")
Back to Journals » OncoTargets and Therapy » Volume 13
Silencing TCF4 Sensitizes Melanoma Cells to Vemurafenib Through Inhibiting GLUT3-Mediated Glycolysis
Authors Liu C, He S, Zhang J , Li S, Chen J, Han C
Received 10 January 2020
Accepted for publication 23 March 2020
Published 29 May 2020 Volume 2020:13 Pages 4905—4915
DOI https://doi.org/10.2147/OTT.S245531
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Can Liu,1 Siqi He,1 Jianfei Zhang,2 Shiyan Li,3 Jian Chen,4 Chaofei Han1
1Department of Burn and Plastic Surgery, The Third Xiangya Hospital of Central South University, Changsha, Hunan 410013, People’s Republic of China; 2Department of Burn and Plastic Surgery, The Second Affiliated Hospital of South China University, Hengyang, Hunan 421001, People’s Republic of China; 3Department of Burns and Plastic Surgery, Nanjing Drum Tower Hospital, The Affiliated Hospital of Nanjing University Medical School, Nanjing, Jiangsu 210008, People’s Republic of China; 4Department of Burns and Plastic Surgery, The First Hospital of Putian City, Putian, Fujian 351100, People’s Republic of China
Correspondence: Chaofei Han
Department of Burn and Plastic Surgery, The Third Xiangya Hospital of Central South University, Tongzipo Road 138, Changsha, Hunan 410013, People’s Republic of China
Tel +86-731-88618215
Email [email protected]
Background: Vemurafenib is a selective BRAF inhibitor with significant early effects in melanoma, but resistance will develop with the duration of treatment. Therefore, overcoming vemurafenib resistance can effectively improve the survival rate of melanoma. The transcriptional activity of TCF4 is necessary to maintain the malignant phenotype of cancer cells. However, the effect of TCF4 on melanoma sensitivity to vemurafenib and the underlying mechanism is unclear.
Methods: Vemurafenib-resistant A375 (A375/Vem) and SK-Mel-28 (SK-Mel-28/Vem) cells were constructed by administering increasing concentrations of vemurafenib, and the expression of TCF4 was examined in parent and vemurafenib-resistant cells. TCF4 loss-function cells models were established in A375/Vem and SK-Mel-28/Vem cells, respectively. Cell survival, clone formation, and cell apoptosis were assessed. The downstream target gene of TCF4 was verified by chromatin immunoprecipitation. Finally, the effect of TCF4 on melanoma cells glycolysis was investigated and were performed.
Results: TCF4 expression was increased in vemurafenib-resistant melanoma cells, and knocking down TCF4 could promote the sensitivity of melanoma cells to vemurafenib. Mechanism investigation revealed that TCF4 could interact with GLUT3 and silencing TCF4 could inhibit GLUT3 expression. In addition, overexpression of GLUT3 reversed the growth and glycolysis of tumor cells that were inhibited by TCF4 knockdown.
Conclusion: Our study demonstrates that TCF4 downregulation sensitizes melanoma cells to vemurafenib through inhibiting GLUT3-mediated glycolysis. These findings support TCF4 as an oncogene and provide new mechanism by which TCF4 confers chemotherapy resistance in melanoma.
Keywords: melanoma, chemotherapy resistance, BRAF inhibitor, glucose transporter, transcription factor 4, Warburg effect
Introduction
Melanoma is a common malignant tumor with an increasing incidence. There are approximately 132,000 new cases of melanoma each year worldwide.1 Approximately 25% of patients with advanced melanoma have a one-year survival time, with an average survival time of only 6.2 months.2 Melanoma is divided into four groups by genotype: BRAF mutation, NRAS mutation, NF-1 deletion, and wild type that have no mutations with these three genes.3 About 50% of melanoma have a point mutation in the BRAF gene. The most common mutation is V600E, which mutates from valine to glutamic acid, activates the MAPK (RAS-RAF-MEK-ERK) signaling pathway, thereby promoting the continued growth of tumor cells.4 At present, there are many marketed drugs for the treatment of melanoma, such as, BRAF inhibitors (vemurafenib, dabrafenib) and MAPK/MEK inhibitors (trametinib, cobimetinib), which significantly improve the prognosis of patients with advanced melanoma.5 Vemurafenib is a selective BRAF inhibitor with significant early effects, but resistance will develop with the duration of treatment, especially after 6–8 months of treatment.6 Therefore, overcoming Vemurafenib resistance can effectively improve the survival rate of melanoma.
Transcription Factor 4 (TCF4) is one of the major proteins in the Wnt/β-catenin pathway.7 TCF-4 is a member of the T cytokine/lymph enhancer (TCF/LEF) family. TCF4 located on the chromosome 10q25.3 and coded by TF7L2 (transcript factor 7 like 2) gene.8 This gene contains 17 exons, has a nuclear localization signal domain (NLS), the exon 1 encoding the β-catenin binding region; exon 10–11 encoded high mobility histone domain (HMG-box) which can recognize specific DNA sequences.8 In many tumors, abnormally activated Wnt pathway leads to nucleus translocation of β-catenin and combination with the relevant domains of TCF4 to form a transcription complex, which promotes the overexpression of downstream target genes and promotes the occurrence and development of tumors.9 Therefore, the transcriptional activity of TCF4 is necessary to maintain the malignant phenotype of cancer cells. Previous studies have shown that inhibiting the binding of β-catenin/TCF4 can promote the radiosensitivity of lung cancer cells,10 and other studies have found that TCF4 can mediate neuroendocrine differentiation and result in the resistance of prostate cancer cells to enzalutamide,11 indicating that TCF4 is associated with drug resistance in cancer cells. In melanoma, TCF4 can be inactivated by PGC1α, which is a transcriptional coactivator that promotes mitochondrial biogenesis, protects against oxidative stress and reprograms melanoma metabolism to influence drug sensitivity and survival.12 Inactive TCF4 causes downregulation of metastasis-related genes. In addition, vemurafenib treatment suppresses metastasis by acting on the PGC1α/TCF4 axis.12 However, the role of TCF4 in vemurafenib-resistant melanoma remains unknown.
