")
Back to Journals » OncoTargets and Therapy » Volume 9
Silencing of carboxypeptidase E inhibits cell proliferation, tumorigenicity, and metastasis of osteosarcoma cells
Authors Fan S, Li X, Li L, Wang L, Du Z, Yang Y, Zhao J, Li Y
Received 23 October 2015
Accepted for publication 13 February 2016
Published 10 May 2016 Volume 2016:9 Pages 2795—2803
DOI https://doi.org/10.2147/OTT.S98991
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Faris Farassati
Shuli Fan,1,* Xu Li,2,* Leiming Li,2 Liguo Wang,2 Zhangzhen Du,2 Yan Yang,2 Jiansong Zhao,3 Yan Li3
1Department of Geriatrics, The First Affiliated Hospital of China Medical University, Shenyang, People’s Republic of China; 2Department of Sports Medicine and Joint Surgery, The First Affiliated Hospital of China Medical University, Shenyang, People’s Republic of China; 3Department of Orthopaedic Surgery, Shengjing Hospital of China Medical University, Shenyang, People’s Republic of China
*These authors contributed equally to this work
Abstract: Carboxypeptidase E (CPE), a prohormone processing enzyme, has been implicated in the progression of multiple malignancies. However, the biological role and molecular mechanisms of CPE in osteosarcoma remain elusive. In this study, we assessed the effects of CPE on cell proliferation, tumorigenicity, migration, and invasion in osteosarcoma. Our results showed that silencing of CPE significantly inhibited cell proliferation, caused cell cycle arrest at G0/G1 phase, decreased the expression levels of cell cycle protein, cyclin D1, and inhibited tumorigenicity in vivo. Additionally, CPE downregulation repressed the migratory and invasive capacities of osteosarcoma cells in vitro. Furthermore, overexpression of CPE-ΔN (a splice variant of CPE) enhanced the cell growth, migration, and invasion of osteosarcoma cells. It is possible that both CPE forms are involved in the tumorigenesis and development of osteosarcoma, and therefore CPE may provide a promising biological target for osteosarcoma therapy.
Keywords: carboxypeptidase E, osteosarcoma, proliferation, tumorigenicity, migration, invasion
Introduction
Osteosarcoma is a common malignant bone tumor that frequently occurs in children and adolescents and is reported to be responsible for 2.4% of all pediatric cancers.1,2 Despite significant progress in the diagnosis and therapy of osteosarcoma, the 5-year survival rate has remained unchanged over the past 20 years, especially for metastatic osteosarcoma with less than 20% overall survival.3,4 Therefore, it is imperative to understand the molecular mechanisms of osteosarcoma and identify new therapeutic targets for metastatic osteosarcoma.
Carboxypeptidase E (CPE) was initially identified as a prohormone processing enzyme, which is involved in various biological processes, such as the synthesis of neuropeptides and hormones.5,6 Recently, accumulated evidence suggests that CPE serves many essential nonenzymatic roles in addtion to its enzymatic function. Deregulation of CPE is associated with a variety of diseases. For instance, CPE knockout mice can more easily exhibit disease states, such as obesity,7 diabetes,8 lower bone mineral density phenotype,9 and behavioral deficiencies.10 Increased CPE expression was shown in many types of cancer, and it was implicated in cancer progression as it regulates the proliferation, invasion, and chemosensitivity of tumor cells.11–14 Recently, an N-terminally truncated splice variant of CPE (CPE-ΔN) was identified and found to be highly expressed in metastatic cancers; its expression was correlated with tumor growth and invasiveness, and it might be a potential biomarker for predicting future metastasis and recurrence.15,16 Yang et al17 have shown that the gene coding for CPE was upregulated in osteosarcoma samples compared to that in the nomal controls, indicating a role for CPE in osteosarcoma development. However, how it affects the development and progression of osteosarcoma remains elusive.
The aim of this study was to explore the functional role of CPE in the tumorigenesis and development of osteosarcoma. Decreased CPE expression by RNA interference significantly inhibited cell growth, tumorigenicity, migration, and invasiveness in osteosarcoma cells. Further examination demonstrated that these effects might be due to both forms of CPE.
Materials and methods
Cell culture
Three human osteosarcoma cell lines MG-63, U2-OS, and Saos-2 (Cell Bank of Chinese Academy of Sciences, Shanghai, People’s Republic of China) were cultured in Dulbecco’s Modified Eagle’s Medium (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS, HyClone, Logan, UT, USA), and maintained in a 37°C incubator with 5% CO2. The cells were digested with 0.25% trypsin for passage when cell reached 80% confluence. The use of human CRC cell lines was approved by the Ethics Committee of China Medical University.
