")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Silencing of circHIPK3 Inhibits Pressure Overload-Induced Cardiac Hypertrophy and Dysfunction by Sponging miR-185-3p
Received 8 January 2020
Accepted for publication 18 October 2020
Published 29 December 2020 Volume 2020:14 Pages 5699—5710
DOI https://doi.org/10.2147/DDDT.S245199
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Xiaohan Xu, Junhong Wang, Xiaowei Wang
Department of Cardiovascular Surgery, The First Affiliated Hospital with Nanjing Medical University, Nanjing 210029, People’s Republic of China
Correspondence: Xiaowei Wang
Department of Cardiovascular Surgery, The First Affiliated Hospital with Nanjing Medical University, 300 Guangzhou Road, Gulou District, Nanjing 210029, People’s Republic of China
Email [email protected]
Background: Cardiac hypertrophy is induced by diverse patho-physiological stimuli and indicates an increase in cardiomyocyte size. Circular RNAs (circRNAs) and microRNAs (miRNAs), members of noncoding RNAs, are involved in several biological processes and cardiovascular diseases (CVD). Here, we investigated the potential role of circHIPK3, which is produced by the third exon of the HIPK3 gene in cardiac hypertrophy.
Methods: qRT-PCR and Sanger sequencing were conducted to identify the expression and characteristics (head-to-tail structure, stability, and location) of circHIPK3 in cardiac hypertrophy; Immunostaining of α-SMA was performed to evaluate the size of the cardiomyocytes; Transverse aortic constriction (TAC) induced hypertrophy models of mice were established to investigate the effect of circHIPK3 in vivo. Bioinformatics analysis and luciferase reporter assays, RNA immunoprecipitation, and fluorescence in situ hybridization (FISH) experiments were conducted to investigate the mechanism of circHIPK3-mediated cardiac hypertrophy.
Results: circHIPK3 is circular, more stable, and mainly located in the cytoplasm. Silencing of circHIPK3 inhibited the TAC induced cardiac hypertrophy, and reversed the effect of TAC on the echocardiograph parameters, such as left ventricular end-diastolic pressure (LVEDPS), left ventricular fraction shortening (LVFS), left ventricular ejection fraction (LVEF), and left ventricular systolic dysfunction (LVSD), and also the heart weight to tibial length (HW/TL). Angiotensin II (Ang II) Ang II-treated cardiomyocytes showed larger size of cardiomyocyte and upregulation of fetal genes, biomarkers of cardiac hypertrophy, peptide hormones, atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP), and myofilament protein, β-myosin heavy chain (β-MHC). These effects were reversed by circHIPK3 knockdown. Mechanically, circHIPK3 sponges miR-185-3p. In addition, miR-185-3p targets CASR. The rescue experiments confirmed the interaction between circHIPK3 and miR-185-3p as well as miR-185-3p and CASR.
Discussion: Our data suggested that circHIPK3 serve as a miR-185-3p sponge to regulate cardiac hypertrophy revealing a potential new target for the prevention of TAC- and Ang-II induced cardiac hypertrophy.
Keywords: cardiac hypertrophy, circHIPK3, angiotensin II, transverse aortic constriction, TAC, miR-185-3p, calcium signaling receptor
Introduction
The heart is a dynamic organ and responds to several internal and external patho-physiological hemodynamic overloads such as hypertension, valve disease, myocardial infractions, or regular physical activity and chronic exercise training which lead to cardiac hypertrophy, eventually causing heart failure, responsible for sudden death worldwide.1,2 Cardiac hypertrophy causes an upregulation in the expression of cardiac fetal genes, such as ANP, BNP and β-MHC, increase in the protein content, and assembly of sarcomeres.1,3
Circular RNAs (circRNAs) are dynamic, abundant, evolutionarily conserved, stable, structurally specific noncoding RNAs having a remarkable continuous closed loop structure by covalently joining 3ʹ and 5ʹ ends;4 due to their diverse presence in the body fluids, such as blood, exosomes, saliva, and seminal plasma, they have been functionally associated with different physiological and pathological processes, including autophagy, apoptosis, cell cycle, and proliferation. They are widely present in humans, rats, and mice hearts.5 circRNAs are multifunctional, acting as miRNA sponges and protein sponges, and regulate alternative splicing, transcriptional and post-transcriptional regulation.6 Emerging evidence suggests the role of circRNAs in many diseases, such as neurologic diseases, cardiovascular diseases, cancers, immune diseases, aging, diabetes, digestive diseases, skin, muscle and bone diseases, hepatopathy, and hypertension.6 Circular RNAs play a pivotal role in cardiovascular diseases.7 The first evidence suggesting the role of circRNA in regulating heart physiology and pathology came from studies of heart-related cirRNAs (CRHR) which act as an endogenous sponge of miR-223, thereby inhibiting the miR-223-induced cardiac hypertrophy and heart failure in mice.8,9 Microarray analyses of dysregulated circRNAs indicate detection of 1,163 circRNAs, out of which 63 circRNAs are differentially expressed in heart tissue of mice with myocardial infarction-induced heart failure.10 Recently, circRNAs act as biomarkers in the detection of acute myocardial injury.11 circRNAs sequencing reveals 401 circRNAs are dysregulated, with 303 circRNAs upregulated and 98 circRNA downregulated in the isoproterenol hydrochloride-induced cardiac hypertrophy.12 One of the most abundant circRNA which has been studied extensively is circRNA homeodomain-interacting protein kinase 3 (circHIPK3).13 circHIPK3 is abundant in heart, lungs, liver, etc; its dysregulation is involved in a number of biological processes, including cardiovascular diseases.14
miRNAs are single stranded, endogenous, small (~22 nucleotides) non-coding RNAs;15 they are key regulators of post-transcriptional gene expression and their deregulations are associated with several diseases; they are used as potential diagnostic and prognostic biomarkers of cardiovascular diseases.16 miRNA can target many mRNAs, and a unique mRNA can be the target of multiple miRNAs.17 miRNAs can positively or negatively regulate cardiomyocyte hypertrophy. For example, miR-1,18 miR-133,19 miR-208a and miR-208b,20,21 miR-212/132 family,22 and miR-23a23 are involved in the progression of hypertrophy.
The underlying molecular mechanism of cardiac hypertrophy remains largely unknown. The present work enhanced our understanding on the role of circHIPK3 in cardiac hypertrophy by sponging miR-185-3p and targeting the downstream signaling molecule CaSR. We have produced conclusive evidence which might support the potential role of the circHIPK3/miR-185-3p/CaSR axis in cardiovascular physiology and pathophysiology.
Materials and Methods
The Establishment of Cardiac Hypertrophy Model
Male C57BL/6 mice (8–10 weeks old) were anesthetized with 2% isoflurane and subjected to thoracotomy to establish the cardiac hypertrophy model using transverse aortic constriction (TAC) method. A 6.0 silk suture was placed across the aorta with a 26G blunt needle to obtain the aorta constriction of 0.46 mm in diameter. Mice in the sham group underwent similar surgery, but without ligation. All animal experiments were approved by the Animal Research Ethics Committee of the First Affiliated Hospital, Nanjing Medical University, China, and were performed according to the Guide for the Care and Use of Laboratory Animals published by the United States National Institute of Health, USA.
