")
Back to Journals » Cancer Management and Research » Volume 11
Silencing of TAZ inhibits the motility of hepatocellular carcinoma cells through autophagy induction
Authors Zhou W, Weng J, Wu K , Xu X, Wang H, Zhang J, Zhao C, Yang J, Zhang Y, Shen W
Received 12 May 2019
Accepted for publication 22 August 2019
Published 26 September 2019 Volume 2019:11 Pages 8743—8753
DOI https://doi.org/10.2147/CMAR.S215466
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Wei Zhou,1,2,* Jiachun Weng,1,* Keyan Wu,1,2,* Xiao Xu,1,* Hui Wang,1,* Jing Zhang,3,* Chengxue Zhao,1 Jie Yang,1 Yu Zhang,1,4,5 Weigan Shen1,4,5
1Department of Cell Biology, School of Medicine of Yangzhou University, Yangzhou, People’s Republic of China; 2Department of Internal Medicine, Affiliated Hospital of Yangzhou University, Yangzhou, People’s Republic of China; 3Department of Internal Medicine, Northern Jiangsu People’s Hospital Affiliated to Yangzhou University, Yangzhou, People’s Republic of China; 4Jiangsu Co-Innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, Yangzhou University, Yangzhou, People’s Republic of China; 5Jiangsu Key Laboratory of Integrated Traditional Chinese and Western Medicine for Prevention and Treatment of Senile Diseases, Yangzhou University, Yangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yu Zhang; Weigan Shen
Department of Cell Biology, School of Medicine of Yangzhou University, No. 136 Middle Jiangyang Street, Yangzhou 225009, Jiangsu, People’s Republic of China
Tel +86 05 148 797 8875
Fax +86 05 148 734 1733
Email [email protected]; [email protected]
Purpose: The aim of the present study was to investigate the effect of knockdown and knockout of the transcriptional co-activator with PDZ-binding motif (TAZ) on the migration, invasion and autophagy of the hepatocellular carcinoma (HCC) cell lines, as well as the functional connection between the autophagy and cell migratory processes induced by loss of TAZ in HCC cell lines.
Methods: HCC cell lines SMMC-7721 and SK-HEP1 stably knockdown and knockout of TAZ were established by the lentiviral-mediated TAZ knockdown and knockout approaches. Reverse transcription-quantitative real-time polymerase chain reaction and Western blotting were performed to examine the expression of TAZ and indicated genes in downstream pathways in HCC cell lines. Transwell assay and autophagic flux assay were used to evaluate the effect of TAZ knockdown and knockout on the motility and the autophagy of HCC cell lines.
Results: We initially found that TAZ exhibited highly abundant and was expressed predominantly in HCC cell lines with different spontaneous metastatic potential. Through performing loss-of-function assays, we demonstrated that both TAZ knockdown and knockout promoted HCC cell autophagy and reduced HCC cell migration, invasion and epithelial-to-mesenchymal transition. In addition, autophagy inhibition in TAZ knockdown and knockout SMMC-7721 and SK-HEP1 cells in the presence of 3-methyladenine or chloroquine partially abrogated the migratory and invasive ability induced by TAZ knockdown and knockout.
Conclusion: Our findings indicated that loss of TAZ in HCC cells suppressed cell motility probably via altering the autophagy, suggesting that TAZ emerges as an important target in regulating cell motility and autophagy in HCC cells, and blocking TAZ may be a novel therapeutic strategy against HCC.
Keywords: TAZ, autophagy, migration, invasion, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is the third leading cause of cancer death worldwide, and the vast majority of HCC-associated deaths are caused by metastasis.1,2 Tumor metastasis is a complex and multi-step process, wherein cancerous cells must detach from the primary tumor, lose their epithelial polarity, degrade and invade through the surrounding tissue, enter and survive in capillary blood, seed a target organ, exit circulation, and finally home in distant organs.3 Therefore, understanding and investigating the cellular mechanisms that modulate metastasis is vital to the development of effective HCC therapies.
Autophagy, a highly conserved intracellular degradation system that utilizes the autophagosome to deliver cytoplasmic unnecessary or dysfunctional components to the lysosome, plays crucial roles in cell development, differentiation, survival, and homeostasis.4 Accumulating evidence indicates that autophagy is involved in the tumorigenesis and metastatic progression of various cancers, including HCC.5–7 Although autophagy has been shown to be involved in modulating tumor cell migration and invasion, differentiation, and epithelial-to-mesenchymal transition (EMT),5 the exact contribution of autophagy into the modulation of HCC cell migration and invasion remains largely unexplored.
The transcriptional co-activator with PDZ-binding motif (TAZ), also known as WW domain containing transcription regulator 1, is one of the Hippo downstream effectors responsible for mediating the vast majority of the physiological functions in the Hippo signaling cascade. The dysregulation of the Hippo tumor suppressor pathway leads to the activation of TAZ, then activating the TEA domain transcription factors to transcribe target genes involved in the hyperproliferation, anti-apoptosis, and enhanced cell migration, which are associated with tumorigenesis and aggressive phenotypes.8–10 Although TAZ has been shown to play critical roles in cell migration, invasion, EMT, and stemness in multiple human cancers,11–14 and the autophagy machinery has been proposed to play a role in cell migration and EMT,15–17 the functional connection between the autophagy and cell migratory processes mediated by TAZ remains to be determined.
In the present study, we generated TAZ knockdown and knockout HCC cell lines by using lentiviral-mediated gene knockdown and clustered regularly interspaced short palindromic repeat/CRISPR associated protein 9 (CRISPR/Cas9) gene editing to investigate the functional connection between the autophagy and cell motility induced by TAZ. We demonstrated that both TAZ knockdown and knockout promoted HCC cell autophagy and reduced HCC cell migration, invasion, and EMT. Importantly, our results suggest that loss of TAZ suppressed cell motility probably via altering the autophagy of HCC cells.
Materials and methods
Cell lines
The human HCC cell lines SMMC-7721, HepG2, SK-HEP1 and human embryonic kidney cell line 293T (HEK293T) were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, People’s Republic of China), and HCC cell line HCCLM3 was obtained from the KeyGEN BioTECH (Nanjing, People’s Republic of China). All cells were routinely cultured in DMEM (Hyclone, Beijing, People’s Republic of China) supplemented with 10% FBS (Gibco, Carlsbad, CA, USA) and 1% penicillin-streptomycin, and maintained at 37°C in a humidified incubator with 5% CO2.