Plasmid construction and stable cell line screening
Short hairpin ribonucleic acid (shRNA) that targets CPE or scramble nonspecific sequence (negative control) was constructed in the pGCsi-H1 Vector (GeneChem, Shanghai, People’s Republic of China). The resulting plasmid was then transfected into MG-63 cells using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Twenty-four hours after transfection, cells were selected with G418 (200 μg/mL, Invitrogen) for stable CPE-silenced clones. The sequences of CPE shRNA are 5′-GATCCCCCGAGACAATTGTCAACCTGTTCAAGAGACAGGTTGACAATTGTCTCGTTTTT-3′ (forward) and 5′-AGCTAAAAACGAGACAATTGTCAACCTGTCTCTTGAACAGGTTGACAATTGTCTCGGGG-3′ (reverse). To obtain CPE-ΔN overexpressed cells, pcDNA3.1 vector with the coding sequence of CPE-ΔN was transfected into the Saos-2 cells using Lipofectamine 2000 Reagent (Invitrogen). The primers of CPE-ΔN were designed according to the reported gene sequences of human CPE (GenBank accession number NM_001873.2).
Real-time PCR
Total RNA from the MG-63, control shRNA, and CPE shRNA cells was extracted using an RNA simple Total RNA Kit (TIANGEN, Beijing, People’s Republic of China) and reverse transcribed into complementary DNA. The primer sequences are as follows: CPE: 5′-TGTAGATGGAACCACCAACGG-3′ (forward) and 5′-ACAAATCCTTTAACTCCTCGG-3′ (reverse); CPE-ΔN: 5′-TGTAGATGGAACCACCAACGG-3′ and 5′-ACAAATCCTTTAACTCCTCGG-3′; and β-actin: 5′-CTTAGTTGCGTTACACCCTTTCTTG-3′ (forward) and 5′-CTGTCACCTTCACCGTTCCAGTTT-3′ (reverse). Expression of CPE was determined using an Exicycler™ 96 real-time (RT) polymerase chain reaction (PCR) machine (Bioneer, Daejeon, South Korea).
Western blot analysis
Total proteins from cultured cells and tissues were extracted using radioimmunoprecipitation assay lysis buffer (Beyotime Institute of Biotechnology, Haimen, People’s Republic of China); the protein concentration was quantified using bicinchoninic acid method (Beyotime). Equal amounts of proteins were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% nonfat milk, followed by incubation with corresponding diluted primary antibodies (anti-CPE, 1:3,000, BD Biosciences; anti-β-actin, 1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA; anti-cyclin D1 and anti-p65, 1:400, Boster, Wuhan, People’s Republic of China) at 4°C overnight. The membranes were subjected to further incubation with horseradish peroxidase-labeled secondary antibody (Beyotime) at 37°C for 45 minutes. Targeted protein was visualized using electrochemiluminescence reagents (7Sea Biotech, Shanghai, People’s Republic of China), and the scanned images were analyzed with Gel-Pro-Analyzer software.
MTT assay
The cell viability was assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, the cells were cultured in 96-well microplates, with each well containing 3×103 cells. MTT (Sigma-Aldrich, St Louis, MO, USA) at a final concentration of 0.2 mg/mL was added to each well at different time points (12, 24, 48, 72, or 96 hours). Cell supernatants were removed after additional 4 hours incubation, and the formazan crystals were dissolved in dimethyl sulfoxide (200 μL/well, Sigma-Aldrich). The optical density at 490 nm was determined with a microplate reader.
Colony formation assay
Five hundred cells per dish were trypsinized, resuspended in culture medium, and seeded into 35 mm dishes; culture medium was changed every 2 or 3 days. After 14 days of incubation, the visible cell colonies were fixed with 4% paraformaldehyde, followed by staining with Giemsa solution (Nanjing Jiancheng, Nanjing, People’s Republic of China) for 8 minutes. The colonies consisting of at least 50 cells were counted under a microscope, and colony formation efficiency was calculated.
Flow cytometry analysis
Effect of CPE on cycle distribution was determined using flow cytometry. Cells were trypsinized and fixed in 70% cold ethanol at 4°C for 2 hours; after incubation with staining buffer (containing propidium iodide and RNase A) for 30 minutes at 37°C in the dark, the cells were then analyzed by flow cytometry in FACSCalibur (BD Diagnostics, Franklin Lakes, NJ, USA).
Tumor xenograft study
Female BABL/c nude mice about 6 weeks old were purchased from Vital River Laboratory Animal Technology Co Ltd (Beijing, People’s Republic of China). Animal care and experimental protocols were approved by the Experimental Animal Ethics Committee of China Medical University, and were in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. To initiate tumor xenograft, 1×106 parental MG-63 cells, MG-63 cells expressing control shRNA, or CPE shRNA were injected subcutaneously into the right armpit flank of nude mice (n=6 each group). Tumor sizes were monitored every 3 days, and the tumor volumes were calculated.
Hematoxylin and eosin staining
To evaluate the histologic alterations, xenograft tissues were fixed in formaldehyde, paraffin embedded, and sliced into 5 μm sections. After staining with hematoxylin and eosin, the pathologic changes in tumor tissues were observed under a light microscope.