The mice were randomly assigned to the following groups: sham, TAC, and TAC+si-circHIPK3. The adenovirus (Biocobio, Tianjin, China) of circHIPK3 was injected into the left ventricle myocardium 1 week before the TAC surgery. Heart tissues were collected for further detection after 4 weeks of TAC treatment.
Cell Culture and Transfection
Neonatal C57BL/6 mice were anesthetized with 5% isoflurane, and mice were sacrificed by cervical dislocation until they lost their righting reflex. The left ventricular tissues were isolated and minced into 1–3 mm3 pieces. Primary neonatal mouse cardiomyocytes (NMCMs) were separated from the tissues with digestion of 0.05% collagenase II (Invitrogen, CA, USA) and 0.5% trypsin (Gibco, USA). HEK-293T cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). NMCMs and HEK-293T cells were maintained at 37°C with 5% CO2in Dulbecco’s Modified Eagle Medium (DMEM, Hyclone Laboratories, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, Grand Island, NY, USA).
When cells reached confluence, NMCMs were removed by trypsin-EDTA and plated in 6-well plates. Adenovirus mediated knockdown of circHIPK3 (ad-si-circHIPK3) and miR-185-3p (ad-si-miR-185-3p), as well as overexpression of CaSR (ad-CaSR) were added to cells for 24 hours, then the culture medium was replaced for an additional 48 hours in the presence of 1 μM angiotensin II (Ang II, Sigma, USA) to induce cardiomyocyte hypertrophy.
Bioinformatics Analysis
The circinteractome software (https://circinteractome.nia.nih.gov/) was used to predict the target gene of circHIPK3. The targetscan7.2 (http://www.targetscan.org/vert_72/) was used to predict the target gene of miR-185-3p.
Luciferase Reporter Assay
The wild type or mutated sequence of circHIPK3 and CASR containing the predicted binding site of miR-185-3p was amplified and inserted into the luciferase reporter vector pMIR-Report (Promega, Madison, WI, USA). HEK-293T cells were co-transfected with luciferase reporter constructs, as well as internal control pRL-TK (Promega) and miR-185-3p or miR-control by using Lipofectamine 2000 (Invitrogen). At 48 hours post-transfection, cells were harvested and luciferase activity was measured using a dual-luciferase Reporter Assay System (Promega). The relative luciferase activity was normalized to renilla luciferase activity.
RNA Extraction and Real-Time PCR Analysis
Total RNA were isolated from heart tissues and cultured cardiomyocytes using Trizol (Invitrogen). Reverse transcription were performed using a EasyScript First-Strand cDNA Synthesis SuperMix kit (Transgen, Beijing, China) according to the manufacturer’s instructions. qRT-PCR were carried out using SYBR Green Master Mix (Thermo Fisher Scientific) in Light Cycler®480 System (Roche). The relative abundance of mRNA was calculated according to the 2−ΔΔCt method. GAPDH or U6 was used as an internal reference for the mRNAs and miRNAs, respectively. The primers used in RT-PCR were shown as follows: forward: CACAGATCTGATGGATTTCAAGA and reverse: CCTCATCTTCTACCGGCATC for ANP; forward: GTCAGTCGTTTGGGCTGTAAC and reverse: AGACCCAGGCAGAGTCAGAA for BNP; forward: CCGAGTCCCAGGTCAACAA and reverse: CTTCACGGGCACCCTTGGA for β-MHC; forward: ATCCATAGCAGCGGATCGACT and reverse: CAGGATAGCATGCAGACTTAGC for circHIPK3; forward: ACTCCACTCACGGCAAATTC and reverse: TCTCCATGGTGGTGAAGACA for GAPDH; forward: CACAGATCTGATGGATTTCAAGA and reverse: CCTCATCTTCTACCGGCATC for U6.
RNase R Digestion and Actinomycin D Treatment
RNA samples were degraded using RNase R (TAKARA, Dalian, China) for 20 minutes at 37°C and then subjected to qPCR. In addition, the cardiomyocytes were treated with 20 ng/mL Actinomycin D (Amresco, USA) to block new RNA synthesis, and then, total RNA was extracted at the time point of 1, 2, 4, 8, and 16 hours for the further qPCR analysis.
RNA Pull Down
The experiment was performed using RNA pull down kit. RNA pull-down was carried out using a specific biotinylated probe that hybridized with target RNA (mRNA or miRNA). It can then be pulled down, reversely transcribed to cDNA, and identified by qRT-PCR.
Immunostaining
Cells for immunostaining were fixed with 4% paraformaldehyde for 20 minutes and permeabilized with 0.5%Triton X-100 for 15 minutes, and then blocked with 3% BSA for 30 minutes. Cells were incubated with the primary antibody (α-SMA, Abcam, Cambridge, UK) at 4 °C overnight. After washing thrice, the blots were incubated with Alexa Fluor-labeled secondary antibody (Invitrogen) for 2 hours followed with DAPI staining for another 10 minutes (Olympus, Japan).
Histological Analysis
Heart tissues fixed with paraformaldehyde were embedded with paraffin and cut into slices of 4 μm. The size and morphologic alterations of the heart was assessed by hematoxylin/eosin (HE) staining and Masson trichrome staining. Sections were mounted and scanned with a fluorescent microscope (Olympus, Japan).
Echocardiography for Mice
Echocardiography of the left ventricle in mice with TAC for 4 weeks was performed using the Vevo 2100 Imaging System. Briefly, mice were anesthetized with 2% isoflurane then maintained under anesthesia with 1.5% isoflurane and examined. The mouse was placed chest up on an examination board and wiped clean to remove all hair in the area of interest. The M-mode measurements of left ventricular (LV) dimensions were conducted using the scan head and calculated from more than three heart cycles. Left ventricular inner diameters during diastole and systole were determined from M-modes along with heart rate. Diastolic and systolic volumes were acquired by applying Simpson’s rule of discs to the serially acquired short axis images. Stroke volume was calculated as: diastolic volume–systolic volume. Ejection fraction was calculated as: (stroke volume/diastolic volume)×100%. Cardiac output was determined by: stroke volume×heart rate.
Western Blot Assay
Cell lysates were prepared and the level of protein expression was measured using Bradford method. Forty micrograms of protein was loaded to the SDS-PAGE for electrophoresis. Then the proteins were transferred to the PVDF membrane and blocked with 5% milk in TBS-T for 1 hour and then incubated with specific primary antibodies (anti-CASR, Abcam, UK; anti-GAPDH, Abcam) at 4°C overnight and secondary antibodies conjugated with horseradish peroxidase at room temperature for 1 hour, respectively. The signals on the membrane were detected using nitrocellulose membrane (NCM) in a chemiluminescence imaging system (Tanon, Beijing, China).