In this study, we knocked down TCF4 in vemurafenib-resistant melanoma cells and examined its effects on melanoma sensitivity to vemurafenib, evaluated by cell colony and cell apoptosis. The underlying mechanism was also explored.
Materials and Methods
Cell Culture and Generation Vemurafenib-Resistant Cells
Melanoma cell lines, A375 and SK-Mel-28, were purchased from National Infrastructure of Cell Line Resource (Beijing, China). Cells were grown in Roswell Park Memorial Institute (RPMI)-1640 medium (GlutaMAX™-I, cat no. 72400120, Gibco). Ten percent fetal bovine serum (Gibco, CA, USA) was added in the medium. Cells were cultured under the condition in a 37°C humidified atmosphere of 5% CO2. The vemurafenib-resistant A375 and SK-Mel-28 cell lines (A375/Vem and SK-Mel-28/Vem) were established in our lab. A375 and SK-Mel-28 cells (1 × 105/mL) were treated with the sequential increases of vemurafenib concentrations, from 0.5 to 6.0 μM every 3 days for 6 weeks. Then, cell colonies were isolated.13 A375/Vem and SK-Mel-28/Vem cells were, respectively, replenished with 1.0 or 2 μM vemurafenib every 3 days.
Cell Transfection
SiRNAs were ordered from GenePharm (Shanghai, China). siRNA knockdown was carried out with two siRNAs targeting the TCF4 cDNA sequence. The sequences as following: TCF4 siRNA-1: 5ʹ-AGAGAAGAGCAAGCGAAAUAC-3ʹ, siRNA-2: 5ʹ- UAGCUGAGUGCACGUUGAAAG-3ʹ. Scramble oligonucleotides were used as a negative control. TCF4 cDNA ORF plasmid was purchased from Sino Biological (Beijing, China). Cells were transfected by Lipofectamine 2000 (Thermo Fisher, USA) following the manufacturers’ instructions. Cells were harvested at 48 h after transfection.
CCK-8 Assay
Cell proliferation rates were measured using Cell Counting Kit-8 (CCK-8) (Beyotime, Hangzhou, China). A total of 0.5×104 cells were seeded in each 96-well plate for 24 h. After treatment, the cells were further incubated for 24 h. Ten microliter CCK-8 reagents were added to each well at 1 h before the endpoint of incubation. OD 570 nm value in each well was determined by a microplate reader.
Immunoblotting
After indicated treatment, the cells were lysed by RIPA lysis buffer (Cell Signaling Technology) for 30 min on ice. The 50 g protein sample was subjected to 10% SDS-PAGE and transferred to PVDF membranes. The membranes were blocked by 5% non-fat milk for 1 h at room temperature. The membranes then were incubated with primary antibodies (anti-MDR, cat no. sc-55510, 1:1000, Santa Cruz Biotechnology; anti-P-gp, cat no. ab103477, 1:1000, Abcam; anti-TCF4, cat no. sc-166699, 1:1000, Santa Cruz Biotechnology; anti-GLUT3, cat no. sc-74399, 1:1000, Santa Cruz Biotechnology; and anti-β-actin, cat no. sc-47778, 1:1000, Santa Cruz Biotechnology) overnight at 4°C. The membranes were washed three times with PBS. After washing, the membranes were incubated with appropriate secondary antibodies for 1 h at 37°C. The bands were visualized by enhanced chemiluminescence.
Apoptosis Analysis
Annexin V-FITC Apoptosis Detection Kit (cat no. CA1020-100T, Solarbio, Beijing, China) was used in this experiment. After treatment, cells were collected and then resuspended in binding buffer. Annexin V-FITC (5 μL) and Propidium Iodide (5 μL) were added and mixed well. The cells were incubated with the mixed solution for 15 min. Finally, flow cytometric analysis was performed in Attune NxT (Thermofisher, Shanghai, China).
Clone Formation Assay
After treatment, the cells were collected and resuspended in the medium at 2000 cells per mL, and then were seeded into a 60 mm culture dish at 1000/dish. The cells were cultured for 1–2 weeks in a 37°C 5% CO2 incubator. When macroscopic clones appeared in the culture dish, the culture was terminated. The supernatant was discarded and washed 3 times with PBS. The cells were fixed with 1 mL of 4% paraformaldehyde for 15 min and then incubated with crystal violet for 20 min. After washing with PBS for 3 times, the clones were counted directly in a microscope (low magnification). Finally, the clone formation rate was calculated as follows: (number of clones/number of cells inoculated) × 100%.
Chromatin Immunoprecipitation (ChIP)
Chromatin Immunoprecipitation (ChIP) assay was performed using the EZ-Magna ChIP kit (EMD Millipore). Cells were fixed with 4% paraformaldehyde and incubated with glycine for 10 min to generate DNA–protein cross-links. Cells were lysed with Cell Lysis Buffer and Nuclear Lysis Buffer, and sonicated to generate chromatin fragments of 400–800 bp. The lysates were immunoprecipitated with Magnetic Protein A Beads conjugated with TCF4 antibody (rabbit monoclonal; Merck, Millipore) or rabbit non-immune IgG (as negative control). The precipitated DNA was analyzed by PCR. The following primers were used: Glut3-F: ATGCACATCCTGTATTATCC, Glut3-R: GAACAGACTGTTACAGTTGG.
Measurement of ATP
ATP Assay Kit (cat no. S0026B) was purchased from Beyontime Biotechnology (Shanghai, China) and used to detect the ATP concentrations in cells according to the manufacturer’s instructions.
Measurement of Glucose Uptake
Glucose Uptake Assay Kit (cat no. KA4085) was purchased from Abnova (Taipei City, Taiwan) and used to detect the glucose uptake in cells according to the manufacturer’s instructions.
Measurement of Lactate Level
D-Lactate Assay Kit (Colorimetric) (cat no. ab83429) was purchased from Abcam (Shanghai, China) and used to detect the lactate level in cells according to the manufacturer’s instructions.
Statistical Analysis
GraphPad Prism software (GraphPad Software Inc., La Jolla, CA) was used for statistical analyses. Student’s t-test for two group and one-way ANOVA with post hoc Bonferroni test for three or more group were used to assess the statistical significance. P < 0.05 was considered statistically significant.