Scratch wound assay
Cells were cultured in 6-well plates in complete medium until they reached confluence. A 200 μL pipette tip was used to scratch a uniform wound on the monolayer cell cultures. The stripped cells were washed away with serum-free culture medium, and the other cells were cultured in medium containing 10% FBS. Images were captured at 0, 12, and 24 hours after scratch, and the distances of cells’ migration into the denuded area were measured. Migration rate was calculated as:
|
|

Matrigel invasion assay
Transwell membrane filter inserts (8 μm pore size, Corning Inc, New York, NY, USA) and the Matrigel (BD Biosciences) were used for Transwell invasion assay. The cells were seeded onto the upper chamber (2×104 cells/well). The lower chamber was filled with 800 μL of culture medium containing 20% FBS. After 24 hours of incubation, cells in the upper chamber were scraped away, and the cells that penetrated through the lower chamber were stained with 0.5% crystal violet dye (Amresco, Solon, OH, USA) for 5 minutes. The average number of cells in five fields per membrane was counted in triplicate with a microscope.
Statistical analysis
Data are presented as the mean ± standard deviation, with analyses carried out using GraphPad Prism 5.0 software (San Diego, CA, USA). One-way analysis of variance was used for the analyses between the groups, and multiple comparisons were performed using Bonferroni post hoc test. P<0.05 was considered statistically significant.
Results
Establishment of stably CPE-silenced osteosarcoma cell line
To investigate the role of CPE in osteosarcoma, we first determined the expression levels of CPE in three osteosarcoma cell lines by Western blot analysis; the MG-63 cells with highest CPE expression (Figure 1A) in tested cell lines were chosen for loss of function study. The efficiency of shRNA-mediated CPE inhibition was determined using RT-PCR and Western blot analysis. As shown in Figure 1B and C, CPE shRNA cells presented low expression of CPE at mRNA and protein levels compared with the control shRNA cells (P<0.001). The molecular weight of CPE was approximately 52 kDa. These results indicated that the stable CPE-silenced osteosarcoma cell line was constructed.
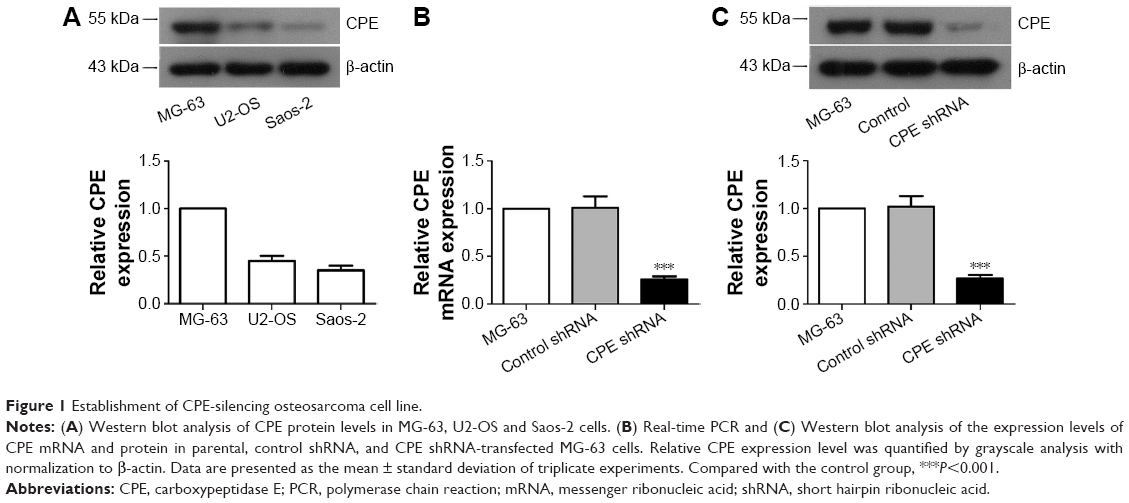 | Figure 1 Establishment of CPE-silencing osteosarcoma cell line. |
Silencing of CPE inhibits the proliferation and induces cell cycle arrest in osteosarcoma cells
The effect of CPE knockdown on cell proliferation was determined using MTT assay. Results showed that silencing of CPE significantly inhibited the proliferation as compared with the control shRNA cells (Figure 2A; P<0.01). Consistently, clone formation assays also showed that less colony numbers existed in CPE-silenced cells compared with the control cells (Figure 2B and C; P<0.01), indicating a positive role of CPE in osteosarcoma cell proliferation. We further demonstrated that CPE inhibition affected the cell cycle progression as detected by flow cytometry (Figure 2D and E). CPE inhibition led to the accumulation of cells in G0/G1 phase (P<0.01) and reduced the S and G2 phase cell population (P<0.05) as compared with the control cells. Meanwhile, the expression level of cyclin D1, which contributes to tumorigenesis through regulating cell cycle G1/S transition,18 was significantly decreased in CPE-silenced cells (Figure 2F; P<0.01). These results suggest that silencing of CPE expression could arrest cell cycle in G0/G1 phase and inhibit cell proliferation of osteosarcoma MG-63 cells.
