In addition, in vivo systemic toxicity is a common concern for nanoparticles applied in biomedicine. Body weight changes in tumor-bearing mice were observed during the antitumor study in MCF7/ADR xenografts (Figure 9B). The significant weight loss was only observed in the mice when they were given free Dox solutions, and the fluctuation in body weight might have reflected the severe toxicity of free Dox. Three HMNs groups exhibited similar body weights to the control group, indicating the excellent safety of HMNs as a result of the controllable release of drugs in blood circulation.
Discussion
The current studies described the development of HMNs that could deliver an NLS-Dox conjugate and anti-miR21 simultaneously to reverse drug resistance and enhance chemotherapeutic effects. The cationic NLS peptides not only guided Dox compounds toward cell nuclei but also electrostatically interacted with anti-miR21 for tumor-specific delivery. The cationic dense complexes were precisely enveloped by anionic lipid membranes, in which Apo AI proteins were embedded to simulate the structural and functional properties of natural HDLs. We demonstrated that codelivery of NLS-Dox and anti-miR21 using HMNs provided significant antitumor effects in drug-resistant MCF7/ADR cell lines, as well as in xenografted nude mouse models.
In this work, NLS-Dox had two effects: the conjugate of NLS peptide was used to translocate Dox into the site of action, the nucleus, and prevent it from the drug efflux of Pgp transporters; on the other hand, the cationic NLS-Dox condensed anti-miR21 to form the cores of HMNs, facilitating the coencapsulation of NLS-Dox and anti-miR21 in one nanoparticle. Although the NLS-Dox had only five cationic charged amino acids, the complexation between NLS-Dox and anti-miR21 was completed at a relatively low N:P ratio of 10:1. Nevertheless, we found that the complexes of NLS peptide and anti-miR21 were not as stable as the corresponding NLS-Dox/anti-miR21 complexes at the same N:P ratio of 10:1, resulting in the release of anti-miR21 bands on the gels (data not shown). Since the increased amounts of NLS-peptide produced denser complexes, we observed that absolute complexation was almost completed at an N:P ratio of 20:1. This discrepancy could be explained by the Dox moiety in the NLS-Dox conjugate interacting with anti-miR21 molecules and subsequently going some way toward stabilization of the formed complexes at a low N:P ratio. In this regard, although the N:P ratio of 10:1 provided complete complexation for NLS-Dox and anti-miR21, we chose the N:P ratio of 20:1 as the optimum ratio for the following preparations of coloaded and control HMNs.
Consistently with previous studies,35 the preparation studies showed that the optimization of HMNs needed to balance several components, including NLS-Dox, anti-miR21, and DMPG lipids. The negatively charged DMPG would compete with anti-miR21 for NLS-Dox, leading to the disassembly of the NLS-Dox/anti-miR21 complex. To address this issue, the neutral lipid DOPE was an essential component for the smooth coating of the NLS-Dox/anti-miR21 complex. Due to DOPE’s preference to adopt an inverted hexagonal (HII) phase, the unstable structure of DMPG–DOPE liposomes easily prompted membrane fusion and then facilitated more rapid enveloping of the complexes under electrostatic interaction compared with rigid DMPG liposomes. The results of gel retardation analysis demonstrated that cationic complexes were completely encapsulated into liposomes at a weight ratio of DMPG:anti-miR21 of 40. With further increased DMPG:anti-miR21 ratios, there was no apparent improvement in the preparation of HMNs (data not shown). However, the high cost of DMPG lipids should be taken into consideration, and thus we eventually selected the optimized DMPG:anti-miR21 ratio as 40. In addition to the ease of coating, the fusogenic DOPE has been reported to destabilize the membrane in acidic endosome environment, thereby allowing the release of NLS-Dox and anti-miR21 into the cytoplasm. This hypothesis was verified by intracellular trafficking results (Figure 5), indicating the beneficial effect of DOPE on targeted codelivery of NLS-Dox and anti-miR21 by HMNs.