Results
TCF4 Is Upregulated in Vemurafenib-Resistant Melanoma Cells
To investigate the role of TCF4 in vemurafenib-resistant melanoma cells, we established the vemurafenib-resistant melanoma cells, A375/vem and SK-Mel-28/vem. The A375/vem and SK-Mel-28/vem and their parental cells were exposed to a series of concentration of vemurafenib. The cell viability was significantly decreased in all cells. However, the rates of descent in A375/vem and SK-Mel-28/vem cells were lower than that of in their parental cells (Figure 1). The IC50 of A375/vem (3.8 μM) and SK-Mel-28/vem (6.1 μM) were significantly higher than in A375 (0.8 μM) and SK-Mel-28 (1.8 μM) (Figure 1). In addition, the markers of drug resistance, MDR and P-gp, were significantly increased in A375/vem and SK-Mel-28/vem cells compared with their parental cells (Figure 2). We then tested the expression of TCF4 by Western blot, the expression of TCF4 was significantly upregulated in A375/vem and SK-Mel-28/vem cells compared with their parental cells (Figure 2), indicating an important role of TCF4 in vemurafenib resistance.
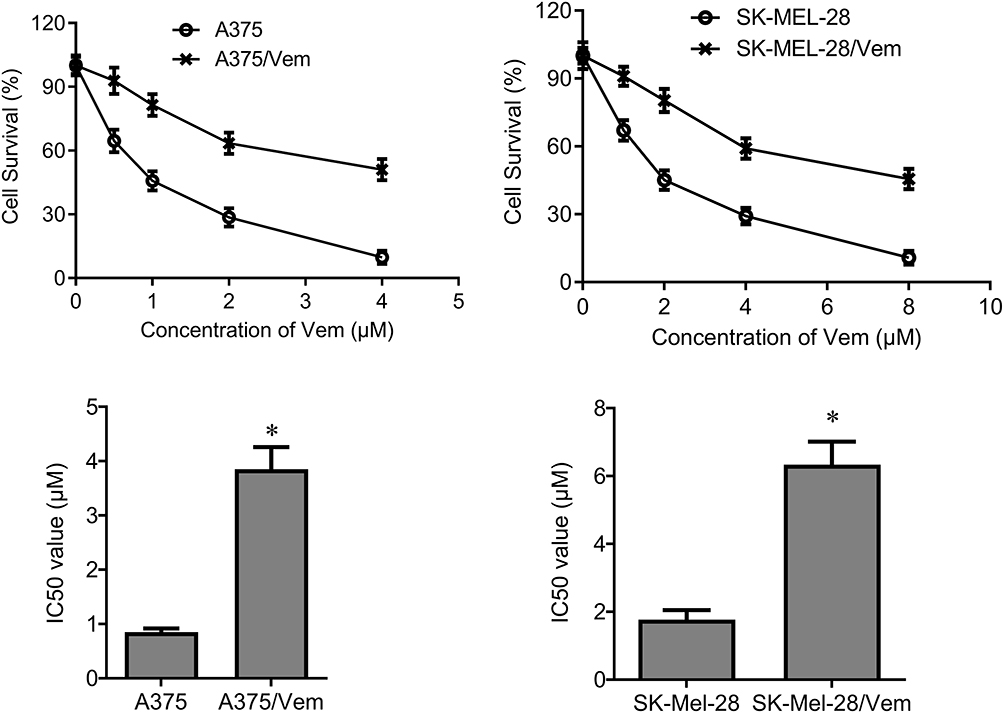 |
Figure 1 IC50 in Vemurafenib-resistant A375 and SK-Mel-28 cells. Vemurafenib-resistant A375 (A375/Vem) and SK-Mel-28 (SK-Mel-28/Vem) cell lines were established. The cells were treated with a series of concentrations of Vemurafenib (Vem). CCK-8 assay was performed to measure the cell viability in A375/Vem and SK-Mel-28/Vem cells after 48-h treatment. IC50 was calculated for A375/Vem and SK-Mel-28/Vem cells and their parental cells. Each experiment was performed three times. *p<0.05. |
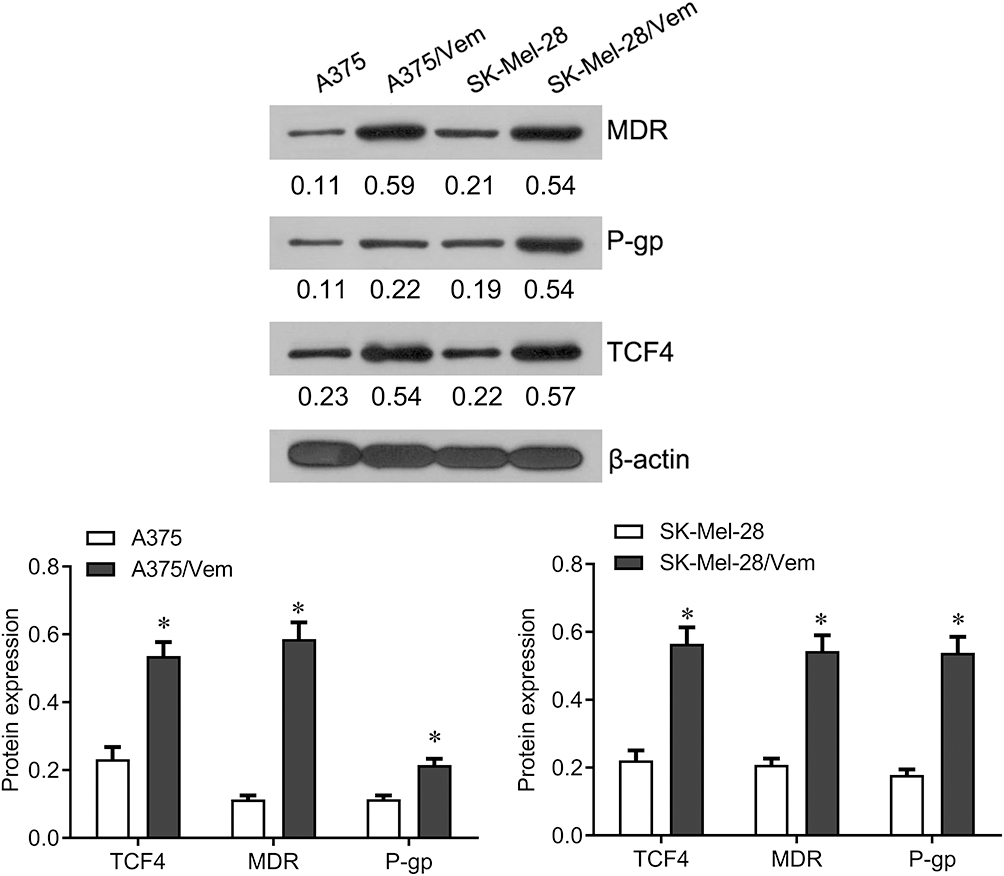 |
Figure 2 The expression of TCF4 in Vemurafenib-resistant A375 and SK-Mel-28 cells. Western blot was performed to examine the expression of TCF4 and the markers of drug resistance, MDR and P-gp in Vemurafenib-resistant A375 and SK-Mel-28 cells and their parental cells. Each experiment was performed three times. Vem, vemurafenib. *p<0.05. |
Downregulation of TCF4 Sensitizes Melanoma Cells to Vemurafenib