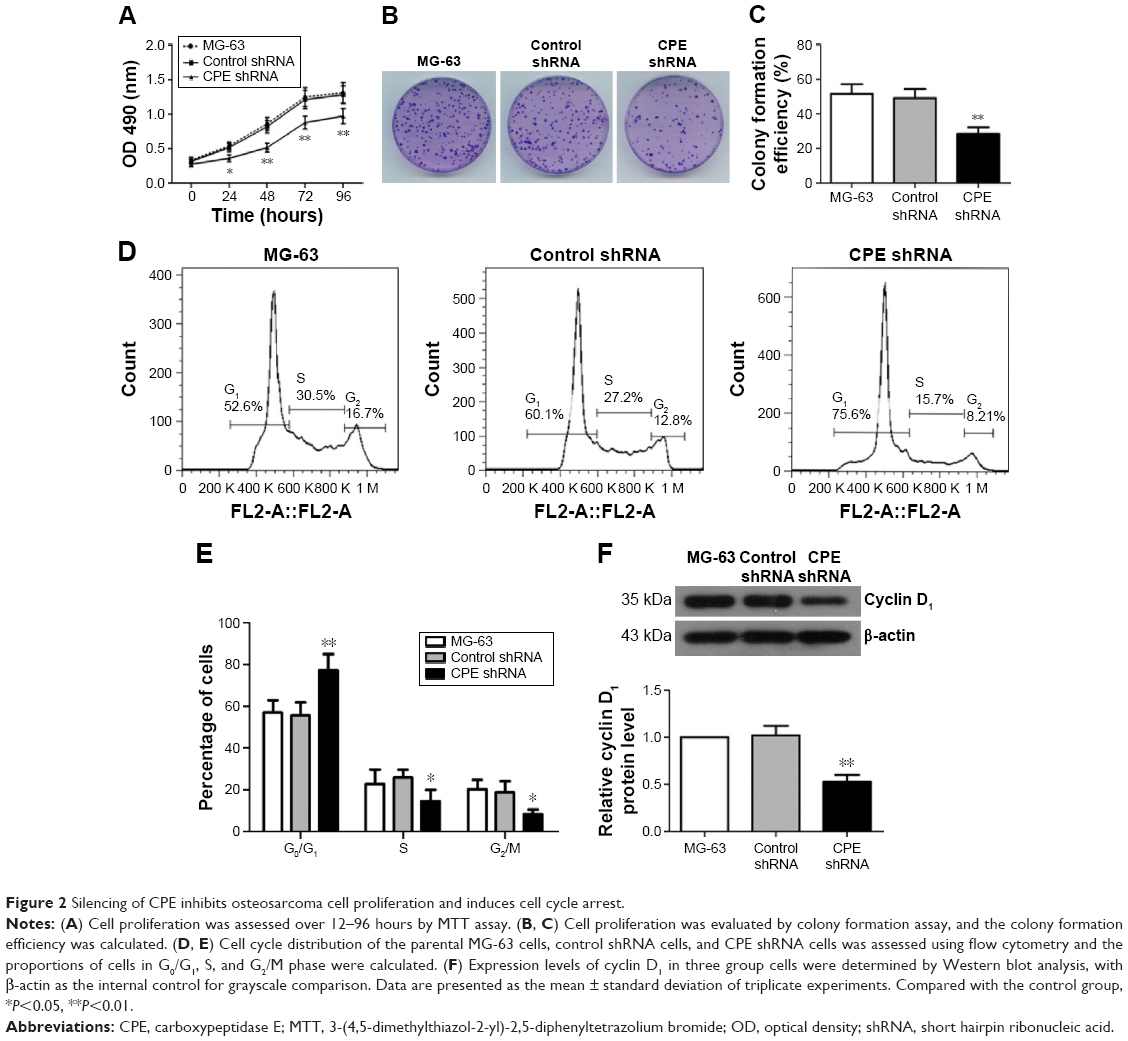 | Figure 2 Silencing of CPE inhibits osteosarcoma cell proliferation and induces cell cycle arrest. |
Silencing of CPE inhibits xenograft osteosarcoma growth in nude mice
In vitro experiments, we have showed that CPE knockdown could inhibit cell proliferation and clonogenicity; we further investigated whether CPE knockdown would reduce osteosarcoma tumor growth in vivo. Parental MG-63 cells and stable CPE shRNA or control-transfected cells were subcutaneously injected into nude mice to initiate tumor xenografts; tumor formation was followed for 35 days. Nude mice bearing control and parental MG-63 cells displayed larger tumor volume and tumor size as shown in Figure 3A and B; however, xenograft tumors derived from CPE-depleted cells were significantly smaller than control or MG-63 group (Figure 3A and B; P<0.01). Western blot results confirmed the decreased expression level of CPE in CPE-silenced cell derived tumors (Figure 3C; P<0.01). Histologic changes of xenograft tumors assessed by HE staining further revealed that control and parental groups showed high-grade malignancy, as characterized by high nuclear-to-cytoplasmic ratio and prominent nucleoli with minimal cytoplasm, while CPE downregulation reversed these features (Figure 3D). These results indicate that silencing of CPE possesses antitumor effects in vivo.
