")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 7
Simeprevir for the treatment of hepatitis C virus infection
Authors Izquierdo L, Helle F, François C, Castelain S, Duverlie G, Brochot E
Received 16 May 2014
Accepted for publication 12 June 2014
Published 14 August 2014 Volume 2014:7 Pages 241—249
DOI https://doi.org/10.2147/PGPM.S52715
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Laure Izquierdo,1 François Helle,1 Catherine François,1,2 Sandrine Castelain,1,2 Gilles Duverlie,1,2 Etienne Brochot1,2
1Virology Research Unit, Jules Verne University of Picardie, 2Department of Virology, Amiens University Hospital, Amiens, France
Abstract: Simeprevir (TMC435, Olysio™), a second-generation hepatitis C virus (HCV) protease inhibitor, has been recently approved for the treatment of genotype 1 chronic hepatitis C in combination with pegylated interferon and ribavirin. This molecule has very different characteristics from first-generation protease inhibitors. Results from trials show that simeprevir is highly effective and safe, with few adverse events. We discuss the specific features of this new treatment option for HCV infection, in terms of in vitro data, pharmacological data, and clinical trials. We also discuss the impact of Q80K polymorphism at baseline. Studies evaluating interferon-free regimens with simeprevir are ongoing. Future combinations of two or more direct-acting antiviral agents, targeting different viral enzymes and with synergistic antiviral effects, will be approved, allowing treatment of pan-genotypic HCV with optimized sustained virologic responses. Simeprevir will undoubtedly be part of future treatment strategies.
Keywords: simeprevir, protease inhibitor, direct-acting antiviral agent, hepatitis C virus
Introduction
Hepatitis C constitutes a worldwide health problem, as more than 150 million people are infected with the hepatitis C virus (HCV).1 The great majority of HCV infections are accompanied by chronic liver disease, leading to fibrosis and cirrhosis, with a risk of complications including ascites and hepatocellular carcinoma. Over recent decades, the standard treatment for HCV genotype 1 chronic infection has been a combination of pegylated interferon (PEG-IFN) and ribavirin (RBV) for 48 weeks or longer. However, this treatment was limited by the low sustained virologic response (SVR) rate, the long duration of treatment, and adverse events. The recent development of direct-acting antiviral (DAA) agents has changed the optimal treatment regimen. The two first-generation NS3/4A protease inhibitors, telaprevir2,3 and boceprevir,4,5 were approved in 2011 for the treatment of genotype 1-infected patients in combination with PEG-IFN and RBV. Simeprevir (TMC435, Olysio™; Janssen Therapeutics, Titusville, NJ, USA) is the third oral protease inhibitor to be approved for use in combination with PEG-IFN and RBV for the treatment of chronic HCV genotype 1 infection in adults with compensated liver disease. While telaprevir and boceprevir belong to the class of a-ketoamide electrophilic trap-containing inhibitors, simeprevir belongs to the macrocyclic class of protease inhibitors.
This review is designed to summarize in vitro, pharmacological, safety profile, and clinical data for simeprevir in terms of improved SVR rates for the treatment of genotype 1 HCV infection.
HCV genotype
Geographic differences in the overall prevalence and distribution of the seven HCV genotypes are well recognized. The occurrence of particular HCV genotypes is different in particular geographic regions of the world. HCV genotypes 1, 2, and 3 are distributed globally, found in the US, South America, Europe, Australia, and Eastern Asia (People’s Republic of China, Japan, and Taiwan). In Japan, the prevalence of subtype 1b is higher (70%) than that of subtype 2a (20%), and the remainder of the population is infected with genotype 2b or other genotypes.6 Genotype 4 is mainly found in Egypt, the Middle East, and Central Africa, whereas genotype 5 is particularly common in Southern Africa. Hepatitis C genotype 6 is endemic in Southeast Asia, in countries such as Vietnam, Thailand, and Myanmar, and is predominant in some countries. However, in a context of globalization and migration of populations, some genotypes are emerging in Western countries; for instance, genotype 4 has recently been reported in France, and genotype 6 has been reported in the US, Canada, and Germany.7
Moreover, the outcome of therapy depends on multiple viral factors such as genotype. HCV genotype represents a strong and independent prognostic factor of treatment efficacy, determining the type and duration of anti-HCV therapy. SVR, defined as undetectable HCV RNA 24 weeks after the end of treatment with PEG-IFN and RBV, can be achieved by 40%–50% of patients infected with HCV genotype 1 and >70% of patients infected with HCV genotypes 2 or 3 in response to PEG-IFN and RBV combination therapy.
HCV genotype 1, which remains the most widespread genotype, has been the focus of most experimental and clinical studies. The dual therapy currently available for chronic HCV infection, consisting of PEG-IFN and RBV, does not provide very high response rates and requires long-term therapy. The addition of DAA agents to the standard of care could improve cure rates and shorten treatment duration.