Antibodies
The antibodies against TAZ (#4883), yes-associated protein (YAP) (#14074), Unc-51 like autophagy activating kinase 1 (ULK1) (#8054), microtubule-associated protein 1 light chain 3 Beta (LC3B) (#3868), SQSTM1/p62 (#39749), β-catenin (#8480), Vimentin (#5741), and horseradish peroxidase (HRP)-linked anti-rabbit IgG (#7074) and anti-mouse IgG (#7076) were purchased from Cell Signaling Technology (Danvers, MA, USA), and antibodies against N-cadherin (#561553) and E-cadherin (#562869) were purchased from BD Biosciences (San Jose, CA, USA). The antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was purchased from KangChen Bio-tech (Shanghai, People’s Republic of China).
Plasmid construction, lentivirus production, and infection
The short hairpin RNA (shRNA) targeting TAZ (sense, 5′-GATCCGCGATGAAT CAGCCTCTGAATTTCAGAGAATTCAGAGGCTGATTCATCGCTTTTTTA-3′ and antisense, 5′-CGCGTAAAAAAGCGATGAATCAGCCTCTGAATTCTCTTGA AATTCAGAGGCTGATTCATCGCG-3′) was cloned into the BamH I and Mlu I sites of the lentiviral shRNA expression vector pLent-U6-Puro empty vector with puromycin resistance (ViGene Biosciences Inc., Rockville, MD, USA). To construct the lentiviral vector for CRISPR/Cas9-mediated TAZ gene knockout, pre-designed sgRNA targeting TAZ (sense, 5′-CACCGAGAAGCCCGCTGCGGAGGAG-3′ and antisense, 5′-AAA CCTCCTCCGCA GCGGGCTTCTC-3′) were cloned into the BsmB I site of the lentiCRISPRv2-puro vector (#52961, Addgene Inc., Cambridge, MA, USA). The non-silencing control shRNA vector and control sgRNA vector were also generated previously from our laboratory, School of Medicine, Yangzhou University. All of the constructs were validated by DNA sequencing. Lentiviruses were packaged in HEK293T cells by transfection with the control vectors, shRNA or sgRNA targeting TAZ constructs together with the lentiviral packaging vector pSPAX2 and pMD2G or pCMV-VSV-G (Addgene Inc.) using EndoFectin Lenti reagent (GeneCopoeia, USA). The viral particles were harvested and purified. For infection, SMMC-7721 and SK-HEP1 cells were transduced with indicated lentiviruses in the presence of polybrene (8 μg/mL). Forty-eight hours after infection, the cells were selected with puromycin (0.5 μg/mL for SMMC-7721 cells and 1.0 μg/mL for SK-HEP1 cells) for 14 days to generate stable TAZ knockdown cell lines or maintained for 6 days followed by monoclonalization to establish stable TAZ knockout cell lines. The knockdown of TAZ was screened by reverse transcription-quantitative real-time polymerase chain reaction (RT-qPCR) and Western blotting, and the knockout of TAZ expression was verified by Western blotting.
Cell migration and invasion assays
Transwell inserts (diameter, 6.5 mm; pore size, 8.0 µm; Corning Incorporated, Corning, NY, USA) were used to perform cell migration and invasion assays as described previously.18 In brief, cells were serum-starved overnight, and 3×104 cells were seeded into the uncoated or matrigel-coated upper chambers of the inserts in 200 μL of serum-free media for migration and invasion assays, respectively. The inserts were then placed into the lower chambers with 600 μL of the complete growth media (containing 10% FBS). After incubation for 24 hrs for SK-HEP1 cells or 48 hrs for SMMC-7721 cells at 37°C in a humidified incubator with 5% CO2, the migrated or invaded cells on the bottom of the inserts were fixed with 4% paraformaldehyde for 20 mins at room temperature and stained with 0.1% crystal violet for 20 mins at room temperature. The phase-contrast images were captured under an inverted microscope and five random fields at 200× magnification were analyzed. All experiments were performed in triplicate from three independent experiments.
RT-qPCR
Total RNA extraction from cell lines was performed using the TRIzol® reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions, and was reverse transcribed to cDNA by using the PrimeScriptTM RT Master Mix (TaKaRa, Dalian, People’s Republic of China). Real-time PCR was carried out using the SYBR Green qPCR system (TaKaRa) on an ABI 7500 Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer’s instruction. Samples were cycled once at 95°C for 5 mins, then 40 cycles of 95°C for 10 s and 60°C for 34 s. Primers used in the present study were as follows: TAZ forward, 5′-CTTGGATGTAGCCAT GACCTT-3′ and reverse, 5′-TCAATCAAAACCAGGCAATG-3′; connective tissue growth factor (CTGF) forward, 5′-AGGAGTGGGTGTGTGACGA-3′ and reverse, 5′-CCAGGCAGTTGGCTCTAATC-3′; cysteine-rich angiogenic inducer 61 (CYR61) forward, 5′-AGCCTCGCATCCTATACAACC-3′ and reverse, 5′-TTCTTTCACAAG GCGGCACTC-3′; N-cadherin forward, 5′-CAGTGTACAGAATCAGTG-3′ and reverse, 5′-CAACAGTAAGGACAAACA-3′; E-cadherin forward, 5′-CAAGTGAC CACCTTAGAG-3′ and reverse, 5′-GAATTTGCAATCCTGCTT-3′; β-catenin forward, 5′-ACCTATACTTACGAAAAACTAC-3′ and reverse, 5′-CCACCAGCTTCTACAA TA-3′; Vimentin forward, 5′-GACGCCATCAAC ACCGAGTT-3′ and reverse, 5′-CTTTGTCGTTGGTTAGCTGGT-3′; LC3B forward, 5′-AGCAGCATCCAACCA AAATC-3′ and reverse, 5′-TGTGTC CGTTCACCAACAG-3′; SQSTM1/p62 (p62) forward, 5′-ATCGGAGGATCCGAGTGT-3′ and reverse, 5′-TGGCTGTGAGCTGC TCTT-3′; ULK1 forward, 5′-AAGATCGCTGACTTCGGCTT-3′ and reverse, 5′-TTC TCGTAGAACAGGCGCAG-3′; and GAPDH forward, 5′-GCACCGTCAAGGCTGA GAAC-3′ and reverse, 5′-TGGTGAAGACGCCAGTGGA-3′; the relative expression of the mRNAs was calculated using the 2−ΔΔCt method with GAPDH expression as an endogenous control.