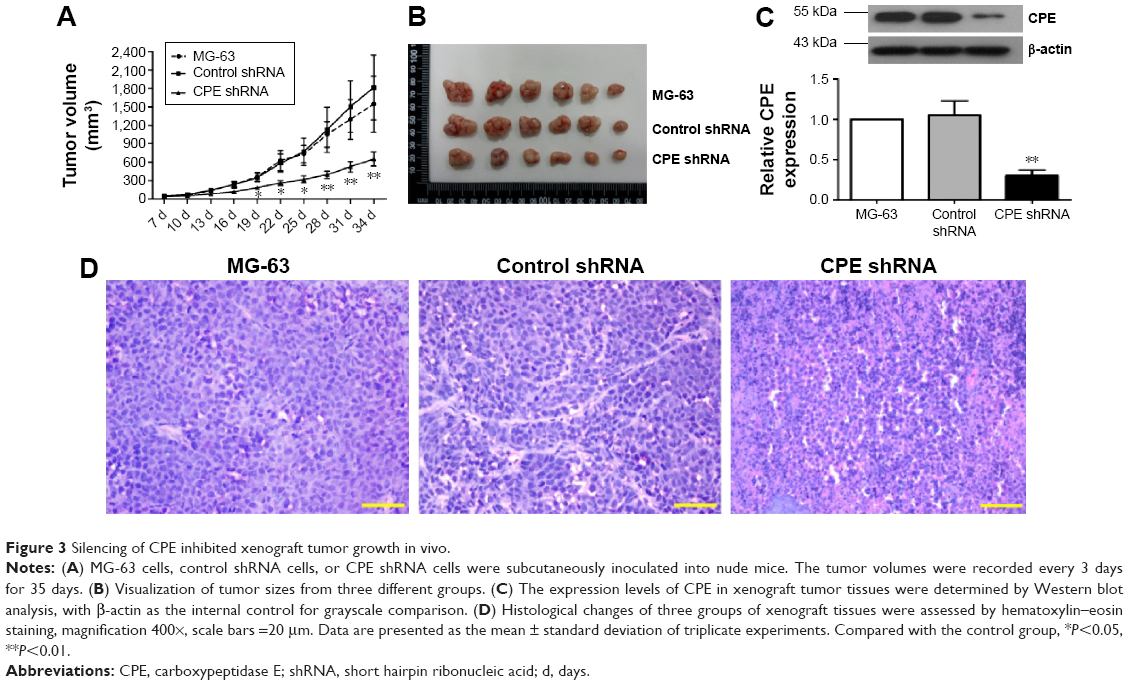 | Figure 3 Silencing of CPE inhibited xenograft tumor growth in vivo. |
Silencing of CPE represses invasion and migration of osteosarcoma cells
Migration and invasion are major steps for malignancy.19 To detect whether CPE downregulation affects the cell motility, we further performed the wound healing assay and Matrigel invasion assay to detect whether CPE downregulation affects cell metastasis. CPE-silenced cells showed lower migration distance compared with the control cells (Figure 4A and B; P<0.05 at 12 hours and P<0.01 at 24 hours), indicating an inhibitory effect of CPE silencing on cell motility. Matrigel invasion assay similarly revealed that less CPE-silenced cells invaded through the Matrigel-coated insert after 24 hours, whereas control and parental cells maintained high invasive capacities (Figure 4C and D; P<0.01). These results indicate that CPE expression is involved in cell migration and invasiveness of osteosarcoma cells.
