")
Back to Journals » Cancer Management and Research » Volume 10
Silencing the expression of copine-III enhances the sensitivity of hepatocellular carcinoma cells to the molecular targeted agent sorafenib
Authors Chen Z, Jiang ZK, Zhang WZ, He BX
Received 9 March 2018
Accepted for publication 15 May 2018
Published 29 August 2018 Volume 2018:10 Pages 3057—3067
DOI https://doi.org/10.2147/CMAR.S167781
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leylah Drusbosky
Zhuo Chen, Zhengkui Jiang, Wenzhou Zhang, Baoxia He
The Affiliated Cancer Hospital of Zhengzhou University/Henan Cancer Hospital, Zhengzhou 450008, People’s Republic of China
Background: The application of the oral targeted therapeutic agent sorafenib provides new hope for patients suffering from advanced stages of hepatocellular carcinoma (HCC), but the prognosis of such patients remains poor due to the rapid development of the multidrug resistance process in cancer pathogenesis. The present work evaluated whether copine-III, a novel cancer regulator encoded by the CPNE3 gene, would be a potential indicator of sorafenib resistance in HCC treatment.
Materials and methods: The endogenous expression of copine-III in clinical specimens was examined by quantitative polymerase chain reaction. Copine-III siRNA was transfected into HCC cells to downregulate copine-III expression. The effect of copine-III on sorafenib’s antitumor activation was identified by in vitro and in vivo experiments (MTT, Transwell, and flow cytometry as well as a nude mice model).
Results: High levels of copine-III in clinical specimens are related to poor prognosis of advanced HCC patients on sorafenib treatment. Infection of Ad-siCPNE3 significantly decreased the endogenous expression of copine-III and enhanced the susceptibility of MHCC97-H cells to sorafenib: the IC50 value decreased from 1.15±0.11 to 0.25±0.05 μmol/L. Moreover, silencing copine-III enhanced the effect of sorafenib on apoptosis, in vitro invasion/migration, and subcutaneous or intrahepatic growth of MHCC97-H cells in nude mice.
Conclusion: Copine-III is a novel potential indicator of prognosis for patients who received sorafenib for advanced HCC treatment.
Keywords: hepatocellular carcinoma, CPNE3, copine-III, molecular targeted agent, sorafenib-resistance
Introduction
Hepatocellular carcinoma (HCC) is one of the most fatal malignancies in the Asia-Pacific region, especially in the People’s Republic of China.1,2 Most patients suffering from advanced HCC at initial diagnosis have poor prognosis due to tumor insensitivity against radiation therapy and chemotherapy.3–5 Application of an oral multi-kinase inhibitor/molecular targeted agent (e.g., sorafenib) has been expected to provide promising effects in HCC treatment; however, only a low proportion of patients (30–40%) were sensitive to sorafenib, and the rapid development of tumor resistance to sorafenib hinders the efficacy of the related therapy.6,7 Therefore, there is an urgent need to identify the indicators for prognosis of patients who receive sorafenib and to develop new strategies to overcome the resistance of HCC patients to sorafenib during treatment.
The copine protein family has nine members and are encoded by CPNE1–CPNE9 genes that are conserved from plants to humans.8 Similar to other family members, copine-III has three major functional domains including two N-terminal C2 domains (C2Ds) and a C-terminal von Willebrand A-like domain/copine A domain.9 Recently, Jung et al provided evidence that the high expression of CPNE3 genes would be a key cause of the cancer stem cell (CSC) process in the pathogenesis of acute myeloid leukemia (AML).10 However, the detailed roles of copine-III in human cancers, and especially in HCC, are still unclear.
In the present work, we aim to reveal the roles of Copine III in regulating HCC and especially in sensitivity of HCC cells to molecular agent. Adenoviral siRNA vector was used to downregulate Copine III’s expression. The sensitivity of HCC to molecular targeted agent sorafenib was examined by in vitro assays, including MTT or transwell experiments; or in vivo assays, e.g. subcutaneous or intrahepatic HCC model in nude mice. Our results indicate that silencing copine-III enhanced the susceptibility of MHCC97-H cells to the antitumor agent sorafenib and, therefore, copine-III can be a novel therapeutic target for HCC treatment.