Overview of the DAA agents market
In the past, optimal treatment for chronic genotype 1 HCV infection was based on PEG-IFN and RBV. However, this therapy presents a number of limitations, such as low SVR rates (40%–50%) and significant adverse effects, which lead to discontinuation of therapy and limit their clinical use.8,9
Major progress has recently transformed the standard treatment regimen for HCV genotype 1 infection, including the availability of DAA agents.10 Each step of the viral cycle can now be targeted by anti-HCV drugs, and all viral enzymes (NS2 and NS3/4 proteases, their cofactor NS4A, NS5A replication complex, and NS5B RNA-dependent RNA polymerase) are potential drug targets. Four major classes of DAA agents, including NS3/A4 protease inhibitors, NS5B polymerase nucleos(t)ide analogs, NS5B polymerase nonnucleoside analogs, and NS5A inhibitors, are under development. Two inhibitors of the HCV NS3/A4 serine protease have currently been approved by the US Food and Drug Administration for the treatment of HCV genotype 1 infection. In 2011, these two first-generation DAA agents, telaprevir and boceprevir, were registered in the US and Europe. The second-generation of NS3/4A protease inhibitors is still being developed, including simeprevir which was approved in 2013. Other new molecules with different viral enzyme targets are also currently being evaluated in clinical trials.11
HCV protease
Boceprevir, telaprevir, and simeprevir specifically target HCV viral replication by inhibiting the HCV NS3/4A protease and are therefore termed “protease inhibitors”. HCV NS3/4A protease is a trypsin-like serine protease, which is essential for the generation of components of the viral RNA replication complex. This protease is responsible for cleaving the polyprotein downstream of the NS3 site, allowing the release of functional proteins and the generation of mature proteins.12 The enzyme is also responsible for cleaving two cellular proteins in the interferon (IFN) cascade, thus blocking the cellular IFN induction pathway.13 These two host proteins are mitochondrial antiviral-signaling proteins (otherwise known as IPS-1, VISA, or Cardif) and toll/interleukin-1 receptor (TIR)-domain-containing adaptor-inducing IFN-β, which are essential adaptor proteins that initiate signaling leading to activation of IFN regulatory factor 3 and IFN-α/β synthesis.14 However, NS3/4A protease inhibitors can restore IFN regulatory factor 3 signaling in HCV-infected cells but only at concentrations far in excess of the antiviral half-maximal effective concentration (EC50). These inhibitors may therefore exert two distinct effects: blocking viral replication by inhibiting viral polyprotein processing, and restoring IFN-signaling pathways that are thought to be disrupted by NS3/4A.
In clinical studies, NS3/4A inhibitors are conventionally grouped into two classes. The first class is that of linear peptidomimetics with ketoamide incorporating a reactive electrophilic ketoamide at the cleavage site that targets the catalytic Ser139 of the active site of the enzyme via a fully reversible mechanism. Examples of this class include telaprevir and boceprevir. The second class is a competitive, reversible, macrocyclic, noncovalent inhibitor. Macrocycles are useful to improve affinity and selectivity for protein targets, while preserving sufficient bioavailability characteristics of small molecules. Simeprevir belongs to this class. The antiviral activity of simeprevir is mediated by noncovalent binding to HCV protease with rapid association and slow dissociation.
Simeprevir
Global data
Simeprevir is a single pill, oral HCV NS3/4A protease inhibitor taken once daily with food, currently approved for the treatment of patients with HCV genotype 1 infection with compensated liver disease. It is classified as a second-generation protease inhibitor with a macrocyclic structure, presenting an advantage in terms of binding affinity and specificity for NS3 protease compared to first-generation protease inhibitor with a linear structure. Due to the difference in structure, the drug resistance profile is somewhat different from that of telaprevir.15 In all patients, treatment with Olysio should be initiated in combination with PEG-IFN-α and RBV and should be administered for 12 weeks. All treatment-naïve and prior relapser patients, including those with cirrhosis, should receive an additional 12 weeks of PEG-IFN-α and RBV after completing 12 weeks of treatment with Olysio. All prior nonresponder patients (including partial and null-responders), including those with cirrhosis, should receive an additional 36 weeks of PEG-IFN-α and RBV after completing 12 weeks of treatment with Olysio, PEG-IFN-α, and RBV (total treatment duration of 48 weeks).
In vitro studies
Available data from biochemical protease assays indicate that simeprevir exhibits potent inhibition of NS3/4A protease of genotypes 1a, 1b, 2, 4, 5, and 6, with a medium inhibitory concentration (IC50) <13 nM for all HCV NS3/4A enzymes tested. However, IC50 for genotype 3 was 37 nM.16 It is suggested that this may be due to the presence of a naturally occurring D168Q polymorphism at baseline, which is present in most genotype 3a isolates. A D168Q mutation alone has been shown to reduce simeprevir activity in a genotype 1b replicon assay by >700-fold.17 In replicon models, different genotype 1a and 1b replicons showed EC50 for simeprevir ranging from 8 nM to 28 nM and half-maximal cytotoxicity concentration values greater than 16 μmol/L for various human cell lines (selectivity indexes greater than 500).16 Activity of simeprevir against a selection of about 140 genotype 1a and genotype 1b chimeric replicons carrying NS3 sequences derived from HCV NS3/4A protease-inhibitor-naïve subjects resulted in a median fold change in EC50 values of 1.4 (interquartile range: 0.8 to 11) and 0.4 (interquartile range: 0.3 to 0.7) compared to reference genotype 1b replicon, respectively. Still, in the replicon model, simeprevir was synergistic with alpha IFN and an NS5B inhibitor and additive with RBV.16
In preclinical studies, simeprevir was found to be more than 1,000-fold less active against 20 other human proteases, including trypsin and human leukocyte elastase, than against HCV NS3/4A protease, suggesting good safety of this molecule.18 However, in vitro studies have identified that simeprevir is an inhibitor of bilirubin transporters OATP1B1 and MRP2, suggesting a likely mechanism for the elevations of serum bilirubin observed in clinical studies.19 Bilirubin clearance from the blood is principally a three-step process involving bilirubin uptake (anion transporter OATP1B1), bilirubin conjugation (enzyme UGT1A1), and subsequent transportation into the bile (transporter MRP2). Simeprevir is a more potent inhibitor of OATP1B1 (median inhibitory concentration: 720 nM), which is primarily responsible for transporting unconjugated bilirubin, than MRP2 (median inhibitory concentration around 10,000 nM), primarily a conjugated bilirubin transporter.