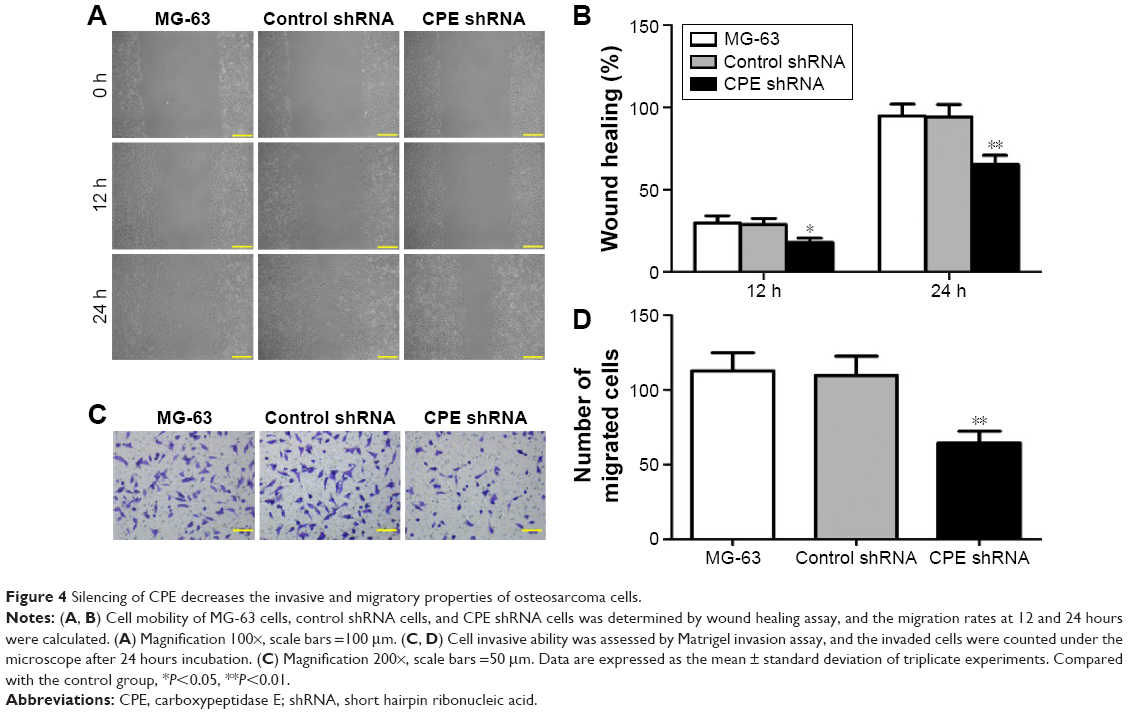 | Figure 4 Silencing of CPE decreases the invasive and migratory properties of osteosarcoma cells. |
Overexpression of CPE-ΔN promotes cell growth, migration, and invasion of osteosarcoma cells
Previous studies have shown that the splice variant form of CPE (CPE-ΔN) plays a key role in tumor metastasis, and the full form of CPE has the opposite effects in metastasis.15,16 Since shRNA targets both CPE forms, we further determined whether these effects are due to CPE-ΔN by constructing CPE-ΔN overexpressing cells. qRT-PCR showed that the CPE-ΔN mRNA level was significantly upregulated in CPE-ΔN-overexpressed Saos-2 cells compared with the vector cells (Figure 5A, P<0.01). Western blot analysis also showed the elevated expression of CPE-ΔN protein in CPE-ΔN-overexpressed cells, and the molecular weight of CPE-ΔN was approximately 46 kDa (Figure 5B). Next, the role of CPE in cell proliferation was assessed by MTT and colony formation assays. As shown in Figure 5C and D, ectopic expression of CPE-ΔN enhanced cell proliferation and formed more colonies as compared to the vector cells. Furthermore, CPE-ΔN exerted a metastatic potential, increased CPE-ΔN expression, enhanced the migratory and invasive capacities of Saos-2 cells compared with the vector cells (Figure 5E and F; P<0.01). It was noteworthy that overexpression of CPE-ΔN had no effect on cell cycle regulation of Saos-2 cells (data not shown). These results indicate that CPE-ΔN promotes cell growth and metastasis in osteosarcoma cells.
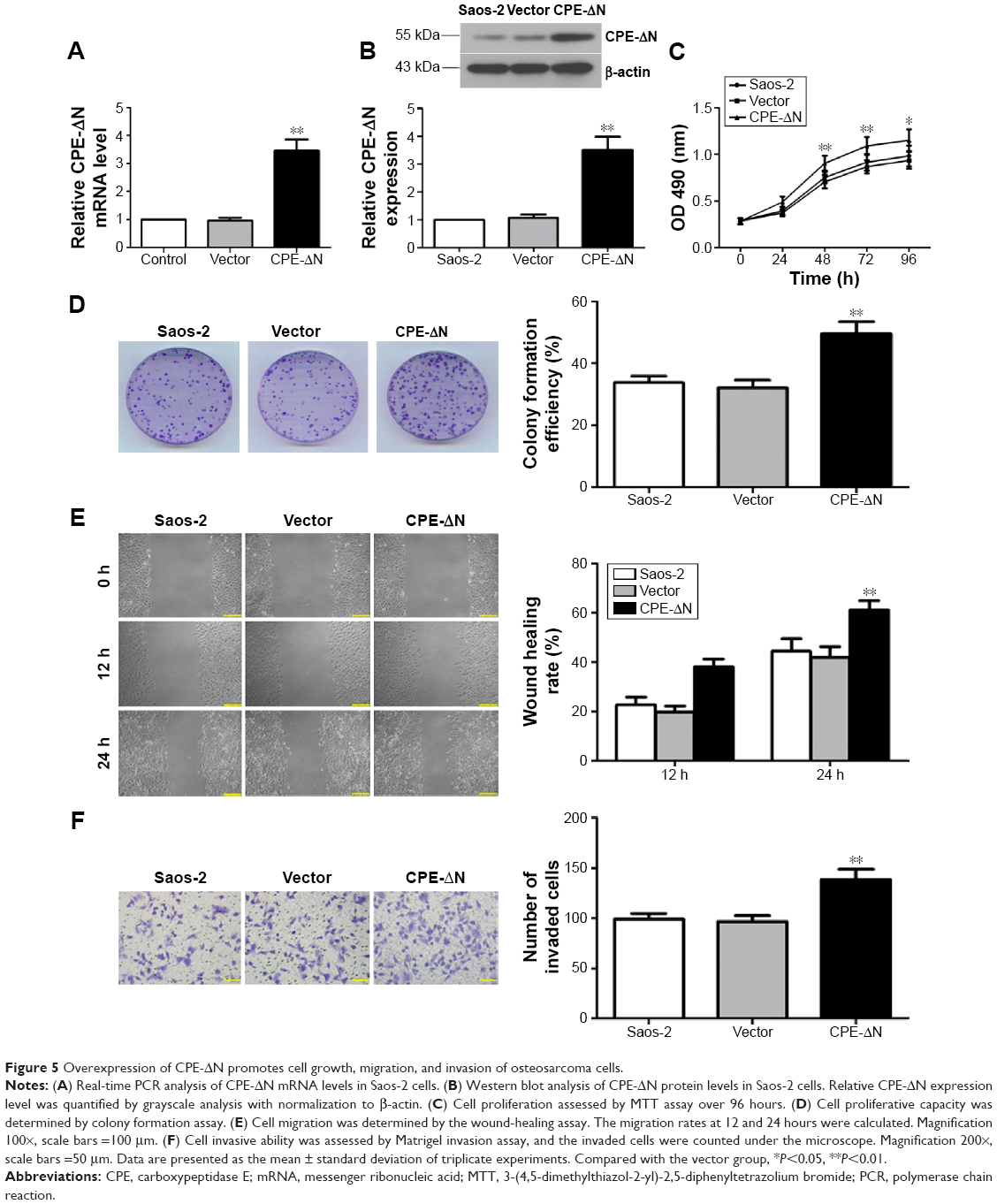 | Figure 5 Overexpression of CPE-ΔN promotes cell growth, migration, and invasion of osteosarcoma cells. |
Discussion
Recurrence and metastasis remain major causes of poor prognosis for osteosarcoma. Finding effective therapies that interfere with this processes are crucial. In this study, we demonstrated that the depletion of CPE inhibited cell proliferation, induced cell cycle arrest in G0/G1 phase, and reduced tumorigenicity in vivo. In addition, silencing of CPE suppressed migration and invasion of osteosarcoma cells. Further overexpression experiments showed that CPE-ΔN was also involved in the regulation of cell growth and migration of osteosarcoma cells. Taken together, CPE may be a promising therapeutic target for osteosarcoma.