We further investigated whether knocking down TCF4 could sensitize melanoma cells to vemurafenib. We designed two siRNA sequences to knock down TCF4 in A375/vem and SK-Mel-28/vem cells and found that the efficiency of siRNA-2 was higher than siRNA-1 (Figure 3A). The siRNA-2 was used for the next experiments. We found that TCF4 knockdown significantly inhibited cell survival compared with scramble control in A375/vem and SK-Mel-28/vem cells under vemurafenib treatment (Figure 3B and C). In addition, TCF4 knockdown suppressed the colony ability and promoted cell apoptosis in A375/vem and SK-Mel-28/vem cells without vemurafenib treatment (Figure 4A and B). Importantly, TCF4 knockdown had great synergistic effect with vemurafenib in suppression of cell colony and promotion of cell apoptosis (Figure 4A and B).
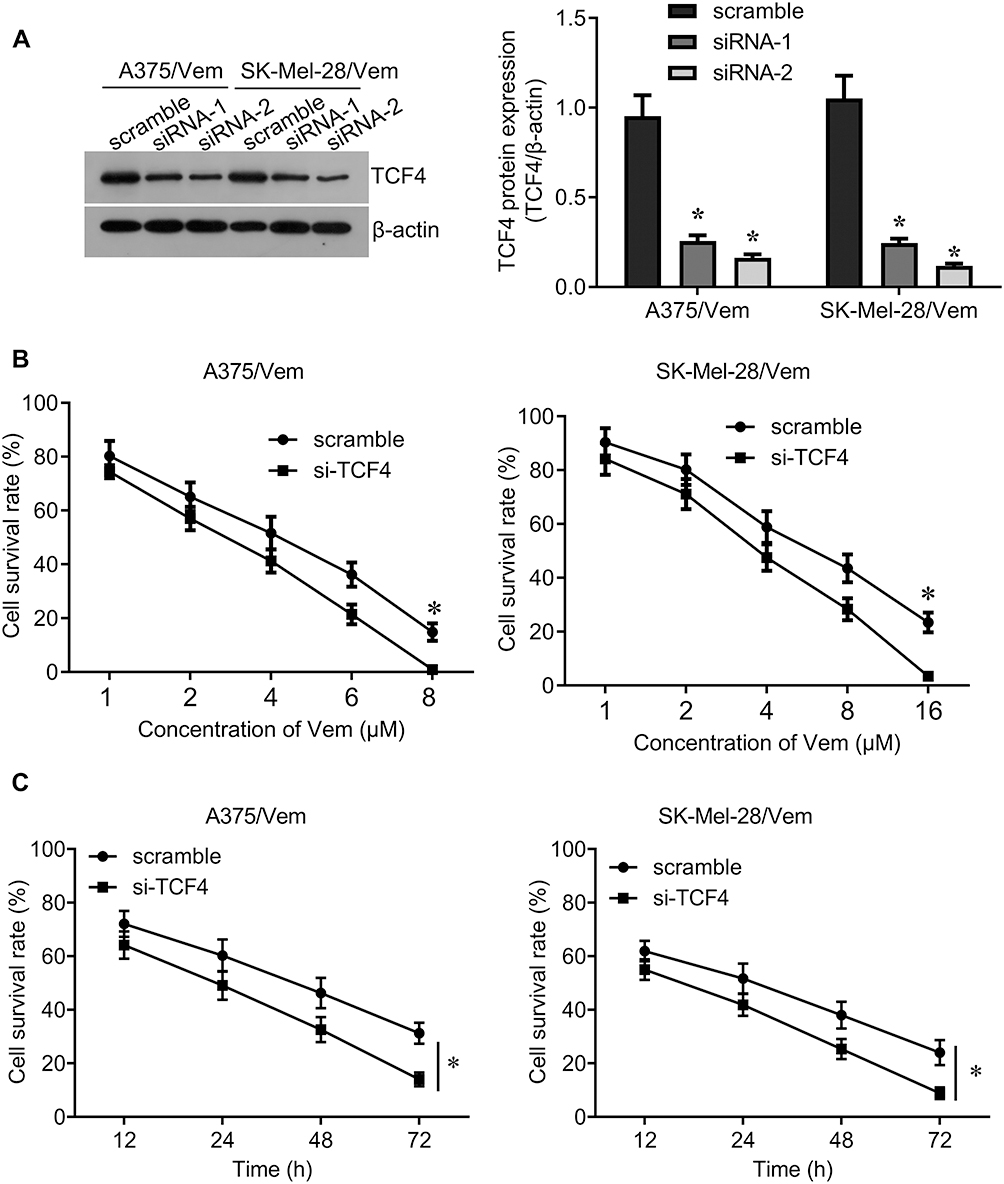 |
Figure 3 Silencing TCF4 sensitizes A375 and SK-Mel-28 cells to Vemurafenib. (A) Western blot was performed to examine the expression of TCF4 in Vemurafenib-resistant A375 and SK-Mel-28 cells after siRNA transfection (left), and quantification of the bands (right). (B) CCK-8 assay was performed to measure the cell viability in A375/Vem and SK-Mel-28/Vem cells after siRNA transfection and Vemurafenib treatment. (C) CCK-8 assay was performed to measure the cell viability at different times after siRNA transfection and vemurafenib treatment (A375/Vem, 3 μM; SK-Mel-28/Vem, 6 μM). Each experiment was performed three times. Vem, vemurafenib. *p<0.05. |
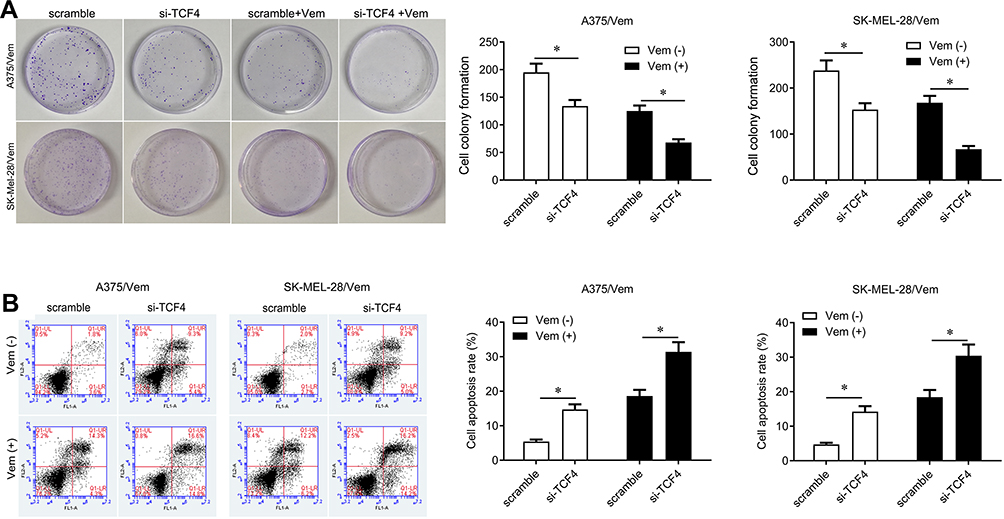 |
Figure 4 Silencing TCF4 inhibits cell colony and promotes cell apoptosis in Vemurafenib-resistant A375 and SK-Mel-28 cells. (A) Colony formation was performed to measure the ability of cell proliferation in A375/Vem and SK-Mel-28/Vem cells after siRNA transfection and Vemurafenib treatment. (B) Cell apoptosis was analyzed to measure the apoptotic rate in A375/Vem and SK-Mel-28/Vem cells after siRNA transfection and Vemurafenib treatment. Each experiment was performed three times. Vem, vemurafenib. *p<0.05. |
GLUT3 Is a Downstream Molecule of TCF4 in Melanoma Cells
We next investigated how TCF4 sensitizes melanoma cells to vemurafenib. GLUT3 plays a key role in glycolysis that promotes drug resistance in cancer cells. TCF4 knockdown reduced the expression of GLUT3 in A375/vem and SK-Mel-28/vem cells (Figure 5A). ChIP results showed that TCF4 interacted with GLUT3 (Figure 5B). We then rescued the expression of GLUT3 in A375/vem and SK-Mel-28/vem cells that transfected with TCF4 siRNA (Figure 