Pharmacology
Absorption
Simeprevir has a relatively long absorption phase, reaching maximum concentration (Cmax) after 4–6 hours. After multiple doses for 5 days, Cmax and area under the plasma concentration-time curve (AUC) after 24 hours (AUC24h) increased more than dose proportionally between 75 mg and 200 mg once daily, suggesting saturation of first-pass metabolism and/or efflux transporters.20 The AUC 24h for the 200 mg once daily dose group was approximately 10 times higher than that for the 100 mg once daily dose. Steady-state is reached after 7 days of once daily dosing.21 Exposure to simeprevir in HCV-uninfected subjects with Child–Pugh B and C cirrhosis was 2.4-fold and 5.2-fold higher, respectively, than in HCV uninfected subjects with normal liver function.20 Plasma Cmax and AUC24h of simeprevir were similar during coadministration of simeprevir with PEG-IFN-α and RBV compared with administration of simeprevir alone. In HCV-infected subjects, the mean steady-state plasma concentration was 1,936 ng/mL, more than 200-fold higher than the EC50 value determined in previous in vitro studies. While plasma exposure dropped to around the EC50 at 24 hours postdosing, the liver concentration remained above the replicon 99% effective concentration (EC99) for up to 31 hours postdosing, thus suggesting the feasibility of once-daily dosing.22 Moreover, in a Phase I study, it was shown that simeprevir exposure was higher in healthy Japanese volunteers than in Caucasian volunteers.23 In Phase III trials, mean plasma simeprevir exposure in Asian subjects (n=14) was 3.4-fold higher than in the general population of this trial. The AUC24h of simeprevir was increased by 61%–69% when administered with food; simeprevir should therefore be taken with food. Finally, simeprevir is a substrate and inhibitor of P-glycoprotein.21
Distribution
Simeprevir is extensively (99.9%) bound to plasma proteins, mainly to albumin. The absolute bioavailability was 44% after a single oral administration. Transport into human hepatocytes is thought to be mediated by OATP1B1/3. In rats, a liver to blood ratio of 29:1 was found, which would mean good distribution to the liver.21 For humans, in preclinical studies, the liver to plasma concentration ratio was high (ratio of 39).16 The highest tissue/plasma AUC ratios were observed in the small intestine (ratio of 128). While tissue simeprevir concentrations reached peak values within 4 hours postdosing, simeprevir concentrations in liver remained above the EC99 for up to 31 hours postdosing, and plasma concentrations were higher than the EC99 at 8 hours and around the EC50 at 24 hours postdosing.16
Metabolism
Simeprevir, like telaprevir and to a lesser extent boceprevir, is metabolized by CYP3A4. It can therefore be the subject of drug–drug interactions with moderate or strong inhibitors and inducers of CYP3A enzymes, with significant increases or decreases in exposure to simeprevir. Unlike boceprevir and telaprevir, simeprevir is an inhibitor of gut cytochrome 3A4 but not hepatic CYP3A4.22 The effect of a low dose (600 mg) of a potent CYP3A inducer, rifampicin, on the pharmacokinetics of simeprevir was evaluated and showed that the combination of rifampicin and simeprevir resulted in a 48% decrease in AUC24h while Cmax was increased by 31%.24 Although simeprevir exposure in subjects with moderate hepatic impairment was higher than in healthy subjects, no dose adjustments are required in patients with moderate hepatic impairment.
Excretion
Simeprevir is eliminated by biliary excretion. After a single dose of 200 mg of simeprevir, approximately 91% of total radioactivity was recovered in feces and less than 1% in urine,21 indicating that simeprevir is eliminated from the body via biliary excretion and renal excretion is irrelevant. The elimination half-life in HCV-infected patients was 41 hours, which is almost 3–4 times longer than that in HCV-uninfected individuals. The pharmacokinetic parameters of simeprevir were also not influenced by renal function and no dose adjustments are necessary in patients with mild, moderate, or severe renal impairment. Safety and efficacy, however, have not been studied in patients with end-stage renal disease or on hemodialysis.