Aberrant expression of CPE has been shown in many neuroendocrine and nonendocrine cancer types.13 For instance, CPE was highly expressed in breast,20 cervical,21 colorectal,11,22 and kidney cancer.23 However, decreased CPE expression was also observed in human fibrosarcoma and glioblastoma cells.14,24 These different results might be dependent on the different tumor environments and the relative expression of CPE and CPE-ΔN in tumor cells.14 In this study, we found that RNA interference-mediated downregulation of CPE resulted in cell cycle arrest in G0/G1 phase, reduced the expression of cell cycle-related protein cyclin D1, and suppressed osteosarcoma cell growth and tumorigenicity activity in vitro and in vivo. Since proliferation and cell cycle are important factors for tumor development and progression,25,26 our results indicate that CPE promotes tumorigenesis in osteosarcoma through regulating cell proliferation and cell cycle progression, which are consistent with prior results found in pancreatic and colorectal cancer.11,12
In addition to these findings, we also observed that silencing of CPE expression resulted in the inhibition of migration and invasion of osteosarcoma cells. Recent studies have demonstrated that the splice variant, CPE-ΔN, is highly expressed in metastatic cancers and is a biomarker for predicting future metastasis.13,16 However, the full form of CPE (WT-CPE) has the opposite effects and has anti-invasion and antimigration properties.14,24 In this study, shRNA interference downregulated both forms of CPE. Thus, the invasion and migration-promoting effects might be due to CPE-ΔN and not WT-CPE. By overexpressing CPE-ΔN in osteosarcoma cells, we found that overexpression of CPE-ΔN significantly enhanced the growth rate of osteosarcoma cells and promoted cell migration and invasiveness, but had no effect on cell cycle regulation. Together, these results indicate that both CPE forms might be involved in the tumorigenesis and development of osteosarcoma. However, there are still some limitations in our results, such as the limited cell lines used. So, the role of CPE on osteosarcoma malignancy in human patients still needs to be demonstrated.
In summary, data from our results demonstrate that silencing of CPE inhibits cell growth in vitro and in vivo, causes cell cycle arrest at G0/G1 phase, and suppresses metastasis of osteosarcoma cells. Overexpression of CPE-ΔN promotes cell proliferation, migration, and invasion. It is possible that both CPE forms are involved in the tumorigenesis and development of osteosarcoma, and therefore CPE may provide a promising biological target for osteosarcoma therapy.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (numbers 30973021 and 81272946).