Western blotting
Cells were washed twice in cold PBS and lysed in RIPA lysis buffer containing 50 mM Tris, 0.15 M NaCl, 1 mM EGTA, 1% NP40, 0.25% SDS (Beyotime Institute of Biotechnology, Haimen, People’s Republic of China), protease inhibitors mix and phosphatase inhibitors from Roche Diagnostics at 4°C for 30 mins. After centrifugation at 4°C at 12,000×g for 15 mins, the supernatants of cell lysis were collected and the protein levels were quantified using BCA method. Cell lysates were further diluted in 4×sample buffer and boiled at 100°C for 10 mins. The samples were subjected to 10% SDS-PAGE separation and transferred onto the polyvinylidene fluoride membrane (Millipore, Billerica, USA). After blocking with 5% non-fat milk powder in TBST for 1 hr at room temperature, the membrane was incubated overnight with primary antibodies at 4°C. After incubation with HRP-linked anti-rabbit or mouse IgG for 2 hrs at room temperature, protein visualization was detected using the Pierce ECL Plus Western blotting substrate (Thermo Fisher Scientific, Inc., USA), and the GAPDH was served as the loading control.
Autophagic flux assay
Cells were retrovirally transduced with GFP-LC3-RFP-LC3ΔG (Addgene Inc.). After incubation in complete medium for 24 hrs, images were obtained under a fluorescence microscope. All image acquisition settings were performed at the same state during the image collection.
Statistical analysis
Data were presented as means±standard deviation. Statistical significance was determined by the Student’s t-test, and P-values <0.05 was considered statistically significant.
Results
Expression of TAZ in HCC cell lines
The expression of TAZ/YAP in HCC cell lines HepG2, SMMC-7721, SK-HEP1 and HCCLM3, four HCC cell lines with different spontaneous metastatic potential, was measured by Western blotting. As shown in Figure 1, TAZ remained highly abundant and was expressed at higher levels than YAP in all four HCC cell lines (Figure 1A), which is consistent with the publication by Hayashi,18 indicating that TAZ was a predominant protein while YAP was a minor protein in these HCC cells. Therefore, we selected low metastatic potential and high metastatic potential cell lines (SMMC-7721 and SK-HEP1, respectively) for subsequent experiments. We assessed the expression of TAZ in both SMMC-7721 and SK-HEP1 cells by using the lentiviral-mediated TAZ knockdown and knockout approaches. The knockdown efficiency of TAZ knockdown (KD TAZ) was confirmed in stable TAZ knocking down SMMC-7721 and SK-HEP1 cells by RT-qPCR and Western blotting (Figure 1B and C), while TAZ knockout (KO TAZ) was generated by using CRISPR/Cas9 gene disruption to generate TAZ null clones in SMMC-7721 and SK-HEP1 cells and confirmed by Western blotting (Figure 1D). No significant changes in YAP abundance were observed in shRNA-mediated TAZ knockdown (KD TAZ) and CRISPR-Cas9-mediated TAZ knockout (KO TAZ) cells (Figure 1C and D). To validate TAZ knockdown and knockout, we further confirmed the decreased mRNA levels of CTGF and CYR61, which are both well-known downstream targets of TAZ/YAP (Figure 1E). Thus, SMMC-7721 and SK-HEP1 cells stably knockdown and knockout TAZ have been well established to perform the subsequent experiments.
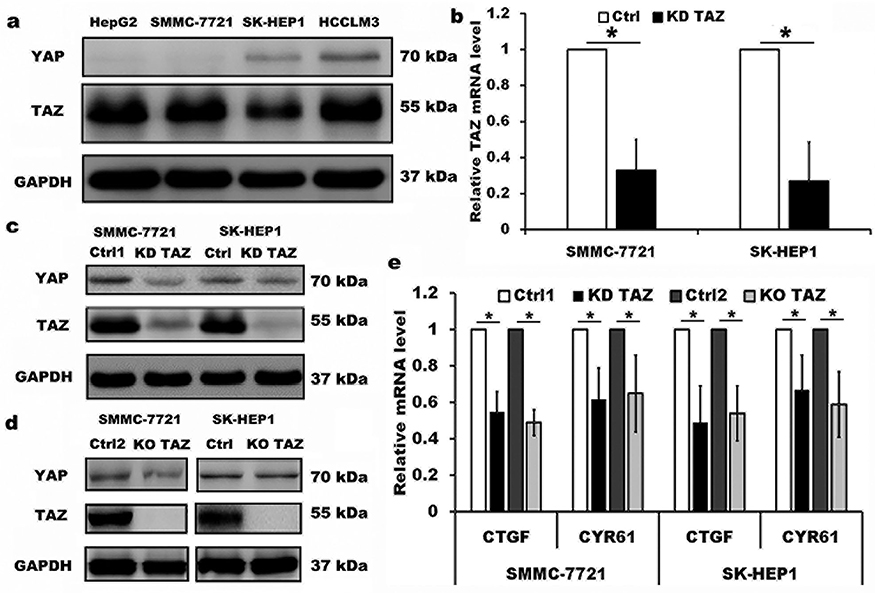 |
Figure 1 Knockdown and knockout of TAZ in the lentiviral-transduced SMMC-7721 and SK-HEP1 cells. Notes: (A) Western blot analysis of TAZ and YAP proteins in four HCC cell lines with different spontaneous metastatic potential. GAPDH served as loading control. Representative blots are shown (n=3). (B) TAZ mRNA levels were evaluated from SMMC-7721 and SK-HEP1 cells stably expressing shRNA specific for TAZ (KD TAZ) and the corresponding vector control (Ctrl) using RT-qPCR, and normalized against GAPDH. Data are the mean±SD (n=4). P-values were obtained from Student’s t test. *P<0.05. (C) Knockdown of TAZ (KD TAZ) was determined by Western blotting in SMMC-7721 and SK-HEP1 cells, and GAPDH served as a loading control. Representative blots are shown (n=3). (D) Knockout of TAZ (KO TAZ) was confirmed by Western blot analysis in SMMC-7721 and SK-HEP1 cells, and GAPDH served as a loading control. Representative blots are shown (n=3). (E) Quantitative analysis of CTGF and CYR61 mRNA in stable TAZ knockdown and knockout SMMC-7721 and SK-HEP1 cells by RT-qPCR, and normalized against GAPDH. Data were shown as the mean ±SD (n=4). P-values were obtained from Student’s t test. *P<0.05.Abbreviations: TAZ, transcriptional co-activator with PDZ-binding motif; YAP, yes-associated protein; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; KD TAZ, knockdown of transcriptional co-activator with PDZ-binding motif; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; KO TAZ, knockout of transcriptional co-activator with PDZ-binding motif; CTGF, connective tissue growth factor; CYR61, cysteine-rich angiogenic inducer 61; HCC, hepatocellular carcinoma. |
Silencing of TAZ inhibits the migration and invasion of HCC cells
It has been reported that TAZ knockdown results in inhibition of HCC cell migration and invasion.14 Considering that TAZ was predominantly expressed in HCC cells as mentioned above, we then performed transwell assays to assess whether TAZ knockout affects the migratory ability of the low metastatic potential SMMC7721 cells and the high metastatic potential SK-HEP1 cells, respectively. As shown in Figure 2, compared with the stably expressing control-shRNA (Ctrl1) and control-sgRNA (Ctrl2) cells, either knockdown or knockout of TAZ in SMMC-7721 and SK-HEP1 cells dramatically decreased the cell migration and invasion. These results suggest that silencing of TAZ inhibits the migration and invasion of HCC cells.