Statistical Analysis
All data are presented as the means±standard deviation (SD). The statistical analyses were performed using one-way ANOVA and Student’s test. The data were analyzed using GraphPad Prism 5.0, and P<0.05 was considered to be statistically significant. Data are expressed as means±SD from three independent experiments.
Results
Identification and Validation of circHIPK3 in Cardiomyocyte
CircHIPK3 are widely present in humans, rats, and mice hearts (Werfel et al, 2016; Wang et al, 2019). CircBase retrieval revealed that 907 bp of CircHIPK3 was derived from the exon 9–13 of the HIPK3 gene. Sanger sequencing further confirmed the head-to-tail splicing of this product (Figure 1A). As we know, circRNAs are stable as they are a ring structure. In order to evaluate the stability of circHIPK3, we evaluated the amount of circHIPK3 in cardiomyocytes under RNase R treatment. The amount of linear mRNA of HIPK3 decreased drastically under the RNase R treatment (Figure 1B). However, no alteration was found in circHIPK3 under the digestion of RNase R. The half-life of the linear and circular HIPK3 was evaluated under Actinomycin D treatment. Our results indicated that circular HIPK3 is much more stable than the linear form of HIPK3 (Figure 1C). Furthermore, cDNA and gDNA were extracted separately from cardiomyocytes and subjected to nucleic acid gel electrophoresis detection. The results indicated that circHIPK3 could be detected in cDNA but not in gDNA (Figure 1D). qRT-PCR analysis of nuclear and cytoplasmic RNAs further showed that CircHIPK3 is mainly localized in the cytoplasm of cardiomyocyte (Figure 1E). Similarly, RNA FISH further confirmed that circHIPK3 was mainly localized in the cytoplasm (Figure 1F).
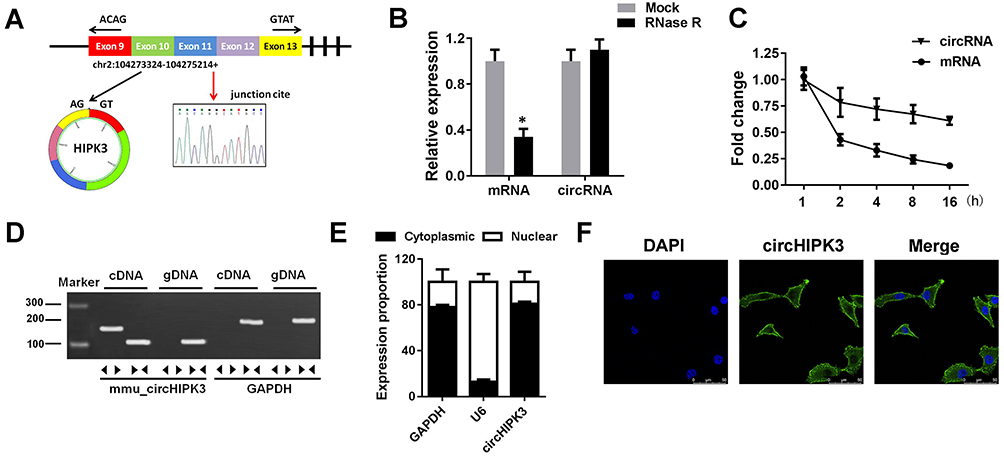 |
Figure 1 The identification of circHIPK3 in cardiomyocytes. (A) circHIPK3 was back-spliced by exons 9–13 of HIPK3 gene as the black arrow showed. The existence of circHIPK3 was confirmed by Sanger sequencing, the red arrow shows the head-to-tail splicing junction site of circHIPK3. (B) qRT-PCR analysis of linear mRNA HIPK3 and circHIPK3 after RNase R treatment. (C) qRT-PCR analysis of circHIPK3 and HIPK3 mRNA after treatment with Actinomycin D. (D) qRT-PCR validation of the existence of circHIPK3. Divergent primers amplified circHIPK3 in cDNA but not in genomic DNA (gDNA). (E) qRT-PCR assay was performed to evaluate the location of circHIPK3. (F) FISH was conducted to indicate the assay with location of circHIPK3. *P<0.05 vs mock group. Scale bar=50 μm. Data are expressed as means±SD from three independent experiments. |
circHIPK3 Up-Regulation Was Observed in Mice with Cardiac Hypertrophy
HE staining revealed that TAC treated hearts have a larger size, suggesting cardiac hypertrophy (Figure 2A). Masson staining showed that TAC treatment significantly induced myocardial fibrosis (Figure 2B). Furthermore, echocardiographic parameters of the mice, including LVEDP, LVFS, LVEF, and LVESD were evaluated with an ultrasound system which further indicated that the cardiac function of the mice subjected to TAC treatment was impaired (Figure 2C). The ratio of heart weight to tibial length (HW/TL) was increased after TAC treatment (Figure 2D). In addition, an in vitro hypertrophy model was established by treating cardiomyocytes with Ang II; immunofluorescence with alpha-smooth muscle actin (α-SMA) displayed larger size of cardiomyocytes treated with ang II as compared to the sham ones (Figure 2E). The levels of fetal genes like ANP, BNP, and β-MHC are used to evaluate the extent of myocardial hypertrophy; the three fetal genes showed upregulation under Ang II treatment (Figure 2F). Real-time PCR analysis revealed that circHIPK3 expression was increased in cardiomyocytes after treatment with Ang II compared to that in the control group (Figure 2G). Furthermore, the level of circHIPK3 was upregulated in cardiac tissue of TAC mice as compared to the sham ones (Figure 2H).