Disclosure
The authors report no conflicts of interest in this work.
References
Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3–13. | ||
He H, Ni J, Huang J. Molecular mechanisms of chemoresistance in osteosarcoma (review). Oncol Lett. 2014;7(5):1352–1362. | ||
Geller DS, Gorlick R. Osteosarcoma: a review of diagnosis, management, and treatment strategies. Clin Adv Hematol Oncol. 2010;8(10): 705–718. | ||
Chou AJ, Geller DS, Gorlick R. Therapy for osteosarcoma: where do we go from here? Paediatr Drugs. 2008;10(5):315–327. | ||
Fricker LD, Snyder SH. Purification and characterization of enkephalin convertase, an enkephalin-synthesizing carboxypeptidase. J Biol Chem. 1983;258(18):10950–10955. | ||
Cawley NX, Wetsel WC, Murthy SR, Park JJ, Pacak K, Loh YP. New roles of carboxypeptidase E in endocrine and neural function and cancer. Endocr Rev. 2012;33(2):216–253. | ||
Naggert JK, Fricker LD, Varlamov O, et al. Hyperproinsulinaemia in obese fat/fat mice associated with a carboxypeptidase E mutation which reduces enzyme activity. Nat Genet. 1995;10(2):135–142. | ||
Cawley NX, Zhou J, Hill JM, et al. The carboxypeptidase E knockout mouse exhibits endocrinological and behavioral deficits. Endocrinology. 2004;145(12):5807–5819. | ||
Cawley NX, Yanik T, Woronowicz A, Chang W, Marini JC, Loh YP. Obese carboxypeptidase E knockout mice exhibit multiple defects in peptide hormone processing contributing to low bone mineral density. Am J Physiol Endocrinol Metab. 2010;299(2):E189–E197. | ||
Woronowicz A, Koshimizu H, Chang SY, et al. Absence of carboxypeptidase E leads to adult hippocampal neuronal degeneration and memory deficits. Hippocampus. 2008;18(10):1051–1063. | ||
Liang XH, Li LL, Wu GG, et al. Upregulation of CPE promotes cell proliferation and tumorigenicity in colorectal cancer. BMC Cancer. 2013;13:412. | ||
Liu A, Shao C, Jin G, et al. Downregulation of CPE regulates cell proliferation and chemosensitivity in pancreatic cancer. Tumour Biol. 2014;35(12):12459–12465. | ||
Murthy SR, Pacak K, Loh YP. Carboxypeptidase E: elevated expression correlated with tumor growth and metastasis in pheochromocytomas and other cancers. Cell Mol Neurobiol. 2010;30(8):1377–1381. | ||
Murthy SR, Dupart E, Al-Sweel N, Chen A, Cawley NX, Loh YP. Carboxypeptidase E promotes cancer cell survival, but inhibits migration and invasion. Cancer Lett. 2013;341(2):204–213. | ||
Lee TK, Murthy SR, Cawley NX, et al. An N-terminal truncated carboxypeptidase E splice isoform induces tumor growth and is a biomarker for predicting future metastasis in human cancers. J Clin Invest. 2011;121(3):880–892. | ||
Zhou K, Liang H, Liu Y, Yang C, Liu P, Jiang X. Overexpression of CPE-deltaN predicts poor prognosis in colorectal cancer patients. Tumour Biol. 2013;34(6):3691–3699. | ||
Yang Z, Chen Y, Fu Y, et al. Meta-analysis of differentially expressed genes in osteosarcoma based on gene expression data. BMC Med Genet. 2014;15:80. | ||
Jirawatnotai S, Hu Y, Michowski W, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 2011;474(7350):230–234. | ||
Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–695. | ||
Du J, Keegan BP, North WG. Key peptide processing enzymes are expressed by breast cancer cells. Cancer Lett. 2001;165(2):211–218. | ||
Bachtiary B, Boutros PC, Pintilie M, et al. Gene expression profiling in cervical cancer: an exploration of intratumor heterogeneity. Clin Cancer Res. 2006;12(19):5632–5640. | ||
Provenzani A, Fronza R, Loreni F, Pascale A, Amadio M, Quattrone A. Global alterations in mRNA polysomal recruitment in a cell model of colorectal cancer progression to metastasis. Carcinogenesis. 2006;27(7):1323–1333. | ||
Cutcliffe C, Kersey D, Huang CC, Zeng Y, Walterhouse D, Perlman EJ. Clear cell sarcoma of the kidney: up-regulation of neural markers with activation of the sonic hedgehog and Akt pathways. Clin Cancer Res. 2005;11(22):7986–7994. | ||
Horing E, Harter PN, Seznec J, et al. The “go or grow” potential of gliomas is linked to the neuropeptide processing enzyme carboxypeptidase E and mediated by metabolic stress. Acta Neuropathol. 2012;124(1):83–97. | ||
Sherr CJ. Cancer cell cycles. Science. 1996;274(5293):1672–1677. | ||
Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411(6835):342–348. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.