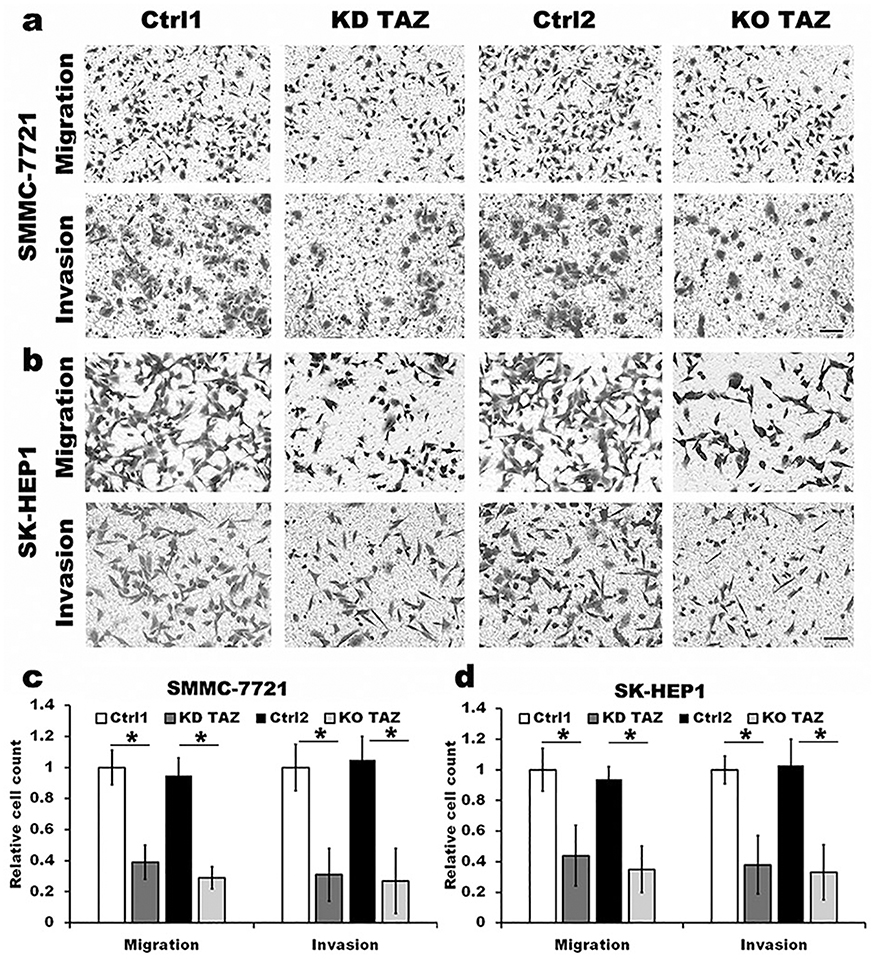 |
Figure 2 TAZ knockdown and knockout attenuated the migration and invasion of SMMC-7721 and SK-HEP1 cells. Notes: (A and B) Migration and invasion of stable TAZ knockdown and knockout SMMC-7721 and SK-HEP1 cells and their corresponding control cells were determined by Transwell assays. Representative images of three independent experiments are shown. Scale bar =50 μm. (C and D) Quantification of relative numbers of migrated and invaded cells in five randomly fields for each replicate was shown. All experiments were performed independently three times and data were shown as mean±SD. P-values were obtained from Student’s t test. *P<0.05.Abbreviations: TAZ, transcriptional co-activator with PDZ-binding motif; KD TAZ, knockdown of transcriptional co-activator with PDZ-binding motif; KO TAZ, knockout of transcriptional co-activator with PDZ-binding motif. |
Despite the studies described that TAZ knockdown plays an important role in EMT of cancer,12–14 the role of TAZ knockout in EMT of HCC remains unclear. To test whether knockout of TAZ affected the EMT, we performed Western blotting assay to examine the expression of EMT markers, including E-cadherin (E-CAD) and β-catenin (β-CAT) (epithelial marker), Vimentin (VIM) and N-cadherin (N-CAD) (mesenchymal markers). The results revealed that both TAZ knockdown (KD TAZ) and knockout (KO TAZ) upregulated the E-CAD and β-CAT, and downregulated the VIM and N-CAD in SMMC-7721 and SK-HEP1 cells, when compared with indicated control cells (Figure 3). Together, these data indicated that either TAZ knockdown or knockout reduces EMT in HCC cells.