Pharmacodynamics
There was no clear pharmacokinetic/pharmacodynamic relationship between simeprevir exposure and antiviral activity with simeprevir doses of 75 mg once daily (QD) or above. Within the range of exposures to simeprevir in Phase III trials, no clear exposure–response relationships for efficacy (rapid virologic response [RVR], SVR, viral breakthrough [VBT] or relapse) were observed.25 During clinical trials with simeprevir, higher exposures to simeprevir have been associated with increased frequency of adverse reactions, including rash and photosensitivity.
Resistance profile
Due to the difference in structure, the drug resistance profile is slightly different from that of first-generation protease inhibitors.
In vitro
In in vitro studies, where resistance profiles were studied with Huh7 cells and the replicon model,26 mutations at positions 43, 80, 155, 156, and 168 alone or in combination were also identified. A substitution at D168 accounted for 80% of sequences. The D168A mutation was most commonly detected in genotype 1a replicons, and the D168V mutation was most commonly detected in genotype 1b replicons. The EC50 was around 2,000-fold higher for replicons with the D168V mutation compared to the wild-type EC50. The R155K mutation was not identified in genotype 1b replicon colonies but was identified in several genotype 1a colonies, which can be partly explained by the fact that two nucleotide changes are required for this mutation in genotype 1b virus. Other substitutions such as Q80G or L, S122G, N, or T did not reduce susceptibility to simeprevir.
In the JFH-1/Huh7 model, sequencing of NS3/4A derived from cells harvested at multiple time points between passages 12 and 19 resulted in the continuous detection of mutations D168N and F43V in cells treated with 500 nM and 4,000 nM simeprevir, respectively.26 Interestingly, the mutations observed in the genotype 2a JFH-1 genome were located at the same positions as those identified in the genotype 1 replicon systems.
Among the various positions identified, mutations at residue Q80 had an impact on the activity of simeprevir, with a less than 10-fold change (7.7) of EC50. This Q80K variant, naturally present in the 1a replicon, had a limited effect on simeprevir activity. However, the resistance barrier for Q80K-carrying variants appears to be lower and facilitates the emergence of additional mutations, resulting in higher treatment failure rates.
Interestingly, in a study evaluating the susceptibilities of 39 baseline clinical isolates determined by a transient-replication assay on a panel of protease inhibitors,27 a number of samples were less sensitive to simeprevir but more sensitive to boceprevir and telaprevir and vice versa. All these samples contained the Q80K polymorphism. These data confirmed that these two classes of HCV protease inhibitors may interact with the HCV protease differently, although they all bind to the same active site.
Clinical studies
In the Phase IIb PILLAR (Protease Inhibitor TMC435 trial assessing the optimaL dose and duration as once daiLy Anti-viral Regimen) study evaluating simeprevir with PEG-IFN and RBV in treatment-naïve genotype 1 patients, all simeprevir-treated patients who experienced VBT and 27 of the 33 patients with viral relapse presented emerging mutations at NS3 amino acid positions 80, 155, and/or 168 at the time of VBT or relapse.28 Differences in the type of emerging mutations were observed between genotype 1a- and 1b-infected patients. For genotype 1a, R155K was detected, either alone or in combination with other mutations at NS3 positions 80 and/or 168 while D168V was the mutation most commonly observed for genotype 1b. In another Phase IIb trial conducted in HCV genotype 1-infected patients who failed to respond to previous PEG-IFN-α/RBV treatment (ASPIRE [Antiviral STAT-C Protease Inhibitor Regimen in Experienced patients]),29 VBT occurred in 42 patients (10.6%). Previously described mutations emerged in 41 of 42 patients at the time of failure, and D168V was primarily observed in genotype-1b isolates while R155K was mainly observed in genotype 1a. Approximately the same data were observed in Phase III trials.30–32 Although simeprevir shows cross-resistance with telaprevir at amino acid positions 155 and 156, most of the resistant mutations, therefore, occur at the simeprevir-specific amino acid position of 168.
A study conducted 18 months later evaluated the presence of mutation variants in five patients previously treated with 200 mg simeprevir for 5 days.33 No variants were detected by virus population sequencing, but low levels of persistence resistant variants were detected when using deep sequencing.
The observed prevalence of NS3 Q80K polymorphic variants at baseline in the overall population of the Phase IIb and Phase III trials was 14% while the observed prevalence of the Q80K polymorphism was 30% in subjects infected with HCV genotype 1a and 0.5% in subjects infected with HCV genotype 1b.28 In the US, the prevalence of this mutation in genotype 1 was higher (around 40%–50%), particularly as the proportion of genotype 1a is also higher. In subjects with HCV genotype 1a with a baseline Q80K amino acid substitution, an emerging R155K substitution was observed most frequently at failure.