Materials and methods
Clinical specimens and quantitative polymerase chain reaction (qPCR)
HCC clinical specimens were obtained by liver biopsy and collected by our laboratory. None of the HCC patients had received any prior treatment. The collection of clinical specimens and all protocols or experiments were undertaken with the written informed consent of patients and with approval for experiments from the Ethics Committee of Zhengzhou University. Our studies were conducted in compliance with the Declaration of Helsinki. The methods were not related to a clinical trial. Moreover, all animal studies were carried out in accordance with the UK Animals (Scientific Procedures) Act, 1986 and associated guidelines. For qPCR experiments, a total of 141 HCC cases (Table 1) were involved. Total RNA samples were extracted from all specimens and assessed by qPCR experiments according to methods described previously.11,12 The qPCR (real-time reverse transcription-polymerase chain reactions) was undertaken in an Applied Biosystems 7500 Detection system using Maxima SYBR Green/ROX qPCR Master Mix Assays (Fermentas, Thermo Fisher, Waltham, MA, USA). β-actin was used as the loading control, and relative mRNA expression of CPNE3 was normalized to the expression of human β-actin mRNA. The primers of CPNE3 used in qPCR were forward sequence, GTTTTGGCGCTCAGATA CCTCC; reverse sequence, GACAAGACCGATA CGCCTCTAC.
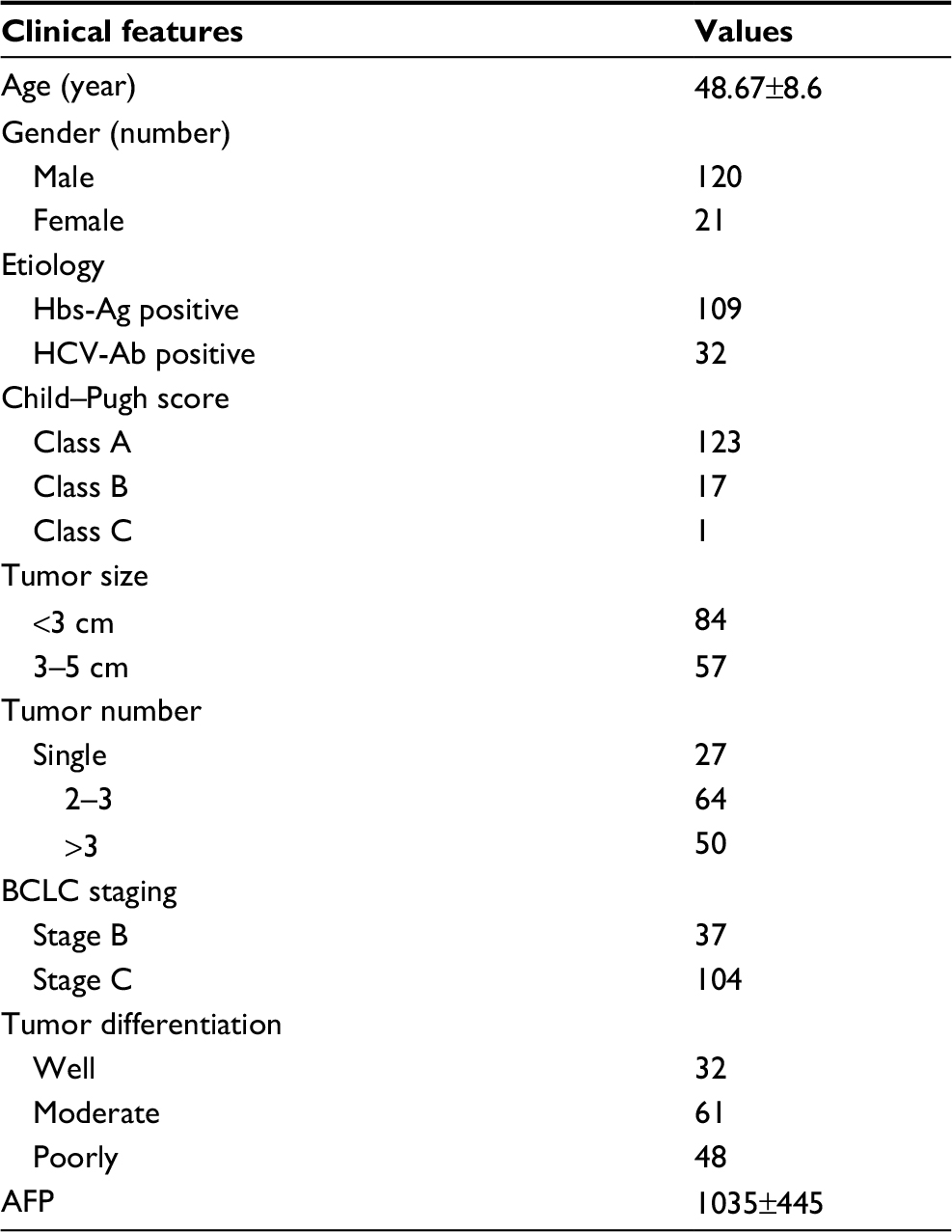 | Table 1 Baseline characteristics of patients in this study Abbreviation: AFP, alpha-feto protein. |
Cell culture and agents