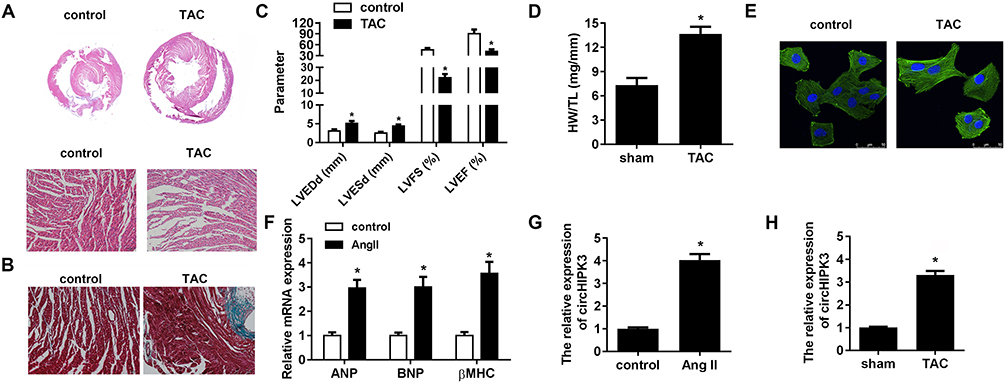 |
Figure 2 Ang II and TAC upregulated the expression of circHIPK3 in cardiomyocyte and myocardium, respectively. (A) The morphology and size of cardiomyocyte subjected to TAC treatment or without TAC treatment were shown using Hematoxylin&eosin (HE) staining. (B) Masson staining was performed to evaluate the myocardial fibrosis. (C) Echocardiographic parameters of the mice, including LVEDP, LVFS, LVEF, and LVESD were measured with a ultrasound system. (D) Quantification of heart weight-to-tibial length ratio was carried out. (E) The morphology and size of cardiomyocyte was detected by immunofluorescence. (F) qRT-PCR analysis was done to evaluate the level of three cardiac fetal genes ANP, BNP, and β-MHC in cardiomyocytes. (G, H) qRT-PCR analysis detected the expression level of miR-185-3p in Ang II-treated cardiomyocytes and TAC-treated myocardium. *P<0.05 vs sham or control group. Scale bar=50 μm. Data are expressed as means±SD from three independent experiments. |
circHIPK3 Silencing Suppresses Cardiac Hypertrophy Induced by TAC and Ang II
Adenovirus vector was used for knocking down circHIPK3 to determine the functional effects of circHIPK3 silencing. Cells were transfected with ad-si-circHIPK3 by intravenously injecting them with ad-si-circHIPK3. We first evaluated the efficiency of the adenovirus and our results indicated that the level of circHIPK3 was significantly reduced both in the myocardium and cardiomyocytes under ad-si-circHIPK3 transduction (Figure 3A and B). circHIPK3 knock down also led to a decrease in the ratio of heart weight to tibial length as compared to the sham ones (Figure 3C). As expected, HE staining also revealed that circHIPK3 knockdown reduced the size of hearts and subsequently reduced the cardiac hypertrophy, protecting cardiac function in TAC-induced cardiac hypertrophy (Figure 3D). In addition, echocardiogram parameters were detected; reduction of LVEDd and LVESd and elevation of LVFS and LVEF under circHIPK3 knockdown (Figure 3E). Afterwards, our in vitro experiments revealed that the cardiomyocytes size was significantly decreased in the ad-si-circHIPK3 group suggesting that circHIPK3 knockdown has a protective role and recovered the Ang II–induced cardiomyocyte phenotype (Figure 3F). Moreover, we found significantly reduced levels of mRNA of ANP, BNP, and β-MHC under ad-circHIPK3 transfection as compared to the sham ones (Figure 3G).
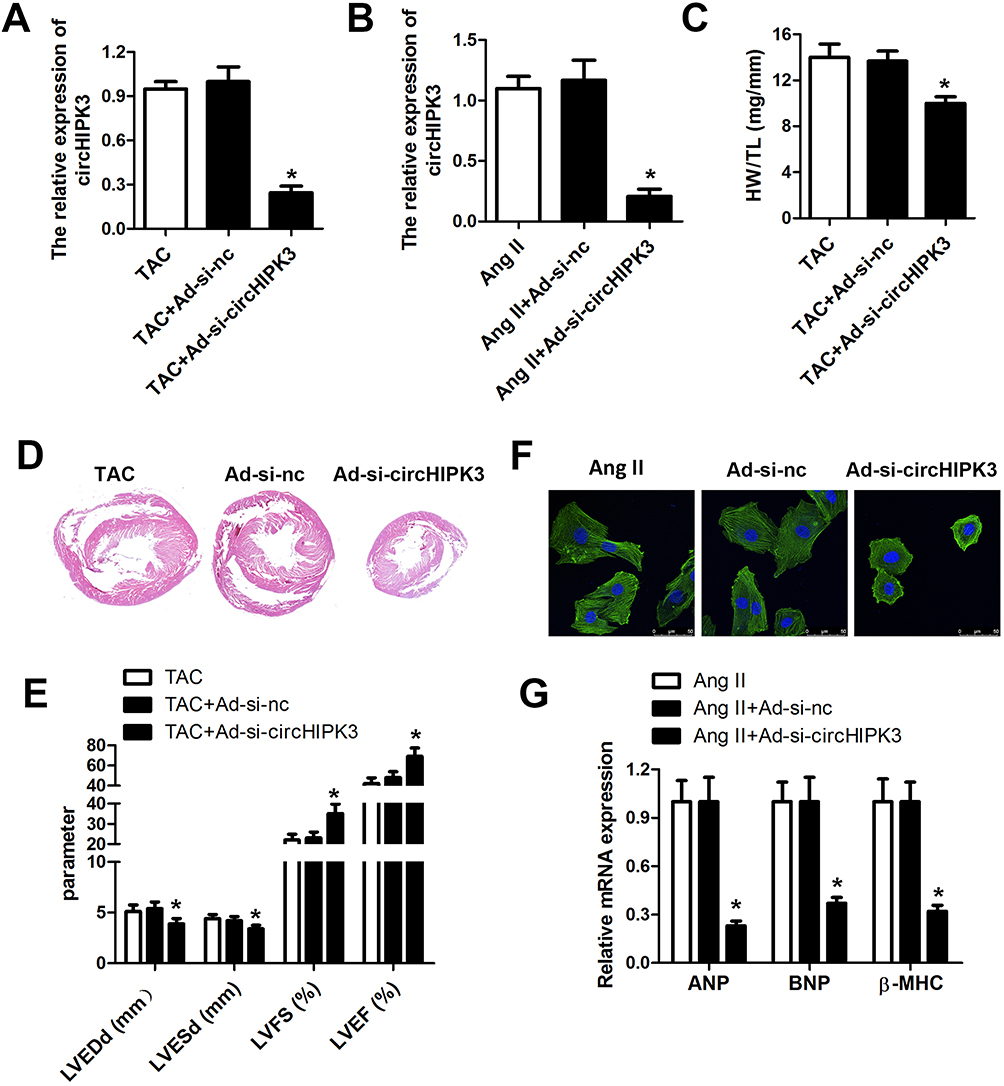 |
Figure 3 Knockdown of circHIPK3 released Ang II- and TAC-induced cardiac hypertrophy. (A, B) qRT-PCR analysis was used to evaluate the efficiency of the adenovirus mediated knockdown of circHIPK3, and circHIPK3 level was significantly reduced in cardiomyocyte and myocardium. (C) Quantification of heart weight-to-tibial length ratio was calculated. (D) Hematoxylin and eosin staining was performed to evaluate the size of the heart under TAC, TAC+ad-nc, and TAC+ad-si-circHIPK3 treatment. (E) Echocardiographic parameters of the mice including LVEDP, LVFS, LVEF, and LVESD were measured with an ultrasound system. (F) The morphology and size of cardiomyocyte subjected to Ang II, Ang II+ad-nc, and Ang II+ad-si-circHIPK3 treatment. (G) qRT-PCR analysis was used to determine the levels of ANP, BNP, and β-MHC. *P<0.05 vs Ang II+ad-nc group or TAC+ad-nc group. Scale bar=50 μm. Data are expressed as means±SD from three independent experiments. |
CircHIPK3 Act as a miR-185-3p Sponge in Cardiac Hypertrophy
To explore the underlying mechanism of circHIPK3 in cardiac hypertrophy, we investigated its potential interacting miRNAs by bioinformatics prediction analysis. We found that the circHIPK3 contains a highly conserved miR-185-3p seed sequence (Figure 4A). To validate the prediction, we cloned the wild-type or mutant sequence containing the predicted targeting region and dual luciferase assay was carried out. Compared to the control mimic group, the mir-185-3p significantly reduced the luciferase activity of pRL-TK-circHIPK3 vector but did not affect the luciferase activities of the mutant vectors (Figure 4B). To further confirm the correlation between miR-185-3p and circHIPK3, we validated the expression of miR-185-3p in response to circHIPK3 overexpression or knockdown. The qPCR results showed that circHIPK3 overexpression significantly reduced the miR-185-3p level in cardiomyocytes while circHIPK3 knockdown notably increased the miR-185-3p level (Figure 4C). Next, we performed FISH to determine whether circHIPK3 and miR-185-3p colocate in cardiomyocytes. The results revealed that circHIPK3 and miR-185-3p colocalize in the cytoplasm of cardiomyocytes (Figure 4D). RNA pull down assay was further carried out indicating that a miR-185-3p probe enriched the expression of circHIPK3 compared to the control probe group (Figure 4E). Pearson correlation coefficient analysis showed an existence of a negative correlation between circHIPK3 and miR-185-3p (Figure 4F). These results demonstrated that circHIPK3 acts as a sponge of miR-185-3p in cardiomyocytes.