Clinical trials
Phase II and III trials with simeprevir
Simeprevir associated with PEG-IFN and RBV has been evaluated for the treatment of HCV genotype 1 patients in four Phase II trials: two Phase IIa trials, DRAGON (Dose and duration Ranging study of Antiviral agent TMC435 in Genotype One HCV treatment-Naïve patients)6 in Japan and OPERA-1 (Optimal Protease inhibitor Enhancement of Response to TherApy 1) in Europe,34 and two international Phase IIb trials, PILLAR28 and ASPIRE.29 Data are available for six Phase III trials conducted in naïve or experienced HCV genotype 1 patients in combination with PEG-IFN and RBV: three trials carried out in Japan, CONCERTO-1,35 CONCERTO-2, and CONCERTO-3;36 and three international trials, QUEST 1 (with PEG-IFN 2a), QUEST 2 (with PEG-IFN 2b),31,32 and PROMISE (PROtease inhibitor TMC435 In patientS who have previously rElapsed on IFN/RBV).30 It is not surprising that many trials are conducted in Japan, not because the pharmacokinetics of the molecule are different in this population as mentioned above, but rather because of the higher proportions of genotype 1b in this country compared to the US or Europe.37 The main findings of these Phase III studies are summarized in Table 1. The simeprevir dose was 100 mg QD for studies conducted in Japan but 150 mg QD for the other studies. Overall, SVR12 (undetectable HCV RNA in serum 12 weeks after the end of treatment) rates were clearly superior for patients treated with simeprevir in each study compared to the placebo arm with PEG-IFN and RBV (Table 1). About 90% of patients are eligible for short treatment duration (24 weeks) with response-guided therapy. The relapse and VBT rates with simeprevir were very low. In the ASPIRE trial, VBT generally occurred before week 12, indicating that it would not be prevented by more prolonged treatment with simeprevir. It should be noted that no data are available from studies conducted outside Japan in nonresponders to prior treatment. The CONCERTO-2 study conducted in this population in Japan has reported poor results, with an SVR rate of 44.3%. The value of simeprevir in combination with PEG-IFN and RBV in this population therefore needs to be reviewed.
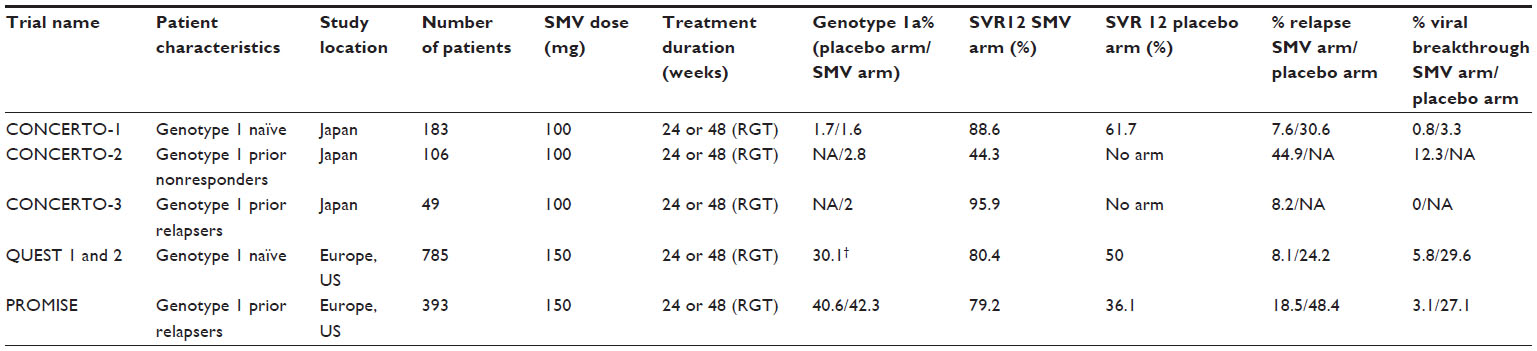 | Table 1 Data from Phase III trials conducted in HCV genotype 1 patients with simeprevir in combination with pegylated interferon and ribavirin |
Q80K mutation
In “Highlights of prescribing information” for Olysio,21 it is recommended to screen patients with HCV genotype 1a infection for the presence of virus with the NS3 Q80K polymorphism at baseline. Alternative therapy should be considered for patients infected with HCV genotype 1a containing the Q80K polymorphism. In Phase III studies, there was no clear benefit for genotype 1a HCV-infected patients with a baseline Q80K mutation treated with simeprevir/PEG-IFN/RBV compared to the Phase III studies concerning SVR rates in the simeprevir arm (total SVR, SVR for HCV genotype 1a with Q80K mutation at baseline, and HCV genotype 1a without Q80K mutation at baseline) and placebo arm receiving PEG-IFN and RBV combination therapy (Figure 1). Studies conducted in Japan with a very low proportion of patients with genotype 1a were obviously not included in this analysis.