Plasmids containing full-length sequences of CPNE3 (cat. no. CH872256) were obtained from Vigene Biosciences Corporation, Jinan, Shandong Province, People’s Republic of China. The adenovirus expression vector of CPNE3 or its siRNA was also designed and constructed by the Vigene Biosciences Corporation. The antitumor agent sorafenib (cat. no. S7397) was purchased from Selleck Company (Houston, TX, USA). MHCC97-H (a highly aggressive HCC cell line), MHCC97-L, HepG2, Hu7, BEL-7402, SMMC-7721, or L-02 cells were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, People’s Republic of China). Cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% FBS added to it at 37°C with 5% CO2.
Western blot analysis
Antibodies against copine-III (cat. no. ab97919), E-cadherin (cat. no. ab1416), N-cadherin (cat. no. ab18203), vimentin (cat. no. ab92547), and GAPDH (cat. no. ab8245) were obtained from Abcam Corporation (Cambridge, MA, USA). Western blotting was conducted by following standard protocols. Total protein was analyzed by SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). Next, blots were blocked with 5% BSA and then incubated with primary antibodies at 37°C for 2 h. Then, blots were incubated with the HRP-conjugated secondary antibodies. Finally, membranes were developed with enhanced chemiluminescence reagents by X-ray films.
Inhibition rate of sorafenib
For cell survival inhibition assays, sorafenib was dissolved in dimethyl sulfoxide (DMSO) with indicated concentrations (10, 3, 1, 0.3, 0.1, 0.03, or 0.01 mmol/L). Then, sorafenib was diluted by DMEM with 0.5% FBS with a dilution of 1:1,000 to the treated cells. The final concentrations of sorafenib were 10, 3, 1, 0.3, 0.1, 0.03, or 0.01 μmol/L, and the DMSO concentration was only 1% DMEM with 0.5% FBS, and 1% DMSO was used as solvent control.