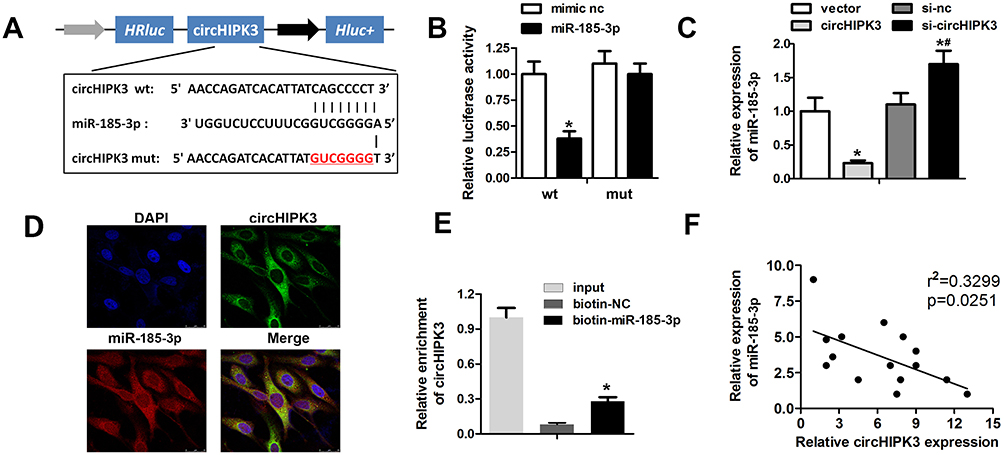 |
Figure 4 circHPIK3 act as a miR-185-3p sponge in cardiomyocytes. (A) Schematic representation of target site between circHIPK3 and miR-185-3p. (B) Luciferase reporter assay was carried out to verify whether miR-185-3p targets circHIPK3 in cardiomyocytes. (C) qRT-PCR analysis was used to detect the expression of miR-185-3p in different groups. (D) FISH assay with specific probes targeting miR-185-3p and circHIPK3 was performed to detect the co-localized miR-185-3p (red) and circHIPK3 (green) in cardiomyocytes. (E) RNA pull down assay with the specific probe of miR-185-3p was used to confirm the interaction between circHIPK3 and miR-185-3p. (F) Pearson correlation coefficient analysis indicated a negative correlation between circHIPK3 and miR-185-3p. *P<0.05 vs mimic nc, vector or biotin-nc group; #P<0.05 vs si-nc group. Scale bar=50 μm. Data are expressed as means±SD from three independent experiments. |
Silencing of miR-185-3p Reversed the Effect of circHIPK3 Knockdown on Cardiac Hypertrophy
To further investigate the correlation between circHIPK3 and miR-185-3p, we detected whether silencing of miR-185-3p could reverse the effect of circHIPK3 knockdown. qRT-PCR was used to evaluate the miR-185-3p expression. The results indicated a significant down-regulation of miR-185-3p in cardiomyocytes subjected to AngII treatment; similarly, circHIPK3 knockdown elevated the level of miR-185-3p. Adenovirus knockdown of miR-185-3p obviously reduced the level of miR-185-3p (Figure 5A). Next, assessment of cardiomyocyte size and the expression of hypertrophic genes including ANP, BNP, and β-MHC were performed. The results demonstrated that the miR-185-3p silencing reversed the effect of circHIPK3 on cardiomyocyte size and the expression of ANP, BNP, and β-MHC (Figure 6C). These findings further confirmed the interaction between miR-185-3p and circHIPK3.
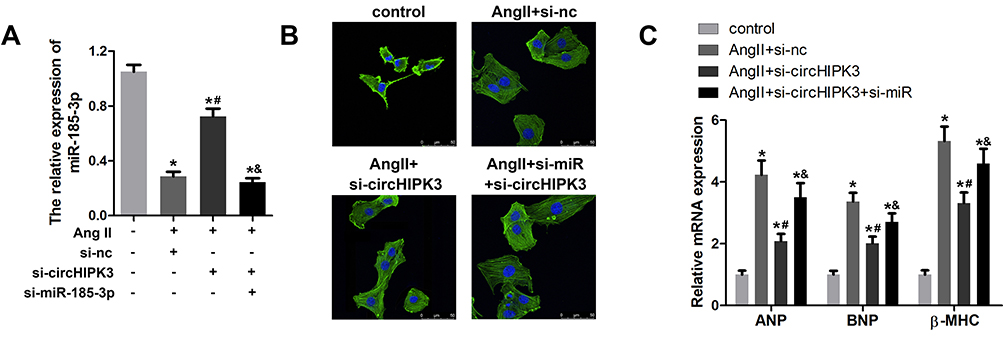 |
Figure 5 miR-185-3p silencing reversed the effect of circHIPK3 in cardiomyocytes. (A) qRT-PCR analysis was used to evaluate the efficiency of the adenovirus mediated knockdown of miR-185-3p. (B) The morphology and size of cardiomyocyte treated with control, Ang II+ad-si-nc, Ang II+ad-si-circHIPK3, and Ang II+ad-si-circHIPK3+ad-si-miR-185-3p. (C) qRT-PCR analysis was used to detect the level of ANP, BNP, and β-MHC. *P<0.05 vs control group, #P<0.05 vs AngII+ad-si-nc group, &P<0.05 vs AngII+ad-si-circHIPK3 group. Scale bar=50 μm. Data are expressed as means±SD from three independent experiments. |
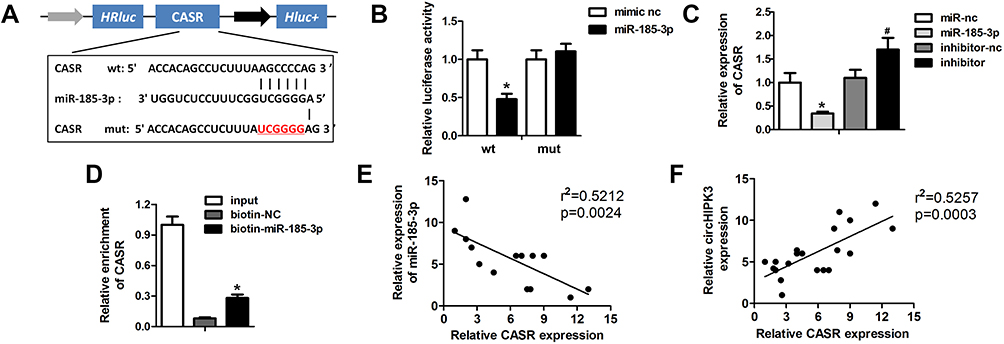 |
Figure 6 miR-185-3p directly target CaSR in cardiomyocytes. (A) Schematic representation of target site between miR-185-3p and CaSR. (B) Luciferase reporter assay was carried out to verify whether miR-185-3p targets CASR in cardiomyocytes. (C) knockdown of miR-185-3p to monitor the expression of CASR was detected by Western blot. (D) Enrichment of CASR. (E, F) Pearson correlation coefficient analysis was performed to investigate the correlation between circHIPK3 and CASR as well as miR-185-3p and CASR. *P<0.05 vs mimic nc, biotin-nc, miR-nc group, #P<0.05 vs inhibitor-nc group. Scale bar=50 μm. Data are expressed as means±SD from three independent experiments. |
Regulatory Effect of miR-185-3p on CaSR