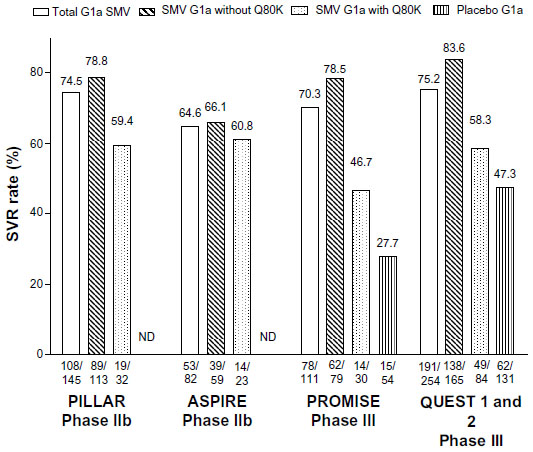 | Figure 1 SVR rates in genotype 1a patients treated with simeprevir in Phase II and III trials. |
Phase III comparison with other DAA agents
Comparison of all SVR data from Phase III randomized, placebo-controlled trials conducted with boceprevir, telaprevir, sofosbuvir, and simeprevir according to virologic characteristics (genotype, viral load) and patient characteristics (IL28B polymorphism, stage of fibrosis) provides a number of conclusions concerning HCV genotype 1 patients. Firstly, simeprevir achieved higher SVR rates in these trials compared to boceprevir and telaprevir but lower rates compared to sofosbuvir in combination with PEG-IFN and RBV. Simeprevir achieved SVR rates equivalent to those obtained with sofosbuvir in treatment-naïve IL28B CC patients (95% and 98%, respectively) and treatment-naïve HCV genotype 1b patients (85% and 82%, respectively). Secondly, simeprevir moderately improved SVR rates compared to telaprevir in treatment-naïve patients with F3–F4 fibrosis (69% and 62%, respectively) and patients with HCV genotype 1a infection (75% and 71%, respectively). In this setting, the drug regimen combining IFN, RBV, and simeprevir is therefore indicated for the treatment of HCV genotype 1 infection. Data on baseline characteristics and treatment responses of naïve and nonnaïve patients included in Phase III study arms with boceprevir, telaprevir, and simeprevir are compiled in Table 2.
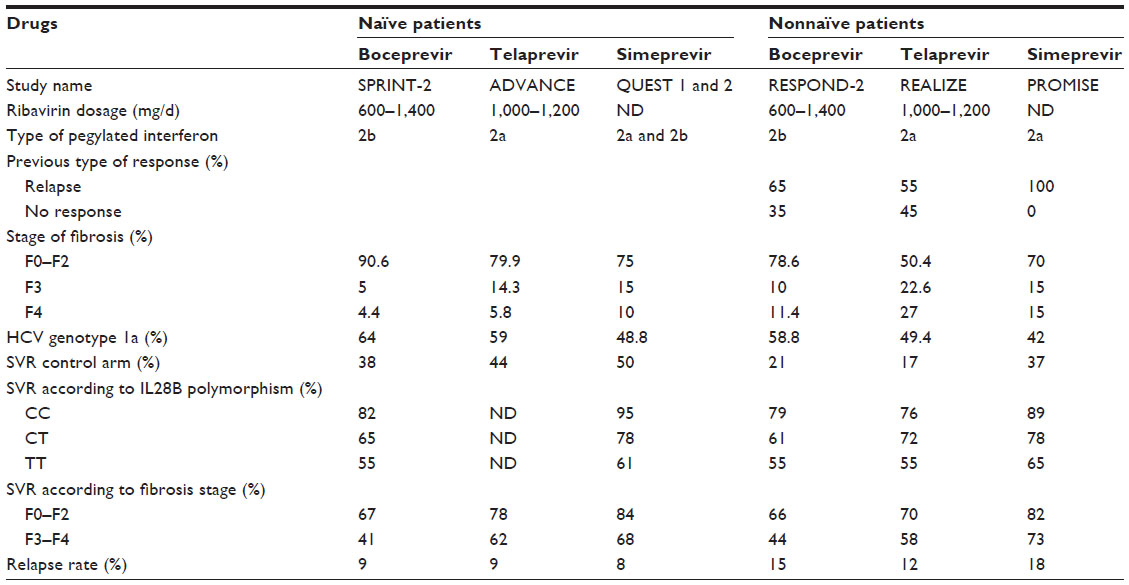 | Table 2 Baseline characteristics and treatment responses of naïve and nonnaïve patients included in Phase III studies arm with boceprevir, telaprevir, and simeprevir |
Adverse events
The safety profile of simeprevir was generally acceptable. The major adverse events described during Phase II/III trials of simeprevir in combination with PEG-IFN and RBV were rash, photosensitivity, pruritus, and nausea. The most common adverse events were associated with PEG-IFN and RBV therapy, including fatigue, influenza-like illness, pruritus, headache, and nausea. An increased frequency and severity of hyperbilirubinemia was associated with simeprevir use. However, no association between elevated bilirubin and clinically relevant hepatotoxicity was demonstrated.30,34 This adverse effect is related to inhibition of bilirubin transporters OATP1B1 and MRP2 by simeprevir,19 causing blockage of bilirubin clearance. Bilirubin levels returned to their baseline values at the end of simeprevir therapy.38 In contrast to first-generation protease inhibitor regimens including boceprevir and telaprevir, no increased frequency of anemia, neutropenia, or skin manifestations was reported. In the pooled Phase III analysis, 14 subjects (2%) in the simeprevir arm and five subjects (1%) in the control arm experienced at least one adverse event leading to discontinuation of simeprevir. In the Phase IIb ASPIRE trial, neutropenia was reported more frequently in the simeprevir arm than in the placebo arm (25.8% versus 16.7%). Nevertheless, as demonstrated in the Compassionate Use of Protease Inhibitors in viral C Cirrhosis (CUPIC) cohort study in France with boceprevir and telaprevir, simeprevir safety data would be reassessed after several months of use in heterogeneous patient populations.39
Conclusion
The challenges for protease inhibitors including simeprevir and new HCV treatment options are to optimize SVR rates in combination with standard care while at the same time, shortening treatment duration. To date, the safety, efficacy, and pharmacokinetics of the newer drugs have not been established in children, and the drugs are not approved for use in patients younger than 18 years of age.