The optimized HMNs were able to mimic the physicochemical properties of naturally occurring spherical HDL nanoparticles while sustaining high coencapsulation efficiency. The Apo AI proteins not only functionalized nanoparticles with tumor-targeting properties but also stabilized and tightened the nanoparticles. The smaller diameter of the HMNs was mainly attributed to the specific functions of Apo AI proteins compared with that of the corresponding coloaded liposomes (Figure 3A). In addition, the diameter of HMNs was in the range of 40–60 nm, which was in agreement with our previous report.35 In comparison to native HDLs, the slightly larger HMNs were still appropriate substrates for the binding and recognition by HDL receptors on the basis of earlier studies.32,48,49 In addition, the different ζ-potentials of HMNs might reflect the exposed NLS-Dox on the surface of nanoparticles, which was used to evaluate roughly the DMPG-coating efficiency of NLS-Dox and anti-miR21 complexes. According to the results (Figure 3B), the ζ-potential of HMNs gradually decreased when the amount of DMPG was increased, and the negatively charged surface of optimized HMNs indicated the absolute shielding of cationic NLS-Dox/anti-miR21 complexes. As such, by this method, NLS-Dox and anti-miR21 could be easily encapsulated in HMNs and protected from degradation by proteases or RNase in the physiological environment.
Following the optimization of coloaded HMNs, cell-based assays were carried out to evaluate the ability of HMNs that could deliver both drugs simultaneously into the tumor cells. The breast cancer cell line MCF7 and drug-resistant MCF7/ADR were chosen, as MCF7 cells have been reported to express HDL receptors highly and have been applied extensively in evaluations of HDL-inspired nanoparticles.42,50–53 The intrinsic fluorescence of Dox enabled convenient investigation for its uptake and intracellular trafficking mechanism using flow cytometry and CLSM. Most substrates for Pgp were possibly recognized within the lipid bilayers. As shown in Figure 4, the low intracellular concentration of Dox in MCF7/ADR indicated that free Dox was rapidly recognized and in turn pumped out from cells by Pgp transporters when it passively diffused into cell membranes. Unlike free Dox, the NLS-Dox might have been transported into its site of action by the delivery of HMNs through their specific pathway, the sequential release into cytoplasm, and nucleus-targeting effect of conjugated NLS peptides. Therefore, the different uptake mechanisms of NLS-Dox made it unlikely to be identified by Pgp and easily removed from the cytoplasm, resulting in the increased concentration inside drug-resistant cells. Furthermore, it was observed that the drug cargo would influence the uptake pathway of HDL-inspired nanoparticles. The drugs physically loaded into HDL nanoparticles were primarily taken up under the regulation of SRB1 receptors and subsequently directly delivered into cytoplasm.34,54 Alternatively, as shown in Figure 5, the drugs electrostatically interacted with HDL-lipid layers and preferred to enter cells following the endocytosis of whole nanoparticles, which was in accord with our previous report.35 This uptake pattern of HMNs has been discovered to be mediated by a cluster of proteins, including the ectopic β-chain of ATP synthase and P2Y13 receptors;55 nevertheless, the exact mechanism is still under study.
In the in vitro cellular assay, the NLS-Dox alone encapsulated in HMNs showed significantly cytotoxicity to the MCF7/ADR cells than free Dox solution, whereas it displayed slightly weaker antitumor effects on wild-type MCF7 cells compared with free Dox treatment, implying that the conjugation of NLS peptide did not impair the antitumor activity of Dox compounds (Figure 6). On the other hand, anti-miR21 was introduced into cells by HMNs and subsequently corrected the abnormal activity of miR21. On the basis of previous reports,56 the escape of anti-iR21 from endo/lysosomes might be mostly attributed to the fusogenicity of DOPE, which induces fusion with endosomal membranes and the succeeding release of anti-miR21 into cytosol. In addition to DOPE, the cationic NLS peptide has some buffering ability, namely, the “proton-sponge effect”, that might also facilitate the disruption of endosomes to some extent. As a result, anti-miR21 significantly reduced the expression of miRNA and the downstream target protein PTEN (Figure 8). Notably, the growth of MCF7 and its drug-resistant MCF7/ADR cells showed moderate inhibition by anti-miR21 alone compared with either free Dox or other HMN formulations. The complicated network of survival-signaling pathways in tumors might contribute to this ineffectiveness of anti-miR21, and highlights the necessity of combination with chemotherapeutic agents in treatment against drug-resistant cancer cells.
With regard to the mechanism of reversal of drug resistance, previous studies have shown that increased levels of miR21 result in loss of the PTEN tumor suppressor, which is the primary brake of the PI3K–Akt pathway. The activated PI3K–Akt pathway is a major signaling pathway regulating multiple cancer processes, such as cell proliferation, cell growth, and drug resistance. In the present study, we found that the miR21 inhibitor reduced the expression of PTEN, implying that miR21 modulates breast cancer chemoresistance through an miR21/PTEN/PI3K/Akt/drug resistance-related signaling pathway (Figure 8).