5C). Rescue of GLUT3 significantly attenuated the effects of TCF4 knockdown on inhibition of cell survival and cell colony and on promotion of cell apoptosis (Figure 6A–C). In addition, TCF4 knockdown inhibited the glycolysis process, evaluated by decrease in glucose uptake, ATP level and lactate production in A375/vem and SK-Mel-28/vem cells (Figure 7). However, rescue of GLUT3 significantly abolished the effects of TCF4 knockdown on glycolysis process (Figure 7).
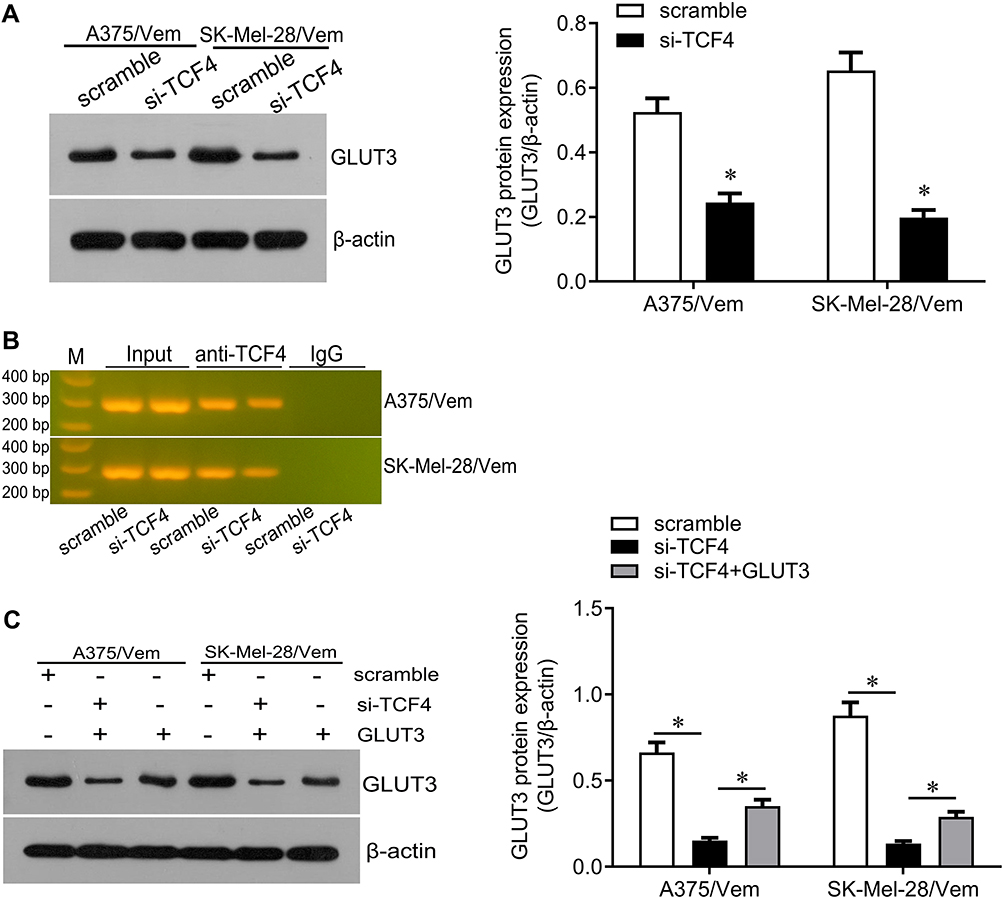 |
Figure 5 TCF4 positively regulates GLUT3. (A) Western blot was performed to examine the expression of GLUT3 in Vemurafenib-resistant A375 and SK-Mel-28 cells after siRNA transfection (left), and quantification of the bands (right). (B) Chip assay was performed to test the interaction between TCF4 and GLUT3. (C) Western blot was performed to examine the expression of GLUT3 in Vemurafenib-resistant A375 and SK-Mel-28 cells after TFC4 siRNA and GLUT3 expressed plasmids transfection (left), and quantification of the bands (right). Each experiment was performed three times. Vem, vemurafenib. *p<0.05. |
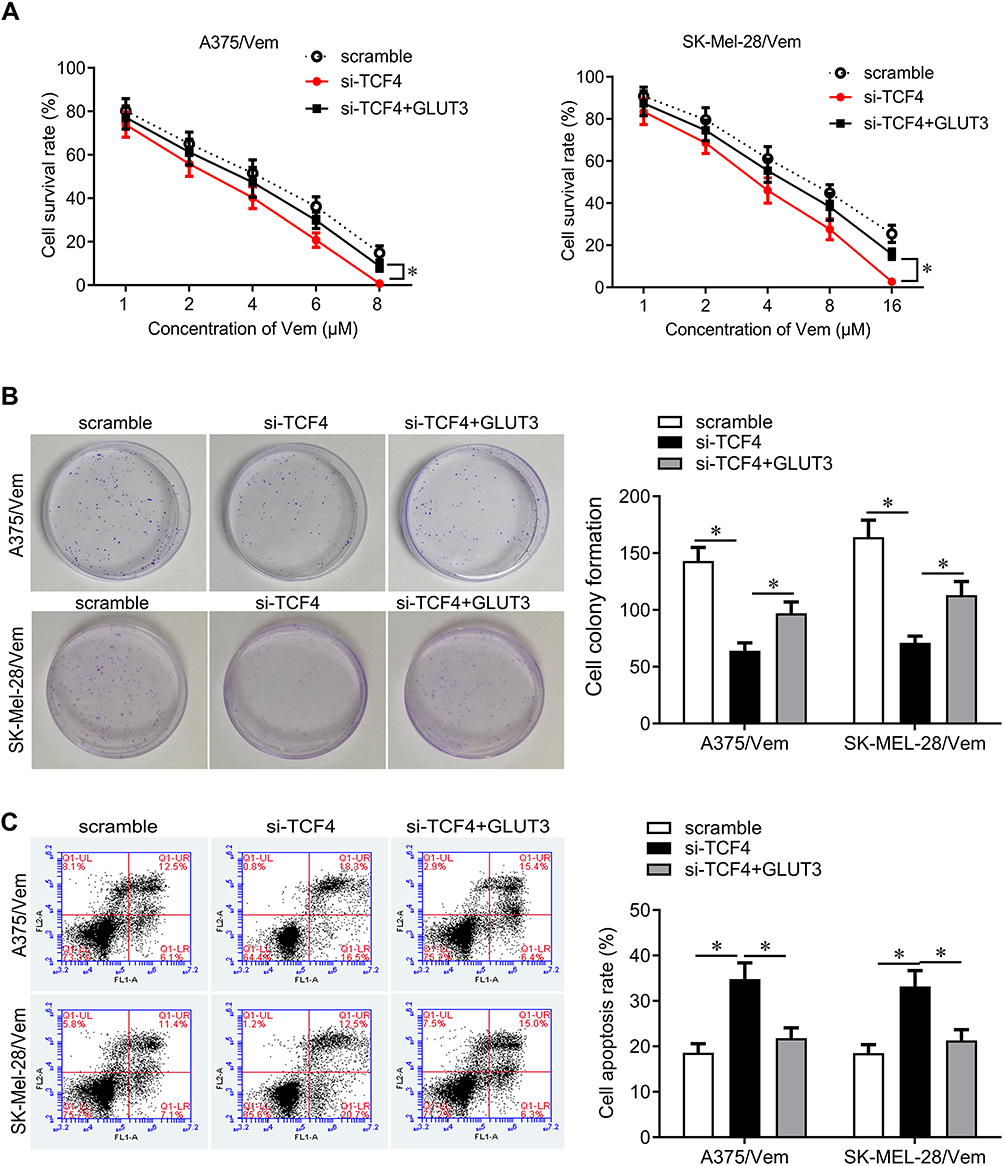 |
Figure 6 GLUT3 reverses TCF4 knockdown-mediated effects on cell proliferation and cell apoptosis. (A) CCK-8 assay was performed to measure the cell viability in A375/Vem and SK-Mel-28/Vem cells after TFC4 siRNA and GLUT3 expressed plasmids transfection. (B) Colony formation was performed to measure the ability of cell proliferation in A375/Vem and SK-Mel-28/Vem cells after TFC4 siRNA and GLUT3 expressed plasmids transfection. (C) Cell apoptosis was analyzed to measure the apoptotic rate in A375/Vem and SK-Mel-28/Vem cells after TFC4 siRNA and GLUT3 expressed plasmids transfection. Each experiment was performed three times. Vem, vemurafenib. *p<0.05. |
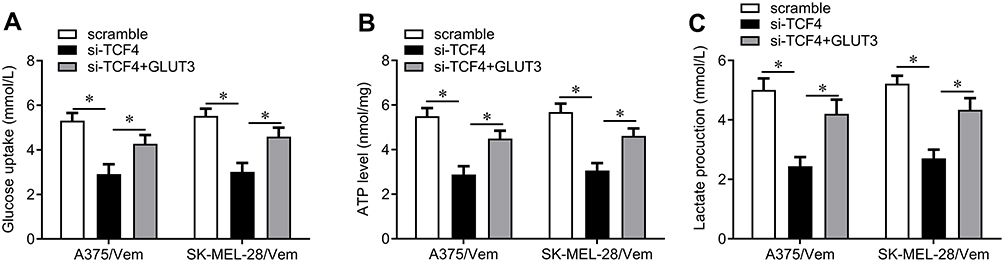 |
Figure 7 The effects of TCF4/GLUT3 on glucose uptake, APT level and lactate production in Vemurafenib-resistant A375 and SK-Mel-28 cells. (A) Glucose Uptake Assay Kit was used to measure the glucose uptake in A375/Vem and SK-Mel-28/Vem cells after TFC4 siRNA and GLUT3 expressed plasmids transfection. (B) ATP Assay Kit was used to measure the ATP level in A375/Vem and SK-Mel-28/Vem cells after TFC4 siRNA and GLUT3 expressed plasmids transfection. (C) D-Lactate Assay Kit was used to measure the lactate production in A375/Vem and SK-Mel-28/Vem cells after TFC4 siRNA and GLUT3 expressed plasmids transfection. Each experiment was performed three times. Vem, vemurafenib. *p<0.05. |