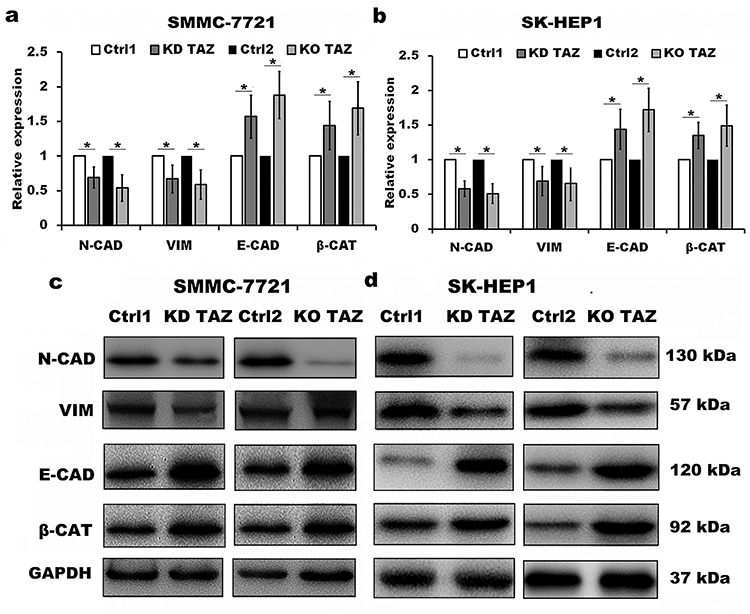 |
Figure 3 TAZ knockdown and knockout reduced epithelial-mesenchymal transition (EMT) of SMMC-7721 and SK-HEP1 cells. Notes: (A and B) Quantitative analysis of mRNA levels of EMT markers in stable TAZ knockdown and knockout SMMC-7721 (A) and SK-HEP1 cells (B) by RT-qPCR, and normalized against GAPDH. Data were shown as the mean±SD (n=4). P-values were obtained from Student’s t test. *P<0.05. (C and D) Protein levels of EMT markers were determined by Western blot in SMMC-7721 (C) and SK-HEP1 cells (D), and GAPDH was used as a loading control. Representative blots are shown (n=3).Abbreviations: TAZ, transcriptional co-activator with PDZ-binding motif; E-CAD, E-cadherin; β-CAT, β-catenin; VIM, Vimentin; N-CAD, N-cadherin; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; KD TAZ, knockdown of transcriptional co-activator with PDZ-binding motif; KO TAZ, knockout of transcriptional co-activator with PDZ-binding motif. |
TAZ regulates autophagy of HCC cells
TAZ has been recognized as an important regulator of cell migration and invasion in HCC cells, however, whether TAZ functions in HCC cell autophagy remains unknown. To identify the relationship between silencing of TAZ and autophagy induction in HCC cells, we firstly performed RT-qPCR and Western blotting to detect the effects of stable TAZ knockdown and knockout on the expression of the autophagy-related genes in SMMC-7721 and SK-HEP1 cells. TAZ knockdown and knockout significantly increased the expression of LC3B and ULK1, two well-established markers of autophagosome formation and autophagy induction, and decreased the levels of SQSTM1/p62 accumulation, a marker indicating low autophagic activity, in SMMC-7721 and SK-HEP1 cells (Figure 4A–D). To verify the role of stable TAZ knockdown and knockout in regulation of autophagic flux, the autophagic flux was measured by using a GFP-LC3-RFP-LC3ΔG reporter construct, which is based on the equimolar release of GFP-LC3 and RFP-LC3∆G upon cleavage by endogenous ATG4 proteases in the transfected cells, and GFP-LC3 is degraded by autophagy, while RFP-LC3ΔG remains in the cytosol, serving as an internal control, thus, the ratiometric GFP/RFP signal ratio inversely correlates with autophagy flux activation. In the ratiometric images, the green or yellow signal represents high GFP/RFP and low autophagic flux, while the red or orange signal represents low GFP/RFP and high autophagic flux.19 Abundant green signals were shown in indicated control cells, while fewer green signals were observed in TAZ knockdown and knockout SMMC-7721 and SK-HEP1 cells. The GFP/RFP ratio visualized by the merge (ratiometric) imaging was decreased in either TAZ knockdown or knockout SMMC-7721 and SK-HEP1 cells, indicating increased autophagic flux (Figure 4E and F). Taken together, these results clearly suggest that silencing of TAZ up-regulates the autophagy activity in HCC cells.
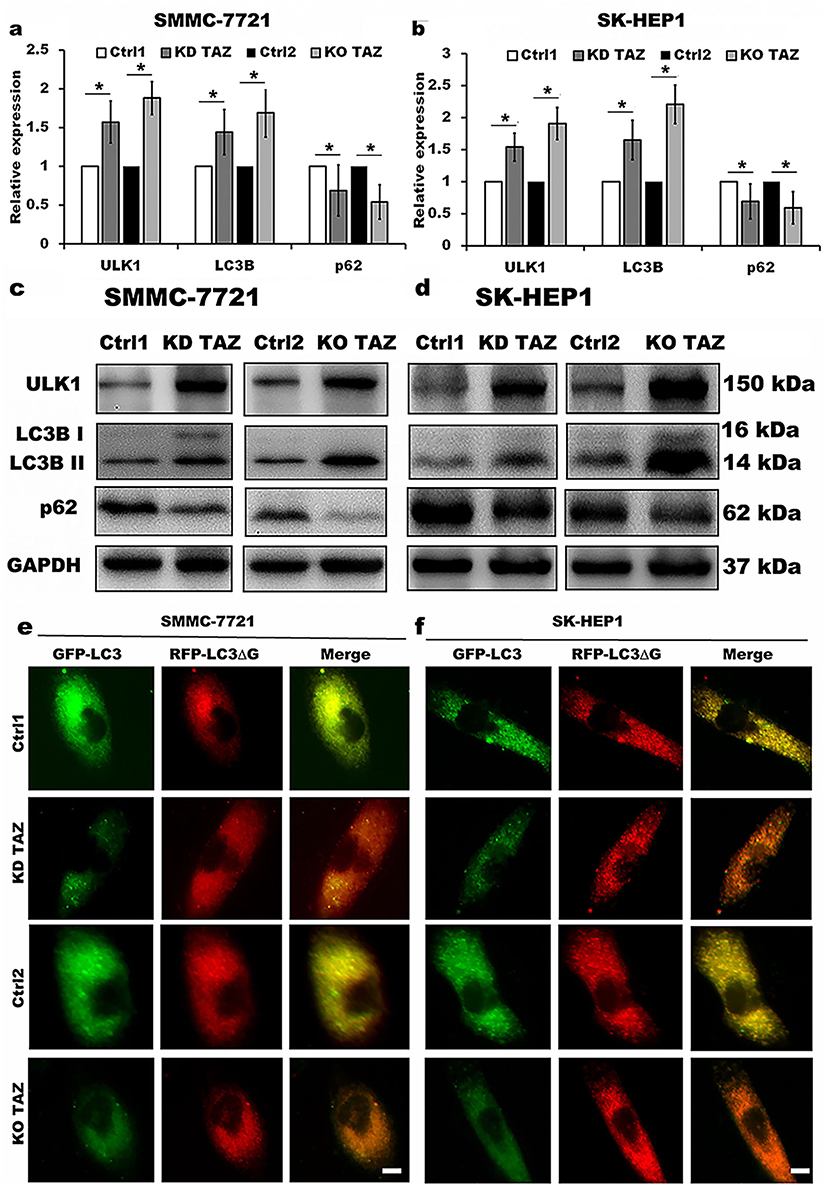 |
Figure 4 TAZ knockdown and knockout stimulated autophagy activity in SMMC-7721 and SK-HEP1 cells. Notes: (A and B) Quantitative analysis of mRNA levels of LC3B, ULK1 and p62 in stable TAZ knockdown and knockout SMMC-7721 (A) and SK-HEP1 cells (B) by RT-qPCR, and normalized against GAPDH. Data were shown as the mean±SD (n=4). P-values were obtained from Student’s t test. *P<0.05. (C and D) Protein levels of LC3B, ULK1 and p62 were determined by Western blot in SMMC-7721 (C) and SK-HEP1 cells (D), and GAPDH was used as a loading control. Representative blots are shown (n=3). (E and F) The autophagic flux was observed in stable TAZ knockdown and knockout SMMC-7721 (E) and SK-HEP1 cells (F) after retrovirally transfected with GFP-LC3-RFP-LC3ΔG reporter construct, and representative images are shown (n=3). Scale bar =10 μm.Abbreviations: TAZ, transcriptional co-activator with PDZ-binding motif; LC3B, microtubule-associated protein 1 light chain 3 beta; ULK1, unc-51 like autophagy activating kinase 1; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; KD TAZ, knockdown of transcriptional co-activator with PDZ-binding motif; KO TAZ, knockout of transcriptional co-activator with PDZ-binding motif. |
Silencing of TAZ inhibits HCC cell migration and invasion by autophagy induction