Our study evaluated the capacity of the coloaded HMNs to overcome drug resistance and inhibit the growth of drug-resistant cancer cells. We observed that NLS-Dox and anti-miR21 loaded together into one single HMN resulted in the best cytotoxicity to MCF7/ADR and parental MCF7 cells, rather than when these components were applied in a separated manner in the same experiments. In vivo, prolonged regression of tumor growth was also demonstrated in treatment with coloaded HMNs. These results suggested that drug resistance was efficiently reversed under anti-miR21 regulation, and subsequently MCF7/ADR tumor cells were sensitized toward Dox treatment again.
Notably, in vivo administration of free Dox solution showed relatively lower yet partly effective suppression of tumor growth than treatment with NLS-Dox alone or the combination of NLS-Dox and anti-miR21. Nevertheless, the administration of free Dox solution in previous reports has shown negligible antitumor effect in MCF7/ADR-xenografted mice.57,58 This discrepancy might be attributed to the high dosage of free Dox in our studies: 7.6 mg/kg compared with 5 mg/kg or 1 mg/kg in other reports. The increased dosages of free Dox or equivalent NLS-Dox were chosen due to the correlation with the required concentration of anti-miR21, 1 mg/kg, at which efficient inhibiting effect on the expression of miR21 could be obtained on the basis of various studies. Accordingly, free Dox solution at this elevated level slightly inhibited the growth of tumor cells in vivo; nevertheless, Dox widely distributed in the whole body also brought about an obvious loss in body weight, which indicated severe systemic toxicity. Conversely, the equivalent NLS-Dox alone or coencapsulated with anti-miR21 in HMNs did not show any visible side effects, further supporting the safety and target-specific delivery capacity of HMNs.
In the case of HMNs, these nanoparticles were developed on the basis of natural HDL, which is considered excellently biodegradable and biocompatible. With regard to the components we used, neutral DOPE and anionic DMPG have been applied frequently in a variety of liposomes and did not demonstrate significant toxicity.59 In addition, recombinant Apo AI proteins expressed from E. coli were shown to be safe and well tolerated in clinical trials.60 From a regulatory perspective, the current application of DMPG, DOPE, and Apo AI proteins indicates that HMNs can be further developed for clinical application with relatively low safety concerns. Moreover, our results suggest that future studies are warranted to investigate the safety of HMNs in detail, in vivo gene-expression profile related to drug resistance, as well as the biodistribution of HMNs.
Conclusion
In summary, we have herein reported the successful codelivery of an NLS-Dox prodrug and miR21 inhibitor by HMNs to treat Dox-resistant cancer cells. The conjugation of NLS peptide to Dox allows it to complex anti-miR21 and to prompt coencapsulation in HMNs. This codelivery strategy demonstrated significantly higher antiproliferative effects against drug-resistant MCF7/ADR breast cancer cells, but no obvious difference on wild-type MCF7 cells when compared to free Dox solutions. As expected, NLS-Dox evaded the efflux transporter and displayed improved intracellular accumulation in MCF7/ADR cells. Additionally, HMNs effectively delivered anti-miR21 into cells for inhibiting overexpressed miR21 that induced cell apoptosis and sensitized MCF7/ADR cells to chemotherapeutic agents in vitro and in vivo. Coloaded HMNs were also found to inhibit tumor growth remarkably and showed prolonged antitumor activity after cessation of treatment in tumor-bearing nude mice. Taken together, these results suggest that the combination of NLS-Dox and anti-miR21 specifically delivered by HMNs might potentiate their synergistic anticancer effects in drug-resistant breast cancer models and might be developed for clinical application in future.
Acknowledgments
The National Natural Science Foundation of China (81402880, 81373897, and 81602656) is gratefully acknowledged for financial support. This work was also supported by the Natural Science Foundation of Jiangsu Province for Youth (BK20130541), the Jiangsu Province Postdoctoral Science Foundation (1402173C), and the Scientific Research Foundation of Jiangsu University (14JDG163).
Disclosure
The authors report no conflicts of interest in this work.