Discussion
Melanoma is highly malignant with an annual growth morbidity of 3% to 5%. About 25% of melanoma patients in Asian countries have BRAF mutations. More than 90% of the BRAF mutations to valine at position 600 are replaced by glutamate (V600E), which cause the mitogen-activated protein kinase (MAPK) signaling pathway to lose control, resulting in uncontrolled cell growth.14 Preclinical studies have shown that inhibiting BRAF by the small molecule RNAs can restore the cell growth cycle, thereby reversing the malignant phenotypes of melanoma cells.15 Vemurafenib is a specific inhibitor of the BRAF gene V600E mutation, which can selectively block RAF/MEK/ERK signaling and suppresses tumor proliferation.16 It has extensive safety and high selectivity. However, long-term single-dose therapy makes patients quickly resistant to Vemurafenib.17 In this study, we found that TCF4 expression was increased in Vemurafenib-resistant melanoma cells, and knocking down TCF4 could promote the sensitivity of melanoma cells to Vemurafenib. Studies have shown that activated TCF4 significantly promotes tumor cell proliferation and metastasis in a variety of tumors. For example, TCF4 gene knockout leads to early apoptosis of stem cells and affects the differentiation and proliferation of large intestinal crypt stem cells through the MAPK signaling pathway.18 The expression level of TCF4 in liver cancer tissues was significantly higher than that in adjacent tissues, and it was higher in metastatic liver cancer tissues. TCF4 showed enhanced transcription activity, promoted the ability of cell clone formation and resistance to chemotherapy in liver cancer cells.19 In addition, TCF4 expression is increased in lung cancer, renal carcinoma, and breast cancer, which is related to the invasion and metastasis of tumor cells.20,21