Simeprevir differs from the other first-generation protease inhibitors in that it is administered once daily and has a different tolerability and resistance profile. This second wave of IFN-based, triple therapy allows shortening treatment duration from 48 weeks or longer to 12 or 24 weeks. Some IFN-RBV-free, all-oral regimens are currently under development and will constitute a major revolution for definitive cure of HCV infection.
Simeprevir is under evaluation for HCV genotype 1 patients in combination with the nucleotide polymerase inhibitor sofosbuvir ± RBV in the COSMOS (Combination of simeprevir and sofosbuvir in HCV genotype 1-infected patients) trial40 and in combination with the NS5a inhibitor daclatasvir ± RBV in the LEAGUE-1 trial.41 Moreover, the preliminary results of a Phase III clinical trial conducted in HCV genotype 4-infected patients were recently reported at the annual meeting of the European Association for the Study of the Liver.42 In April 2014, Janssen Pharmaceuticals, Inc. (Titusville, NJ, USA) announced initiation of the Phase III OPTIMIST (optimal treatment with a simeprevir and sofosbuvir therapy) trials, evaluating the safety and efficacy of simeprevir and sofosbuvir without IFN or RBV for the treatment of chronic genotype 1 HCV infection. Future combinations of two or more DAA agents, targeting different viral enzymes and with synergistic antiviral effects, will be approved, which will allow potent pan-genotypic treatment of HCV with optimized SVR and safety, heightened barrier to resistance, and shortened treatment duration.
Acknowledgment
We thank Anthony Saul for English corrections.
Disclosure
The authors report no conflicts of interest in this work.
References
Lavanchy D. The global burden of hepatitis C. Liver Int. 2009; 29(Suppl 1):74–81. | |
Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364(25):2405–2416. | |
Zeuzem S, Andreone P, Pol S, et al; REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med. 2011;364(25):2417–2428. | |
Bacon BR, Gordon SC, Lawitz E, et al; HCV RESPOND-2 Investigators. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med. 2011;364(13):1207–1217. | |
Poordad F, McCone J Jr, Bacon BR, et al; SPRINT-2 Investigators. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364(13):1195–1206. | |
Hayashi N, Seto C, Kato M, Komada Y, Goto S. Once-daily simeprevir (TMC435) with peginterferon/ribavirin for treatment-naïve hepatitis C genotype 1-infected patients in Japan: the DRAGON study. J Gastroenterol. 2014;49(1):138–147. | |
Bunchorntavakul C, Chavalitdhamrong D, Tanwandee T. Hepatitis C genotype 6: a concise review and response-guided therapy proposal. World J Hepatol. 2013;5(9):496–504. | |
Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347(13):975–982. | |
McHutchison JG, Lawitz EJ, Shiffman ML, et al; IDEAL Study Team. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med. 2009;361(6):580–593. | |
Asselah T, Marcellin P. Direct acting antivirals for the treatment of chronic hepatitis C: one pill a day for tomorrow. Liver Int. 2012; 32(Suppl 1):88–102. | |
Sarrazin C, Hézode C, Zeuzem S, Pawlotsky JM. Antiviral strategies in hepatitis C virus infection. J Hepatol. 2012;56(Suppl 1):S88–S100. | |
Kim CW, Chang KM. Hepatitis C virus: virology and life cycle. Clin Mol Hepatol. 2013;19(1):17–25. | |
Bartenschlager R, Lohmann V, Penin F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol. 2013;11(7):482–496. | |
Liang Y, Ishida H, Lenz O, et al. Antiviral suppression vs restoration of RIG-I signaling by hepatitis C protease and polymerase inhibitors. Gastroenterology. 2008;135(5):1710–1718. e2. | |
Sarrazin C, Zeuzem S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology. 2010;138(2):447–462. | |
Lin TI, Lenz O, Fanning G, et al. In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor. Antimicrob Agents Chemother. 2009;53(4):1377–1385. | |
Lenz O, Vijgen L, Moreno C. Virologic response and characterization of HCV genotypes 2 to 6 under TMC435 monotherapy (study TMC435-C202). Poster presented at: International Workshop on HIV and Hepatitis Virus Drug Resistance and Curative Strategies; June 7–11, 2011; Los Cabos, Mexico. Poster 16. | |
Tanwar S, Trembling PM, Dusheiko GM. TMC435 for the treatment of chronic hepatitis C. Expert Opin Investig Drugs. 2012;21(8):1193–1209. | |
Huisman MT Snoeys J, Monbaliu J, et al. In vitro studies investigating the mechanism of interaction between TMC435 and hepatic transporters. Poster presented at: 61st Annual Meeting of the American Association for the Study of Liver Diseases; October 29–November 2, 2010; Boston, MA. Poster 278. | |
Reesink HW, Fanning GC, Farha KA, et al. Rapid HCV-RNA decline with once daily TMC435: a phase I study in healthy volunteers and hepatitis C patients. Gastroenterology. 2010;138(3):913–921. | |