miRNAs bind and suppress the expression of their target genes to participate in the regulation of pathological process (Kim et al, 2015). To explore the precise mechanism of circHIPK3 and miR-185-3p, we investigated the potential targets of miR-185-3p. Bioinformatics analysis was carried out to determine the potential target of miR-185-3p and it was found to be CaSR (Figure 6A). To validate the bioinformatics prediction analysis, we cloned the wild-type or mutant sequence containing the predicted targeting region into the luciferase reporter gene system. miR-185-3p significantly reduced the activity of LUC-CaSR, while no change was noticed in the mutant (Figure 6B). To further confirm the correlation between miR-185-3p and CaSR, we validated the expression of CaSR in response to miR-185-3p overexpression or knockdown. The qPCR results showed a dramatic downregulation of CaSR under circHIPK3 overexpression in cardiomyocytes. On the contrary, circHIPK3 knockdown notably increased the level of miR-185-3p (Figure 6C). RNA pull down assay was carried out which indicated that the miR-185-3p probe enriched the expression of circHIPK3 as compared to the control probe (Figure 6D). Pearson correlation coefficient analysis showed a negative correlation between miR-185-3p and CaSR and a positive correlation between circHIPK3 and CASR (Figure 6E and F). Together, these data indicated that miR-185-3p specifically targets CASR in cardiomyocytes.
Restoration of CaSR Reversed the Effect of circHIPK3 Knockdown on Cardiac Hypertrophy
To confirm the correlation between circHIPK3 and CaSR, we carried out the rescue experiments to detect whether the restoration of CaSR apparently reverses the effect of circHIPK3 knockdown. qRT-PCR was used to evaluate the CaSR expression. The results indicated a significant up-regulation of CaSR in cardiomyocytes subjected to Ang II treatment; circHIPK3 knockdown suppressed the level of CASR. Adenovirus overexpressing CaSR notably promoted the expression of CaSR (Figure 7A). Afterwards, we examined cardiomyocyte size and the expression of hypertrophic genes including ANP, BNP, and β-MHC. The results demonstrated that the overexpression of CaSR reversed the effect of circHIPK3 silencing on cardiomyocyte size and the expression of ANP, BNP, and β-MHC (Figure 7C). These findings indicated the existence of a relationship among circHIPK3, competing endogenous RNA (ceRNA) and CaSR in pressure overload-induced cardiac hypertrophy and cardiac dysfunction.
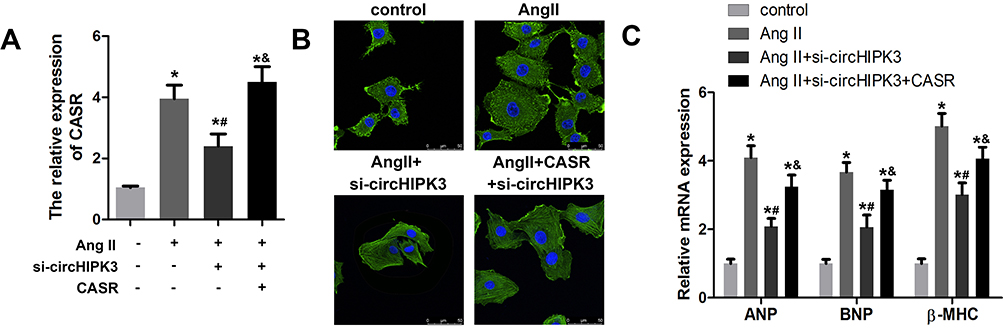 |
Figure 7 Restoration of CaSR reversed the effect of circHIPK3 in cardiomyocytes. (A) qRT-PCR was used to evaluate the efficiency of the adenovirus mediated knockdown of CaSR. (B) The measurement of morphology and size of cardiomyocyte treated with control, AngII+ad-si-nc, AngII+ad-si-circHIPK3, and Ang II+ad-si-circHIPK3+ad-CASR. (C) qRT-PCR analysis revealed the levels of ANP, BNP, and β-MHC. *P<0.05 vs control group, #P<0.05 vs Ang II+ad-si-nc group, &P<0.05 vs Ang II+ad-si-circHIPK3 group. Scale bar=50 μm. Data are expressed as means±SD from three independent experiments. |
Discussion
Cardiac hypertrophy may be due to the physiological or pathological stimuli and prolonged hypertrophic growth is associated with adverse cardiac anomalies that consequently lead to heart failure and sudden death worldwide.1,24 Our observations indicated that circHIPK3 expression was markedly increased under both TAC and Ang II-treated mice hearts. The underlying mechanistic link showed that circHIPK3 regulates pressure overload induced cardiac hypertrophy by sponging miR-185-3p and modulating CaSR. Furthermore, circHIPK3 silencing attenuates cardiac hypertrophy. Our data suggested that circHIPK3 might be a potential therapeutic target for treating cardiac hypertrophy.