In melanoma, TCF4 is preferentially expressed by dedifferentiated/invasive phenotype cells.22 TCF4 is associated with epithelial–mesenchymal transition and invasion, induces the transcription of miR-125b to promotes melanoma cell invasion,23 indicating that TCF4 is associated with a more malignant phenotype. Previous study demonstrated that glucose transporter (GLUT) 3 was a direct TCF4/β-catenin target gene in hepatoblastoma, which is a Wnt/β-catenin-driven malignancy.24 This study found that TCF4 could interact with GLUT3 and silencing TCF4 could inhibit GLUT3 expression. In addition, overexpression of GLUT3 reversed the growth and glycolysis of tumor cells that were inhibited by TCF4 knockdown. GLUT is a major carrier that mediates glucose transport inside and outside the cell, mainly distributed on the cell membrane.25 There are 9 types of GLUT, GLUT1-GLUT9. GLUT3 is responsible for the uptake of glucose for the nervous system.26 More and more studies have found that GLUT3 is overexpressed in various solid tumors, including ovarian cancer, gastric cancer, and non-small cell lung cancer.27 This is because the rapid proliferation of tumor cells makes them in an anoxic environment. Under anaerobic conditions, ATP produced by glycolysis is more than 10 times less than aerobic metabolism. Therefore, tumor cells need to express a large number of GLUTs to meet the excess glucose uptake.28 Melanoma cells have an obvious aerobic glycolytic phenotype, ie, Warburg effect.29 GLUT3 is highly expressed in melanoma and may become a potential biomarker.30 In addition, previous experiments have shown that some GLUTs transport inhibitors have shown good effects in inhibiting tumor growth in mice, making GLUTs a potential target for tumor therapy.31
Summary, this recent study demonstrates that TCF4 downregulation sensitizes melanoma cells to Vemurafenib through inhibiting GLUT3-mediated glycolysis. These findings support TCF4 as an oncogene and provide new mechanism by which TCF4 confers chemotherapy resistance in melanoma.
Acknowledgment
This work was supported by the New Xiangya Talent Project of the Third Xiangya Hospital of Central South University (JY201607).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Garbe C, Amaral T, Peris K, et al. European consensus-based interdisciplinary guideline for melanoma. Part 2: treatment - update 2019. Eur J Cancer. 2019.
2. Bello DM, Panageas KS, Hollmann T, et al. Survival outcomes after metastasectomy in melanoma patients categorized by response to checkpoint blockade. Ann Surg Oncol. 2019.