Evidence is accumulating that autophagy plays a direct role in key aspects of tumor cell motility and invasion.15,20,21 To confirm that stable TAZ knockdown and knockout inhibited the migration and the invasion of the HCC cells through autophagy promotion, 3-methyladenine (3-MA), a class I phosphoinositide 3-kinase and class III phosphatidylinositol 3-kinase inhibitor used widely as a pharmacological inhibitor in autophagy studies, and chloroquine (CQ), a lysosomotropic agent that neutralizes the acidic pH of lysosomes and inhibits the autophagolysosome formation, were given to cell motility assays. Data from transwell assays demonstrated that treatment with 3-MA or CQ partially abrogated the migratory and invasive ability of the cells induced by TAZ knockdown and knockout (Figure 5). As expected, treatment with 3-MA displayed a slight accumulation, whereas treatment with CQ significantly increased, the amount of SQSTM1/p62 and LC3B-II in SMMC-7721 and SK-HEP1 cells stably expressing control-shRNA or control-sgRNA. Notably, addition of CQ obviously increases the amount of LC3B-II and SQSTM1/p62 induced by either TAZ knockdown or knockout in SMMC-7721 and SK-HEP1 cells (Figure 6). Considering that both TAZ knockdown and knockout reduced EMT in HCC cells as mentioned above, in order to gain an insight into the molecular mechanisms involved in the autophagy-mediated modulation of cell migration and invasion in stable TAZ knockdown and knockout SMMC-7721 and SK-HEP1 cells, we further performed Western blotting to evaluate the expression of EMT markers in the presence of 3-MA and CQ. Treatment with both 3-MA and CQ significantly reduced TAZ-knockdown- and knockout-mediated E-CAD and β-CAT (epithelial marker) accumulation, and a meaningful increase in VIM and N-CAD proteins (mesenchymal markers) was also observed (Figure 6). Taken together, these data further support a role of silencing of TAZ inhibits HCC cell migration and invasion may be attributed at least in part to the autophagy induction.
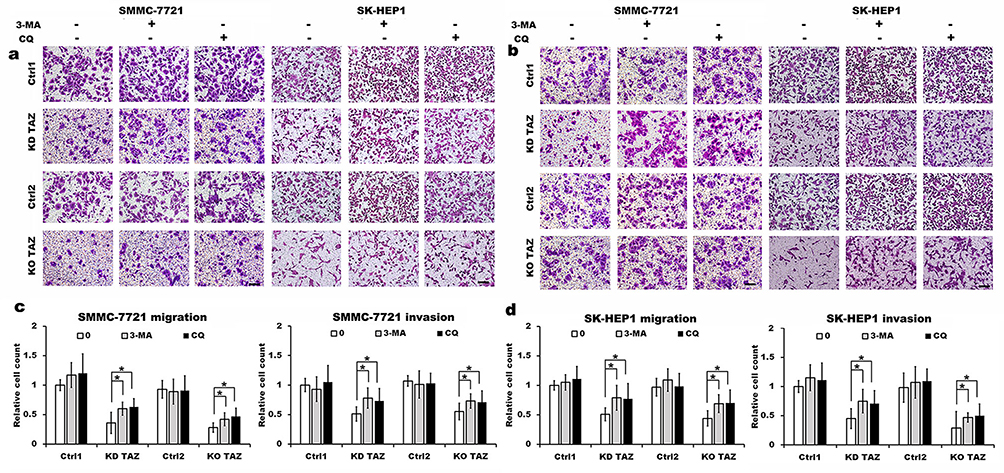 |
Figure 5 TAZ knockdown and knockout reduced the migratory and invasive ability of SMMC-7721 and SK-HEP1 cells through autophagy induction. Notes: (A and B) Migration and invasion of stable TAZ knockdown (A) and knockout (B) SMMC-7721 and SK-HEP1 cells and their corresponding control cells in the presence or absence of 10 mM 3-MA or 30 μM CQ were determined by Transwell assays. Representative images of three independent experiments are shown. Scale bar =50 μm. (C and D) Quantification of relative numbers of migrated and invaded cells in five randomly fields for each replicate was shown. All experiments were performed independently three times and data were shown as mean±SD. P-values were obtained from Student’s t test. *P<0.05.Abbreviations: TAZ, transcriptional co-activator with PDZ-binding motif; 3-MA, 3-methyladenine; CQ, chloroquine; KD TAZ, knockdown of transcriptional co-activator with PDZ-binding motif; KO TAZ, knockout of transcriptional co-activator with PDZ-binding motif. |
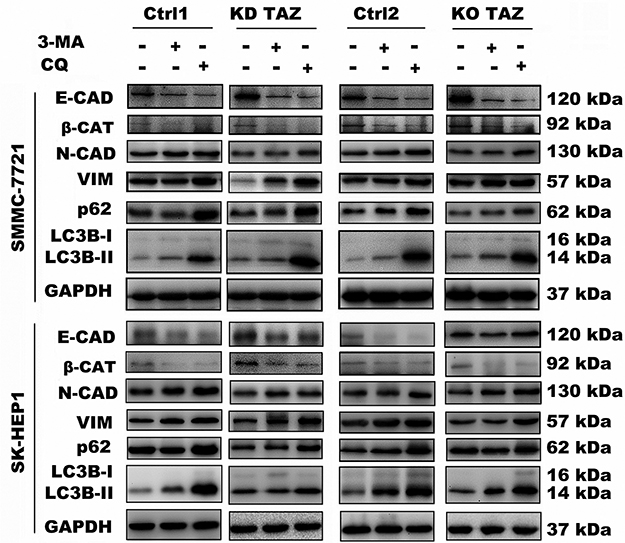 |
Figure 6 Effect of autophagy inhibition on the expression of the indicated genes in stable TAZ knockdown and knockout SMMC-7721 and SK-HEP1 cells. Notes: The indicated proteins in cellular extracts were determined by Western blot from SMMC-7721 and SK-HEP1 cells in the presence or absence of 3-MA (10 mM) or CQ (30 μM). GAPDH was used as a loading control. Representative blots are shown (n=3).Abbreviations: EMT, epithelial-mesenchymal transition; TAZ, transcriptional co-activator with PDZ-binding motif; 3-MA, 3-methyladenine; CQ, chloroquine; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; E-CAD, E-cadherin; β-CAT, β-catenin; VIM, Vimentin; N-CAD, N-cadherin; KD TAZ, knockdown of transcriptional co-activator with PDZ-binding motif; KO TAZ, knockout of transcriptional co-activator with PDZ-binding motif; LC3B, microtubule-associated protein 1 light chain 3 beta. |
Discussion
TAZ and YAP have often been described to equivalent downstream transcriptional co-activators of the Hippo tumor suppressor pathway, however, current study demonstrated that TAZ exhibited highly abundant and was expressed predominantly over YAP in selected four HCC cell lines with different spontaneous metastatic potential, suggesting that TAZ might play an important role in these HCC cells. Accumulating evidence has suggested that TAZ has oncogenic roles in human cancers via promoting cell proliferation, migration, and EMT,11–14 and autophagy is involved in the liver dysfunction and tumorigenesis.17,22,23 The findings presented in this study provide a new link between cell motility and autophagy induced by TAZ knockdown and knockout in HCC cell lines.