CircRNAs sponge miRNAs in cardiac hypertrophy and other cardiac remodeling. For example, sex-determining region Y (Syr) circRNA has 16 binding sites for miR-138,25 heart related circRNA (HRCR) act as an endogenous sponge of niR-223,8 Mitochondrial fission and apoptosis-related circRNA (MFACR) downregulates miR-652-3p-dependent mitochondrial protein 18 (MTP18) activation,26 circRNA_000203 sponges miR-26b-5p and specifically increase the expressions of fibrosis-associated genes, alpha-smooth muscle actin (αSma), collagen Type I Alpha 2 Chain (col1a2) and collagen Type III Alpha 1 chain (Col3a1),27 circRNAs sodium/calcium exchanger 1 (NCX1) targets miR-133a-3p, Sodium-calcium exchanger gene derived circular RNA (circslc8a1) mediates miR-133a-3p.28 circHIPK3 is one of the most abundant circRNA and sponge miRNA in cardiac anomalies. However, the underlying mechanism of circHIPK3 in cardiac hypertrophy is limited. The mechanistic link between circHIPK3 and miR-185-3p was required to be explored. How this is related to CaSR pathway. In the present investigation, we demonstrated that circHIPK3 promotes cardiac hypertrophy and echocardiograph parameters, such as left ventricular end-diastolic pressure (LVEDPS), left ventricular fraction shortening (LVFS), left ventricular ejection fraction (LVEF), and left ventricular systolic dysfunction (LVSD), and the heart weight to tibial length (HW/TL) were found to be impaired in TAC-treated hearts and significantly enhanced mRNA expression of three standard molecular markers of cardiac hypertrophy, ANP, BNP, and β-MHC under the Ang II treatment as compared to the sham ones. Ang II induces cardiac dysfunction by several mechanisms, including vascular inflammation, oxidative stress, remodeling, and so on.29 Previously, circHIPK3 aggravates myocardial ischemia-reperfusion injury by sponging mi124-3p; circHIPK3 sponges miR-124-3p to inhibit proliferation and enhances apoptosis of cardiomyocyte after myocardial ischemia/reperfusion (I/R) injury.30 circHIPK3 is significantly upregulated during post-myocardial infarction (MI) by sponging miR-17-3p and regulates the activity of adenylate cyclase 6 (ADCY6) and also affects the presence of Ca2+ in the cytoplasm.31 Also, circHIPK3 induces proliferation, migration, and development of cardiac fibroblast by sponging miR-29b-3p in angiotensin (AngII)-induced cardiac fibrosis.32 Exosomal circHIPK3 released from hypoxia-pretreated cardiomyocytes become functionally activated in the cardiac microvascular endothelial cells (CMVECs) under oxidative stress conditions and regulate CMVECs via the miR-29a/Insulin-like growth factor 1 (IGF-1) pathway.33
Presently, the bioinformatics analysis showed the potential miRNAs that interact with circHIPK3. To identify the miRNA that interacts with circHIPK3, we have conducted a dual luciferase reporter assay and validated that miR-185-3p significantly reduced the activity of LUC-circHIPK3 among those miRNAs. In this study, immunofluorescence with α-SMA demonstrated larger size of cardiomyocytes with Ang II treatment than sham ones. Importantly, circHIPK3 knockdown (ad-si-circHIPK3) increased the anti-hypertrophic effect of miR-185-3p, but overexpression of circHIPK3 reversed this activity. In addition, miR-185-3p silencing abolished the effect of ad-si-circHIPK3. These observations suggested that circHIPK3 act as a miR-185-3p sponge to regulate cardiac hypertrophy. Previously, the miR-185 family was observed in several cardiac dysfunctions. In a cardiac specific gene set analysis (GSA), out of 18 miRNAs, miR-185 plays a pivotal role in cardiac hypertrophy by negatively targeting multiple factors of calcium ion signaling, calcium/calmodulin dependent protein kinase II Delta (Camk2d), sodium/calcium exchanger 1 (Ncx1), and nuclear factor of activated T-Cell 3 (Nfatc3).34 Moreover, miR-185 regulates dilated cardiomyopathy (DCM) (a leading cause of heart failure) by reducing the levels of anti-β1-AR antibody and TNF-α secreting β cells; 51 DCM patients are examined. Patients with high miR-185 at 1-year follow-up showed a drastic improvement in indexes of cardiac function, such as improvement in left ventricular ejection fraction, left ventricular end diastolic diameter, and a prohormone, N-terminal (NT)-proB-type natriuretic peptide (NT-proBNP), as well as a significant reduction in cardiovascular mortality and total admission for heart failure hospitalization as compared to patents with low miR-185.35 In another study, miR-185 reduction regulates hypoxia-induced myocardial infarction by targeting cathepsin K (catK), and promotes angiogenesis, as well as accelerating cardiac function recovery in mice.36
In the present report, the bioinformatics analysis revealed a potential target of miR-185-3p was CaSR and we further confirmed this association by using dual luciferase reporter assay by knockdown or overexpression of miR-185-3p. Additionally, Pearson correlation showed a negative correlation between miR-185-3p and CaSR, whereas positive regulation was revealed between circHIPK3 and CaSR. Overexpression of CaSR might reverse the effect of circHIPK3 on cardiac hypertrophy which was indicated by the size of cardiomyocyte and the expression of ANP, BNP, and β-MHC. Previously, it has been shown that CaSR play a crucial role in cardiac physiology and pathophysiology.37–39 Moreover, the expression level of CaSR was markedly increased in cardiac hypertrophy, which has been confirmed in our study.40
In conclusion, using in vitro and in vivo hypertrophy models we have shown that circHIPK3 was up-regulated in pressure-overload induced cardiomyocytes and inhibition of circHIPK3 exerts a protective role against cardiac hypertrophy through sponging of a miR-185-3p. Furthermore, it was shown that miR-185-3p in turn targets the calcium sensing receptor (CaSR) gene in cardiomyocytes and that miR-185-3p knockdown or CaSR over-expression can reverse the effect of circHIPK3. Our study has improved our understanding of the biological roles of circRNAs and miRNAs in the physio-pathology of cardiac hypertrophy. circHIPK3/miR-185-3p/CaSR axis acts as a potential therapeutic target for overload-induced cardiac hypertrophy.
Funding
This work was supported by the National Natural Science Foundation of China (grant number:81573234), National Natural Science Foundation of China (grant number:81773445) and “333” Project Of Jiangsu Province (grant number:LGY2016006).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15(7):387–407.
2. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–262. doi:10.1016/j.yjmcc.2016.06.001
3. Semsarian C, Healey MJ, Fatkin D, et al. A polymorphic modifier gene alters the hypertrophic response in a murine model of familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33(11):2055–2060. doi:10.1006/jmcc.2001.1466
4. Chen LL, Yang L. Regulation of circRNA biogenesis. RNA Biol. 2015;12(4):381–388. doi:10.1080/15476286.2015.1020271
5. Capel B, Swain A, Nicolis S, et al. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell. 1993;73(5):1019–1030. doi:10.1016/0092-8674(93)90279-Y
6. Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–338. doi:10.1038/nature11928
7. Ding S, Zhu Y, Liang Y, Huang H, Xu Y, Zhong C. Circular RNAs in vascular functions and diseases. Adv Exp Med Biol. 2018;1087:287–297.