References
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–726. | ||
Flemming A. Anticancer drugs: finding the perfect combination. Nat Rev Drug Discov. 2015;14(1):13. | ||
Wicki A, Mandalà M, Massi D, et al. Acquired resistance to clinical cancer therapy: a twist in physiological signaling. Physiol Rev. 2016;96(3):805–829. | ||
Eckford PD, Sharom FJ. ABC efflux pump-based resistance to chemotherapy drugs. Chem Rev. 2009;109(7):2989–3011. | ||
Kunjachan S, Rychlik B, Storm G, Kiessling F, Lammers T. Multidrug resistance: physiological principles and nanomedical solutions. Adv Drug Deliv Rev. 2013;65(13–14):1852–1865. | ||
Abbasi M, Lavasanifar A, Uludag H. Recent attempts at RNAi-mediated P-glycoprotein downregulation for reversal of multidrug resistance in cancer. Med Res Rev. 2013;33(1):33–53. | ||
McKeegan KS, Borges-Walmsley MI, Walmsey AR. Structural understanding of efflux-mediated drug resistance: potential routes to efflux inhibition. Curr Opin Pharmacol. 2004;4(5):479–486. | ||
Darby RA, Callaghan R, McMahon RM. P-glycoprotein inhibition: the past, the present and the future. Curr Drug Metab. 2011;12(8):722–731. | ||
Carvalho C, Santos RX, Cardoso S, et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. 2009;16(25):3267–3285. | ||
Aller SG, Yu J, Ward A, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009; 323(5922):1718–1722. | ||
Mao X, Si J, Huang Q, et al. Self-assembling doxorubicin prodrug forming nanoparticles and effectively reversing drug resistance in vitro and in vivo. Adv Healthc Mater. 2016;5(19):2517–2527. | ||
Cheng H, Zhu JY, Xu XD, et al. Activable cell-penetrating peptide conjugated prodrug for tumor targeted drug delivery. ACS Appl Mater Interfaces. 2015;7(29):16061–16069. | ||
Zhang YC, Zhang XW, Liu HM, Cai SH, Wu BJ. Mixed nanomicelles as potential carriers for systemic delivery of Z-GP-Dox, an FAPα-based doxorubicin prodrug: formulation and pharmacokinetic evaluation. Int J Nanomedicine. 2015;10:1625–1636. | ||
Chen F, Gerion D. Fluorescent CdSe/ZnS nanocrystal-peptide conjugates for long-term, nontoxic imaging and nuclear targeting in living cells. Nano Lett. 2004;4(10):1827–1832. | ||
Tkachenko AG, Xie H, Liu Y, et al. Cellular trajectories of peptide-modified gold particle complexes: comparison of nuclear localization signals and peptide transduction domains. Bioconjug Chem. 2004;15(3):482–490. | ||
Bremner KH, Seymour LW, Logan A, Read ML. Factors influencing the ability of nuclear localization sequence peptides to enhance nonviral gene delivery. Bioconjug Chem. 2004;15(1):152–161. | ||
Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133(2):647–658. | ||
Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR-21-mediated tumor growth. Oncogene. 2007;26(19):2799–2803. | ||
Hong L, Han Y, Zhang Y, et al. MicroRNA-21: a therapeutic target for reversing drug resistance in cancer. Expert Opin Ther Targets. 2013;17(9):1073–1080. | ||
Ma J, Dong C, Ji C. MicroRNA and drug resistance. Cancer Gene Ther. 2010;17(8):523–531. | ||
Wang ZX, Lu BB, Wang H, Cheng ZX, Yin YM. MicroRNA-21 modulates chemosensitivity of breast cancer cells to doxorubicin by targeting PTEN. Arch Med Res. 2011;42(4):281–290. | ||
van der Aa MA, Koning GA, d’Oliveira C, et al. An NLS peptide covalently linked to linear DNA does not enhance transfection efficiency of cationic polymer based gene delivery systems. J Gene Med. 2005;7(2):208–217. | ||