In the present study, we clearly observed that TAZ exhibited higher expression than YAP in selected four HCC cell lines with different spontaneous metastatic potential, which was consistent with the results in other studies,14,18,24,25 implying that TAZ might act as an oncogene involved in regulation of HCC cell motility. Hence, we firstly selected low metastatic potential SMMC-7721 cell line and high metastatic potential SK-HEP1 cell line in this study to test the effect of TAZ knockout by CRISPR/Cas9 and TAZ knockdown on the cell migration and invasion by Transwell migration and invasion assays. As expected and in agreement with previous reports that knockdown of TAZ reduced cell motility, knockout of TAZ in SMMC-7721 cells and SK-HEP1 cells also markedly decreased cell migration and invasion. The data further indicated the role of loss of TAZ resulting in the inhibition of cell migration and invasion in HCC cells. TAZ has been reported to contribute to HCC by regulating cell proliferation and EMT.14 Therefore, we next evaluated the effect of TAZ knockout and knockdown on the EMT-related markers. Our study showed that both TAZ knockout and knockdown increased the expression of epithelial markers E-CAD and β-CAT, and decreased the expression of mesenchymal markers and VIM and N-CAD at both the mRNA and protein levels. These results further explain the role of TAZ knockout and knockdown in HCC cell motility, probably by reduction of the EMT process.
Autophagy (or autophagocytosis) is an evolutionary conserved, natural, and regulated mechanism of the cell that disassembles unnecessary, dysfunctional components or damaged organelles. Accumulating evidence has emphasized the role of autophagy both as a tumor suppressor as well as a tumor promoter in tumor cell survival.26 On one hand, autophagy has been proposed to have a protective role for tumor by limiting genome instability and promoting survival of tumor cells. On the other hand, autophagy also acts as a tumor suppressor by preventing tumor initiation during the early stages of tumorigenesis.27 Considering that autophagy has been shown as an important modulator of the migration and the invasion in cancer cells,28 and TAZ has been emerged as a vital regulator of HCC cell migration and invasion, this prompted us to investigate whether TAZ knockout and knockdown in HCC cells had any effect on autophagy. We clearly presented confirmatory evidence that loss of TAZ in HCC cells promoted autophagy, which was supported by the observation including measuring LC3B-II, ULK1 and SQSTM1/p62 protein levels, detecting GFP-LC3-RFP-LC3ΔG fluorescence and colocalization (autophagic flux). Notably, we clearly observed that both TAZ knockdown and knockout in SMMC-7721 and SK-HEP1 cells obviously increased the accumulation of LC3B-II and ULK1, two well-established markers of autophagosome formation and autophagy induction, and decreased the amount of SQSTM1/p62, a marker indicating low autophagic activity. Furthermore, we also revealed that TAZ knockdown and knockout in SMMC-7721 and SK-HEP1 cells markedly induced autophagic flux. These findings provided evidence that loss of TAZ is linked with the autophagic degradation machinery in HCC cell lines.
Autophagic activity has been shown to be involved in cell migration and invasion,15,21,22 however, the functional connection between autophagy and cell motility induced by TAZ knockdown and knockout remains unexplored. In this study, we provide evidence that autophagy inhibition stimulated the HCC cell migration and invasion as well as a slight accumulation of SQSTM1/p62 and LC3B-II in the presence of autophagy inhibitor 3-MA, a class III PtdIns3K inhibitor, and obvious increased LC3B-II formation and the amount of SQSTM1/p62 in the presence of CQ, a drug known to inhibit autophagosome to lysosome fusion and widely used to monitor the autophagic flux. As expected, the reduction in the migration and the invasion of HCC cells following TAZ knockdown and knockout was partially reversed by treatment with 3-MA and CQ. Given that autophagy can modulate EMT,16,29 which is important in regulating cell migration and invasion, our results further demonstrated that TAZ knockdown and knockout resulted in autophagy activation, and reduced EMT as well as cell motility in HCC cells, and that, autophagy inhibition after treatment with 3-MA or CQ partially abrogated the same process among TAZ knockdown and knockout cells. Thus, our data clearly demonstrated that loss of TAZ induced autophagic activity limits the capability of HCC cell migration and invasion.