8. Wang K, Long B, Liu F, et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. 2016;37(33):2602–2611. doi:10.1093/eurheartj/ehv713
9. Yang L, Li Y, Wang X, et al. Overexpression of miR-223 tips the balance of pro- and anti-hypertrophic signaling cascades toward physiologic cardiac hypertrophy. J Biol Chem. 2016;291(30):15700–15713. doi:10.1074/jbc.M116.715805
10. Wu HJ, Zhang CY, Zhang S, Chang M, Wang HY. Microarray expression profile of circular RNAs in heart tissue of mice with myocardial infarction-induced heart failure. Cell Physiol Biochem. 2016;39(1):205–216. doi:10.1159/000445617
11. Schulte C, Joshi A, Mayr M. Response by schulte et al to letter regarding article, “comparative analysis of circulating noncoding RNAs versus protein biomarkers in the detection of myocardial injury”. Circ Res. 2019;125(4):e22–e23.
12. Yang MH, Wang H, Han SN, et al. Circular RNA expression in isoproterenol hydrochloride-induced cardiac hypertrophy. Aging (Albany NY). 2020;12(3):2530–2544. doi:10.18632/aging.102761
13. Zheng Q, Bao C, Guo W, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215.
14. Wen J, Liao J, Liang J, Chen XP, Zhang B, Chu L. Circular RNA HIPK3: a key circular RNA in a variety of human cancers. Front Oncol. 2020;10:773. doi:10.3389/fonc.2020.00773
15. Kleaveland B, Shi CY, Stefano J, Bartel DPA. Network of noncoding regulatory RNAs acts in the mammalian brain. Cell. 2018;174(2):350–362. doi:10.1016/j.cell.2018.05.022
16. Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(3):193–205. doi:10.1038/nrm2327
17. van Rooij E, Liu N, Olson EN. MicroRNAs flex their muscles. Trends Genet. 2008;24(4):159–166. doi:10.1016/j.tig.2008.01.007
18. Diniz GP, Lino CA, Moreno CR, Senger N, Barreto-Chaves M. MicroRNA-1 overexpression blunts cardiomyocyte hypertrophy elicited by thyroid hormone. J Cell Physiol. 2017;232(12):3360–3368. doi:10.1002/jcp.25781
19. Care A, Catalucci D, Felicetti F, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13(5):613–618. doi:10.1038/nm1582
20. Wang F, Yuan Y, Yang P, Li X. Extracellular vesicles-mediated transfer of miR-208a/b exaggerate hypoxia/reoxygenation injury in cardiomyocytes by reducing QKI expression. Mol Cell Biochem. 2017;431(1–2):187–195. doi:10.1007/s11010-017-2990-4
21. Diniz GP, Takano AP, Barreto-Chaves ML. MiRNA-208a and miRNA-208b are triggered in thyroid hormone-induced cardiac hypertrophy - role of type 1 angiotensin II receptor (AT1R) on miRNA-208a/alpha-MHC modulation. Mol Cell Endocrinol. 2013;374(1–2):117–124. doi:10.1016/j.mce.2013.04.010
22. Ucar A, Gupta SK, Fiedler J, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3(1):1078. doi:10.1038/ncomms2090
23. Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci U S A. 2009;106(29):12103–12108.
24. Samak M, Fatullayev J, Sabashnikov A, et al. Cardiac hypertrophy: an introduction to molecular and cellular basis. Med Sci Monit Basic Res. 2016;22:75–79. doi:10.12659/MSMBR.900437
25. Wang Y, Wang Y, Li Y, et al. Decreased expression of circ_0020397 in intracranial aneurysms may be contributing to decreased vascular smooth muscle cell proliferation via increased expression of miR-138 and subsequent decreased KDR expression. Cell Adh Migr. 2019;13(1):220–228. doi:10.1080/19336918.2019.1619432
26. Wang K, Gan TY, Li N, et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. 2017;24(6):1111–1120. doi:10.1038/cdd.2017.61
27. Li H, Xu JD, Fang XH, et al. Circular RNA circRNA_000203 aggravates cardiac hypertrophy via suppressing miR-26b-5p and miR-140-3p binding to Gata4. Cardiovasc Res. 2020;116(7):1323–1334.
28. Li M, Ding W, Tariq MA, et al. A circular transcript of ncx1 gene mediates ischemic myocardial injury by targeting miR-133a-3p. Theranostics. 2018;8(21):5855–5869. doi:10.7150/thno.27285
29. Kim GR, Cho SN, Kim HS, et al. Histone deacetylase and GATA-binding factor 6 regulate arterial remodeling in angiotensin II-induced hypertension. J Hypertens. 2016;34(11):2206–2219. doi:10.1097/HJH.0000000000001081
30. Hu D, Zhang Y. Circular RNA HIPK3 promotes glioma progression by binding to miR-124-3p. Gene. 2019;690:81–89. doi:10.1016/j.gene.2018.11.073
31. Deng Y, Wang J, Xie G, Zeng X, Li H. Circ-HIPK3 strengthens the effects of adrenaline in heart failure by MiR-17-3p - ADCY6 axis. Int J Biol Sci. 2019;15(11):2484–2496. doi:10.7150/ijbs.36149
32. Ni H, Li W, Zhuge Y, et al. Inhibition of circHIPK3 prevents angiotensin II-induced cardiac fibrosis by sponging miR-29b-3p. Int J Cardiol. 2019;292:188–196. doi:10.1016/j.ijcard.2019.04.006
33. Wang Y, Zhao R, Liu W, et al. Exosomal circHIPK3 released from hypoxia-pretreated cardiomyocytes regulates oxidative damage in cardiac microvascular endothelial cells via the miR-29a/IGF-1 pathway. Oxid Med Cell Longev. 2019;2019:7954657.
34. Kim JO, Song DW, Kwon EJ, et al. miR-185 plays an anti-hypertrophic role in the heart via multiple targets in the calcium-signaling pathways. PLoS One. 2015;10(3):e122509.
35. Yu M, Liang W, Xie Y, et al. Circulating miR-185 might be a novel biomarker for clinical outcome in patients with dilated cardiomyopathy. Sci Rep. 2016;6(1):33580. doi:10.1038/srep33580
36. Li CC, Qiu XT, Sun Q, et al. Endogenous reduction of miR-185 accelerates cardiac function recovery in mice following myocardial infarction via targeting of cathepsin K. J Cell Mol Med. 2019;23(2):1164–1173. doi:10.1111/jcmm.14016
37. Diaz-Soto G, Rocher A, Garcia-Rodriguez C, Nunez L, Villalobos C. The calcium-sensing receptor in health and disease. Int Rev Cell Mol Biol. 2016;327:321–369.
38. Toka HR, Pollak MR. The role of the calcium-sensing receptor in disorders of abnormal calcium handling and cardiovascular disease. Curr Opin Nephrol Hypertens. 2014;23(5):494–501. doi:10.1097/MNH.0000000000000042
39. Brancaccio D, Cozzolino M. Cardiovascular effects of VDR and CaSR activation. G Ital Nefrol. 2009;26(Suppl 49):S18–S22.
40. Lu M, Leng B, He X, Zhang Z, Wang H, Tang F. Calcium sensing receptor-related pathway contributes to cardiac injury and the mechanism of astragaloside IV on cardioprotection. Front Pharmacol. 2018;9:1163. doi:10.3389/fphar.2018.01163
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.