Medivir AB. Olysio™ (simeprevir) capsule. Highlights of Prescribing Information [prescribing information]. Stockholm: Medivir AB: 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/205123s001lbl.pdf. Accessed December 1, 2013. | |
You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother. 2013;14(18):2581–2589. | |
Verloes R, Shishido A. Phase I safety and PK of TMC435 in healthy volunteers and safety, PK and short-term efficacy in chronic hepatitis C infected individuals. Paper presented at: Japanese Hepatology Congress; June 4–5, 2009; Kobe, Japan. Abstract O-32 4–5. | |
Sekar V, Verloes R, Meyvisch P, Spittaels K, Akuma SH, Smedt GD. Evaluation of metabolic interactions for TMC435 via cytochrome P450 (CYP) enzymes in healthy volunteers. Poster presented at: 45th Annual Meeting of the European Association for the Study of the Liver; April 14–18; 2010; Vienna, Austria. | |
Janssen Research and Development LLC. Simeprevir, Application Number 205123Orig1s000. Clinical Pharmacology and Biopharmaceutics Review(s); 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/205123Orig1s000ClinPharmR.pdf. Accessed September 1, 2013. | |
Lenz O, Verbinnen T, Lin TI, et al. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob Agents Chemother. 2010;54(5):1878–1887. | |
Bae A, Sun SC, Qi X, et al. Susceptibility of treatment-naive hepatitis C virus (HCV) clinical isolates to HCV protease inhibitors. Antimicrob Agents Chemother. 2010;54(12):5288–5297. | |
Fried MW, Buti M, Dore GJ, et al. Once-daily simeprevir (TMC435) with pegylated interferon and ribavirin in treatment-naïve genotype 1 hepatitis C: the randomized PILLAR study. Hepatology. 2013;58(6):1918–1929. | |
Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology. 2014;146(2):430–441. e6. | |
Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a Phase 3 trial. Gastroenterology. 2014;146(7):1669–1679. e3. | |
Jacobson IM, Dore GJ, Foster GR, et al. Simeprevir with pegylated interferon alfa 2a plus ribavirin in treatment-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-1): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet. Epub June 3 2014. | |
Manns M, Marcellin P, Poordad F, et al. Simeprevir with pegylated interferon alfa 2a or 2b plus ribavirin in treatment-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-2): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. Epub June 3 2014. | |
Lenz O, de Bruijne J, Vijgen L, et al. Efficacy of re-treatment with TMC435 as combination therapy in hepatitis C virus-infected patients following TMC435 monotherapy. Gastroenterology. 2012;143(5):1176–1178. e1. | |
Manns M, Reesink H, Berg T, et al. Rapid viral response of once-daily TMC435 plus pegylated interferon/ribavirin in hepatitis C genotype-1 patients: a randomized trial. Antivir Ther. 2011;16(7):1021–1033. | |
Hayashi N, Izumi N, Kumada H, et al. Simeprevir with peginterferon/ribavirin for treatment-naïve hepatitis C genotype 1 patients in Japan: CONCERTO-1, a phase III trial. J Hepatol. Epub April 12, 2014. | |
Izumi N, Hayashi N, Kumada H, et al. Once-daily simeprevir with peginterferon and ribavirin for treatment-experienced HCV genotype 1-infected patients in Japan: the CONCERTO-2 and CONCERTO-3 studies. J Gastroenterol. 2014;49(5):941–953. | |
Toyoda H, Kumada T, Takaguchi K, Shimada N, Tanaka J. Changes in hepatitis C virus genotype distribution in Japan. Epidemiol Infect. 2014:1–5. | |
Maekawa S, Enomoto N. Once-daily simeprevir in combination with pegylated-interferon and ribavirin: a new horizon in the era of direct-acting antiviral agent therapy for chronic hepatitis C. J Gastroenterol. 2014;49(1):163–164. | |
Hezode C, Fontaine H, Dorival C, et al; CUPIC Study Group. Effectiveness of telaprevir or boceprevir in treatment-experienced patients with HCV genotype 1 infection and cirrhosis. Gastroenterology. Epub April 3, 2014. | |
Sulkowski M, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 Prior null responders with metavir f0–2: COSMOS study subgroup analysis. J Hepatol. 2014;60(Suppl 1):S4. | |
Zeuzem S, Hezode C, Bronowicki J-PP, et al. Daclatasvir in combination with simeprevir ± ribavirin for hepatitis C virus genotype 1 infection. Abstract presented at: 21st Conference on Retroviruses and Opportunistic Infections. Topics Antiviral Med. 2014;22(Issue e-1):15. | |
Moreno C, Hezode C, Marcellin P, et al. Once-daily simeprevir (TMC435) with peginterferon/ribavirin in treatment-naive or treatment-experienced chronic HCV genotype 4-infected patients: final results of a phase III trial. Poster presented at: 49th Annual meeting of the European Association for the Study of the Liver. J Hepatology. 2014;60(Suppl 1):S535. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.