3. Mahendraraj K, Sidhu K, Lau CS, McRoy GJ, Chamberlain RS, Smith FO. Malignant melanoma in African-Americans: a population-based clinical outcomes study involving 1106 African-American patients from the Surveillance, Epidemiology, and End Result (SEER) database (1988–2011). Medicine. 2017;96(15):e6258. doi:10.1097/MD.0000000000006258
4. Betancourt LH, Szasz AM, Kuras M, et al. The hidden story of heterogeneous B-raf V600E mutation quantitative protein expression in metastatic melanoma-association with clinical outcome and tumor phenotypes. Cancers. 2019;11:12. doi:10.3390/cancers11121981
5. Hauschild A, Ascierto PA, Schadendorf D, et al. Long-term outcomes in patients with BRAF V600-mutant metastatic melanoma receiving dabrafenib monotherapy: analysis from Phase 2 and 3 clinical trials. Eur J Cancer. 2020;125:114–120. doi:10.1016/j.ejca.2019.10.033
6. Kim A, Cohen MS. The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma. Expert Opin Drug Discov. 2016;11(9):907–916. doi:10.1080/17460441.2016.1201057
7. Ravindranath A, O’Connell A, Johnston PG, El-Tanani MK. The role of LEF/TCF factors in neoplastic transformation. Curr Mol Med. 2008;8(1):38–50. doi:10.2174/156652408783565559
8. Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40(1):14–21. doi:10.1016/j.biocel.2007.01.020
9. Yan M, Li G, An J. Discovery of small molecule inhibitors of the Wnt/beta-catenin signaling pathway by targeting beta-catenin/Tcf4 interactions. Exp Biol Med. 2017;242(11):1185–1197. doi:10.1177/1535370217708198
10. Zhang Q, Gao M, Luo G, et al. Enhancement of radiation sensitivity in lung cancer cells by a novel small molecule inhibitor that targets the beta-catenin/Tcf4 interaction. PLoS One. 2016;11(3):e0152407. doi:10.1371/journal.pone.0152407
11. Lee GT, Rosenfeld JA, Kim WT, et al. TCF4 induces enzalutamide resistance via neuroendocrine differentiation in prostate cancer. PLoS One. 2019;14(9):e0213488. doi:10.1371/journal.pone.0213488
12. Luo C, Lim JH, Lee Y, et al. A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis. Nature. 2016;537(7620):422–426. doi:10.1038/nature19347
13. Faiao-Flores F, Alves-Fernandes DK, Pennacchi PC, et al. Targeting the hedgehog transcription factors GLI1 and GLI2 restores sensitivity to vemurafenib-resistant human melanoma cells. Oncogene. 2017;36(13):1849–1861. doi:10.1038/onc.2016.348
14. Carr S, Smith C, Wernberg J. Epidemiology and risk factors of melanoma. Surg Clin North Am. 2020;100(1):1–12. doi:10.1016/j.suc.2019.09.005
15. Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508(7494):118–122. doi:10.1038/nature13121
16. Salton M, Kasprzak WK, Voss T, Shapiro BA, Poulikakos PI, Misteli T. Inhibition of vemurafenib-resistant melanoma by interference with pre-mRNA splicing. Nat Commun. 2015;6:7103. doi:10.1038/ncomms8103
17. Ajiro M, Zheng ZM. Vemurafenib-resistant BRAF selects alternative branch points different from its wild-type BRAF in intron 8 for RNA splicing. Cell Biosci. 2015;5(1):70. doi:10.1186/s13578-015-0061-7
18. van Es JH, Haegebarth A, Kujala P, et al. A critical role for the Wnt effector Tcf4 in adult intestinal homeostatic self-renewal. Mol Cell Biol. 2012;32(10):1918–1927. doi:10.1128/MCB.06288-11
19. Jiang Y, Zhou XD, Liu YK, Wu X, Huang XW. Association of hTcf-4 gene expression and mutation with clinicopathological characteristics of hepatocellular carcinoma. World J Gastroenterol. 2002;8(5):804–807. doi:10.3748/wjg.v8.i5.804
20. Yang LH, Xu HT, Han Y, et al. Axin downregulates TCF-4 transcription via beta-catenin, but not p53, and inhibits the proliferation and invasion of lung cancer cells. Mol Cancer. 2010;9:25. doi:10.1186/1476-4598-9-25
21. Zhang M, Tang M, Fang Y, et al. Cumulative evidence for relationships between multiple variants in the VTI1A and TCF7L2 genes and cancer incidence. Int J Cancer. 2018;142(3):498–513. doi:10.1002/ijc.31074
22. Eichhoff OM, Weeraratna A, Zipser MC, et al. Differential LEF1 and TCF4 expression is involved in melanoma cell phenotype switching. Pigment Cell Melanoma Res. 2011;24(4):631–642. doi:10.1111/j.1755-148X.2011.00871.x
23. Rambow F, Bechadergue A, Luciani F, et al. Regulation of melanoma progression through the TCF4/miR-125b/NEDD9 cascade. J Invest Dermatol. 2016;136(6):1229–1237. doi:10.1016/j.jid.2016.02.803
24. Crippa S, Ancey PB, Vazquez J, et al. Mutant CTNNB1 and histological heterogeneity define metabolic subtypes of hepatoblastoma. EMBO Mol Med. 2017;9(11):1589–1604. doi:10.15252/emmm.201707814
25. Dura M, Nemejcova K, Jaksa R, et al. Expression of Glut-1 in malignant melanoma and melanocytic nevi: an immunohistochemical study of 400 cases. Pathol Oncol Res. 2019;25(1):361–368. doi:10.1007/s12253-017-0363-7
26. Cosset E, Ilmjarv S, Dutoit V, et al. Glut3 addiction is a druggable vulnerability for a molecularly defined subpopulation of glioblastoma. Cancer Cell. 2017;32(6):856–868. doi:10.1016/j.ccell.2017.10.016
27. Ancey PB, Contat C, Meylan E. Glucose transporters in cancer - from tumor cells to the tumor microenvironment. FEBS J. 2018;285(16):2926–2943. doi:10.1111/febs.14577
28. Zhao M, Zhang Z. Glucose transporter regulation in cancer: a profile and the loops. Crit Rev Eukaryot Gene Expr. 2016;26(3):223–238. doi:10.1615/CritRevEukaryotGeneExpr.2016016531
29. Kamenisch Y, Baban TSA, Schuller W, et al. UVA-irradiation induces melanoma invasion via the enhanced warburg effect. J Invest Dermatol. 2016;136(9):1866–1875. doi:10.1016/j.jid.2016.02.815
30. Ruby KN, Liu CL, Li Z, Felty CC, Wells WA, Yan S. Diagnostic and prognostic value of glucose transporters in melanocytic lesions. Melanoma Res. 2019;29(6):603–611. doi:10.1097/CMR.0000000000000626
31. Guo Z, Cheng Z, Wang J, et al. Discovery of a potent GLUT inhibitor from a library of rapafucins by using 3D microarrays. Angewandte Chemie. 2019;58(48):17158–17162. doi:10.1002/anie.201905578
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.