Collectively, data from this study indicate that HCC cells exhibited a TAZ-dominant expression rather than a YAP-dominant expression, and that an inhibitory role of silencing of TAZ by promoting autophagy and subsequent inhibiting the migration and the invasion of HCC cells. Further study of the underlying mechanism as to how TAZ depletion, YAP/TAZ double knockout and knockdown coordinate the autophagic activity and the connection between the autophagy and cell motility is ongoing; however, the findings presented in the current study may provide important clues for developing novel therapeutic strategies targeting the autophagy process to reduce the HCC metastasis.
Acknowledgments
This work was supported by the National Nature Science Foundation of China (no. 81672336), and the Training Program of Innovation and Entrepreneurship for College Students in Jiangsu Province (no. 201711117038Z).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. doi:10.1016/S0140-6736(18)30302-7
2. Xu X, Lou Y, Tang J, et al. The long non-coding RNA Linc-GALH promotes hepatocellular carcinoma metastasis via epigenetically regulating Gankyrin. Cell Death Dis. 2019;10(2):86. doi:10.1038/s41419-019-1300-3
3. Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Nat Acad Sci USA. 2012;109(37):E2441–E2450. doi:10.1073/pnas.1212021109
4. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell. 2016;166(2):288–298. doi:10.1016/j.cell.2016.05.051
5. Mowers EE, Sharifi MN, Macleod KF. Autophagy in cancer metastasis. Oncogene. 2017;36(12):1619–1630. doi:10.1038/onc.2016.333
6. Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. Autophagy in liver diseases. J Hepatol. 2010;53(6):1123–1134. doi:10.1016/j.jhep.2010.07.006
7. Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–1075.
8. Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev. 2013;27(4):355–371.
9. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol. 2017;18(12):758–770.
10. Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30(1):1–17. doi:10.1101/gad.274027.115
11. Zhou X, Lei QY. Regulation of TAZ in cancer. Protein Cell. 2016;7(8):548–561. doi:10.1007/s13238-016-0288-z
12. Shen S, Huang K, Wu Y, et al. A miR-135b-TAZ positive feedback loop promotes epithelial-mesenchymal transition (EMT) and tumorigenesis in osteosarcoma. Cancer Lett. 2017;407:32–44. doi:10.1016/j.canlet.2017.08.005
13. Ge L, Li DS, Chen F, Feng JD, Li B, Wang TJ. TAZ overexpression is associated with epithelial-mesenchymal transition in cisplatin-resistant gastric cancer cells. Int J Oncol. 2017;51(1):307–315. doi:10.3892/ijo.2017.3998
14. Xiao H, Jiang N, Zhou B, Liu Q, Du C. TAZ regulates cell proliferation and epithelial-mesenchymal transition of human hepatocellular carcinoma. Cancer Sci. 2015;106(2):151–159. doi:10.1111/cas.12587
15. Tuloup-Minguez V, Hamaï A, Greffard A, Nicolas V, Codogno P, Botti J. Autophagy modulates cell migration and β1 integrin membrane recycling. Cell Cycle. 2013;12(20):3317–3328. doi:10.4161/cc.26298
16. Qiang L, He YY. Autophagy deficiency stabilizes TWIST1 to promote epithelial-mesenchymal transition. Autophagy. 2014;10(10):1864–1865. doi:10.4161/auto.32171
17. Lan SH, Wu SY, Zuchini R, et al. Autophagy suppresses tumorigenesis of hepatitis B virusassociated hepatocellular carcinoma through degradation of micro-RNA-224. Hepatology. 2014;59(2):505–517. doi:10.1002/hep.26659
18. Wu W, Zhang X, Qin H, et al. Modulation of tumor cell migration, invasion and cell-matrix adhesion by human monopolar spindle-one-binder 2. Oncol Rep. 2015;33(5):2495–2503. doi:10.3892/or.2015.3855
19. Hayashi H, Higashi T, Yokoyama N, et al. An imbalance in TAZ and YAP expression in hepatocellular carcinoma confers cancer stem cell-like behaviors contributing to disease progression. Cancer Res. 2015;75(22):4985–4997. doi:10.1158/0008-5472.CAN-15-0291
20. Kaizuka T, Morishita H, Hama Y, et al. An autophagic flux probe that releases an internal control. Mol Cell. 2016;64(4):835–849. doi:10.1016/j.molcel.2016.09.037
21. Coly PM, Perzo N, Le Joncour V, et al. Chemotactic G protein-coupled receptors control cell migration by repressing autophagosome biogenesis. Autophagy. 2016;12(12):2344–2362. doi:10.1080/15548627.2016.1235125
22. Catalano M, D’Alessandro G, Lepore F, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol. 2015;9(8):1612–1625. doi:10.1016/j.molonc.2015.04.016
23. Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24(1):69–79. doi:10.1038/cr.2013.161
24. Wu SY, Lan S-H, Wu S-R, et al. Hepatocellular carcinoma-related cyclin D1 is selectively regulated by autophagy degradation system. Hepatology. 2018;68(1):141–154. doi:10.1002/hep.29781
25. Higashi T, Hayashi H, Ishimoto T, et al. miR-9-3p plays a tumour-suppressor role by targeting TAZ (WWTR1) in hepatocellular carcinoma cells. Br J Cancer. 2015;113(2):252–258. doi:10.1038/bjc.2015.170
26. Higashi T, Hayashi H, Kitano Y, et al. Statin attenuates cell proliferative ability via TAZ (WWTR1) in hepatocellular carcinoma. Med Oncol. 2016;33(11):123. doi:10.1007/s12032-016-0845-6
27. Choi KS. Autophagy and cancer. Exp Mol Med. 2012;44(2):109–120. doi:10.3858/emm.2012.44.2.033
28. Li X, Xu HL, Liu YX, An N, Zhao S, Bao JK. Autophagy modulation as a target for anticancer drug discovery. Acta Pharmacol Sin. 2013;34(5):612–624. doi:10.1038/aps.2013.23
29. Kenific CM, Debnath J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015;25(1):37–45. doi:10.1016/j.tcb.2014.09.001
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.