Nagasaki T, Myohoji T, Tachibana T, Futaki S, Tamagaki S. Can nuclear localization signals enhance nuclear localization of plasmid DNA? Bioconjug Chem. 2003;14(2):282–286. | ||
Li Y, Liu RY, Yang J, Ma GH, Zhang ZZ, Zhang X. Dual sensitive and temporally controlled camptothecin prodrug liposomes codelivery of siRNA for high efficiency tumor therapy. Biomaterials. 2014;35(36):9731–9745. | ||
Andey T, Marepally S, Patel A, et al. Cationic lipid guided short-hairpin RNA interference of annexin A2 attenuates tumor growth and metastasis in a mouse lung cancer stem cell model. J Control Release. 2014;184:67–78. | ||
Deng ZJ, Morton SW, Ben-Akiva E, Dreaden EC, Shopsowitz KE, Hammond PT. Layer-by-layer nanoparticles for systemic codelivery of an anticancer drug and siRNA for potential triple-negative breast cancer treatment. ACS Nano. 2013;7(11):9571–9584. | ||
Gao SQ, Tian HY, Guo Y, et al. miRNA oligonucleotide and sponge for miRNA-21 inhibition mediated by PEI-PLL in breast cancer therapy. Acta Biomater. 2015;25:184–193. | ||
Li W, Peng J, Tan L, et al. Mild photothermal therapy/photodynamic therapy/chemotherapy of breast cancer by Lyp-1 modified docetaxel/IR820 co-loaded micelles. Biomaterials. 2016;106:119–133. | ||
Kumar V, Mondal G, Slavik P, Rachagani S, Batra SK, Mahato RI. Codelivery of small molecule Hedgehog inhibitor and miRNA for treating pancreatic cancer. Mol Pharm. 2015;12(4):1289–1298. | ||
Thaxton CS, Rink JS, Naha PC, Cormode DP. Lipoproteins and lipoprotein mimetics for imaging and drug delivery. Adv Drug Deliv Rev. 2016;106(Pt A):116–131. | ||
Simonsen JB. Evaluation of reconstituted high-density lipoprotein (rHDL) as a drug delivery platform: a detailed survey of rHDL particles ranging from biophysical properties to clinical implications. Nanomedicine. 2016;12(7):2161–2179. | ||
Lacko AG, Sabnis NA, Nagarajan B, McConathy WJ. HDL as a drug and nucleic acid delivery vehicle. Front Pharmacol. 2015;6:247. | ||
Rui M, Tang H, Li Y, Wei X, Xu Y. Recombinant high density lipoprotein nanoparticles for target-specific delivery of siRNA. Pharm Res. 2013;30(5):1203–1214. | ||
Kingwell BA, Chapman MJ, Kontush A, Miller NE. HDL-targeted therapies: progress, failures and future. Nat Re Drug Discov. 2014;13(6):445–464. | ||
Duffy D, Rader DJ. Emerging therapies targeting high-density lipoprotein metabolism and reverse cholesterol transport. Circulation. 2006;113(8):1140–1150. | ||
Zamanian-Daryoush M, DiDonato JA. Apolipoprotein A-I and cancer. Front Pharmacol. 2015;6:265. | ||
Kuzu OF, Noory MA, Robertson GP. The role of cholesterol in cancer. Cancer Res. 2016;76(8):2063–2070. | ||
Llaverias G, Danilo C, Mercier I, et al. Role of cholesterol in the development and progression of breast cancer. Am J Pathol. 2011;178(1):402–412. | ||
Kim J, Lee KB, Park JW. Expression of a HDL receptor scavenger receptor class B, type I in prostate cancer cells. Biol Reprod. 2008;78 Suppl 1:108. | ||
Danilo C, Gutierrez-Pajares JL, Mainieri MA, Mercier I, Lisanti MP, Frank PG. Scavenger receptor class B type I regulates cellular cholesterol metabolism and cell signaling associated with breast cancer development. Breast Cancer Res. 2013;15(5):R87. | ||
Oda MN, Bielicki JK, Berger T, Forte TM. Cysteine substitutions in apolipoprotein A-I primary structure modulate paraoxonase activity. Biochemistry. 2001;40(6):1710–1718. | ||
Ryan RO, Forte TM, Oda MN. Optimized bacterial expression of human apolipoprotein A-I. Protein Expr Purif. 2003;27(1):98–103. | ||
Forte TM, Nordhausen RW. Electron microscopy of negatively stained lipoproteins. Methods Enzymol. 1986;128:442–457. | ||
Devulapally R, Sekar TV, Paulmurugan R. Formulation of anti-miR-21 and 4-hydroxytamoxifen co-loaded biodegradable polymer nanoparticles and their antiproliferative effect on breast cancer cells. Mol Pharm. 2015;12(6):2080–2092. | ||
Ma X, Kumar M, Choudhury SN, et al. Loss of the miR-21 allele elevates the expression of its target genes and reduces tumorigenesis. Proc Natl Acad Sci U S A. 2011;108(25):10144–10149. | ||
Wang RN, Gu XC, Zhou JP, et al. Green design “bioinspired disassembly-reassembly strategy” applied for improved tumor-targeted anticancer drug delivery. J Control Release. 2016;235:134–146. | ||
Luthi AJ, Lyssenko NN, Quach D, et al. Robust passive and active efflux of cellular cholesterol to a designer functional mimic of high density lipoprotein. J Lipid Res. 2015;56(5):972–985. | ||
El Roz A, Bard JM, Huvelin JM, Nazih H. LXR agonists and ABCG1-dependent cholesterol efflux in MCF-7 breast cancer cells: relation to proliferation and apoptosis. Anticancer Res. 2012;32(7):3007–3013. | ||
Cao WM, Murao K, Imachi H, et al. A mutant high-density lipoprotein receptor inhibits proliferation of human breast cancer cells. Cancer Res. 2004;64(4):1515–1521. | ||
Kader A, Pater A. Loading anticancer drugs into HDL as well as LDL has little affect [sic] on properties of complexes and enhances cytotoxicity to human carcinoma cells. J Control Release. 2002;80(1–3):29–44. | ||
Zhang F, Wang X, Xu X, Li M, Zhou J, Wang W. Reconstituted high density lipoprotein mediated targeted co-delivery of HZ08 and paclitaxel enhances the efficacy of paclitaxel in multidrug-resistant MCF-7 breast cancer cells. Eur J Pharm Sci. 2016;92:11–21. | ||
Connelly MA, Williams DL. Scavenger receptor BI: a scavenger receptor with a mission to transport high density lipoprotein lipids. Curr Opin Lipidol. 2004;15(3):287–295. | ||
Martinez LO, Najib S, Perret B, Cabou C, Lichtenstein L. Ecto-F1-ATPase/P2Y pathways in metabolic and vascular functions of high density lipoproteins. Atherosclerosis. 2015;238(1):89–100. | ||
Dominska M, Dykxhoorn DM. Breaking down the barriers: siRNA delivery and endosome escape. J Cell Sci. 2010;123(8):1183–1189. | ||
Han M, Lv Q, Tang XJ, et al. Overcoming drug resistance of MCF-7/ADR cells by altering intracellular distribution of doxorubicin via MVP knockdown with a novel siRNA polyamidoamine-hyaluronic acid complex. J Control Release. 2012;163(2):136–144. | ||
Shieh MJ, Hsu CY, Huang LY, Chen HY, Huang FH, Lai PS. Reversal of doxorubicin-resistance by multifunctional nanoparticles in MCF-7/ADR cells. J Control Release. 2011;152(3):418–425. | ||
Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomedicine. 2012;7:49–60. | ||
Kuai R, Li D, Chen YE, Moon JJ, Schwendeman A. High-density lipoproteins (HDL): nature’s multi-functional nanoparticles. ACS Nano. 2016;10(3):3015–3041. |
Supplementary material
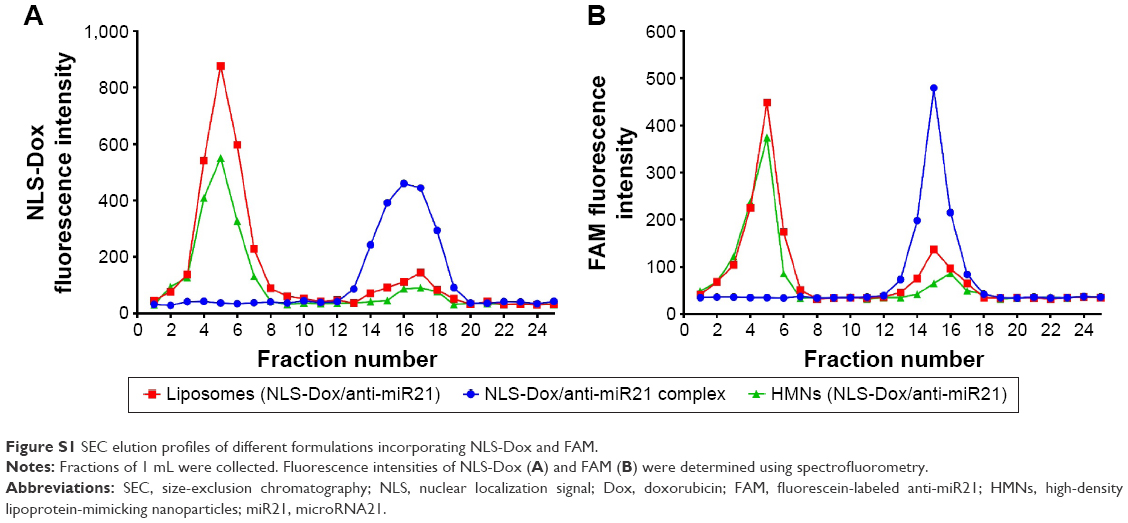 | Figure S1 SEC elution profiles of different formulations incorporating NLS-Dox and FAM. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.