L-02 cells were infected by control or Ad-CPNE3, whereas MHCC97-H were infected by control or Ad-siCPNE3. Cells were analyzed by the inhibition-analysis experiments conducted according to methods described previously.13,14 Sorafenib concentrations were selected as: 10, 3, 1, 0.3, 0.1, 0.03, or 0.01 μmol/L. The inhibition rates of sorafenib on HCC cells were calculated as (OD 490 control group − OD 490 administration group)/(OD 490 control group − OD 490 blank group) × 100%.15,16
Flow cytometry for cell apoptosis
MHCC97-H cells, which were treated with the IC50 concentration of sorafenib for 48 h, were harvested for flow cytometric analysis. For apoptosis analysis, cells were labeled with 7-aminoactinomycin D and fluorescein isothiocyanate-Annexin V by following the manufacturer’s instructions (BD Biosciences, Franklin Lakes, NJ, USA). Then, cells were collected and analyzed by a FACS Calibur Flow Cytometer (BD Biosciences).17,18
In vitro invasion and migration experiments
MHCC97-H cells, which were infected with vectors or treated with agents, were harvested for Transwell assays conducted in a 24-well plate chamber fitted with a polyethylene terephthalate filter membrane with 8-μm pores (Corning Incorporated, Corning, NY, USA). For measuring in vitro invasion, membrane undersurfaces were preincubated with 30 μL extracellular matrix (ECM) mixed with serum-free Dulbecco’s Modified Eagle Medium (DMEM, Sigma Corporation, St. Louis, MO, USA) in 1:5 dilution for 4 h at 37°C. The top chambers of the transwells were filled with a 100 μL cell suspension at 5×105 cells per milliliter density diluted in serum-free medium, and the bottom chambers were filled with 400 μL Dulbecco’s modified eagle medium containing 10% FBS. For the migration assay, the cells were incubated in chambers without ECM coating. Then, cells were incubated in chambers for 12–16 h (in vitro invasion) or 4–6 h (in vitro migration).19,20
In vivo experiments
All animal studies were approved by the Animal Care Committee of Zhengzhou University. The animals (severe combined immune-deficient mice/nude mice) were provided by the Laboratory Animal Center of Zhengzhou University. For the subcutaneous tumor model, MHCC97-H cells, which were infected with control or siCPNE3, were injected into nude mice. The in vivo experiments were conducted according to the protocol described earlier.21,22 Mice received solvent control phosphate buffer saline (PBS) or sorafenib (1.5 mg/kg) treatment. The solvent control (PBS) or sorafenib was given via oral administration every 2 days for 3 weeks. Results were shown as represented by a photograph or tumor weight.
For the intrahepatic tumor model, MHCC97-H cells infected with control or siCPNE3 were directly injected into the right lobe of mice liver. After 4–6 days, mice received solvent control (PBS) or sorafenib (1.5 mg/kg) treatment. Solvent control (PBS) or sorafenib was administered orally every 2 days for 3 weeks. Next, mice were analyzed by MicroPET/CT analysis. The radioactivity examination of liver to blood was carried out according to methods described by Xu et al and Feng et al.23,24
Statistical analysis
The results were represented as the average from triplicate experiments and expressed as the mean ± standard deviation, and Bonferroni’s correction with or without two-way ANOVA was used to determine statistical significance among groups. Analysis of survival rates was conducted by the Kaplan–Meier method and compared by the log-rank test.
Results
Highly endogenous expression of copine-III is associated with poor clinical outcome of sorafenib treatment
To identify the potential roles of copine-III in regulating advanced HCC treatment, we measured the expression of copine-III in HCC samples, according to the median value of CPNE3 mRNA levels, we divided the patients who were selected into two groups: copine-III low group (n=73) and copine-III high group (n=68; Figure 1A and B). The mRNA level of CPNE3 or representative DNA bands of each group are shown in Figure 1A and B. Next, results showed that the CPNE3-high group had poorer prognosis with sorafenib treatment than the CPNE3-low group (Table 2; Figure 1C and D): the time to progress (TTP) of the CPNE3-high group of patients (median TTP: 8.0 months; 95% CI: 7.2–10.8 months) was shorter than that of patients in the CPNE3-low group (median TTP: 11.0 months; 95% CI: 10.9–13.1 months, log-rank P=0.0025); and the median OS (median OS: 10.0 months; 95% CI: 8.1–12.1 months) in the high-CPNE3 group was shorter than that in the CPNE3-low group (median OS: 14.0 months; 95% CI: 12.7–15.3 months; log-rank P=0.0004).
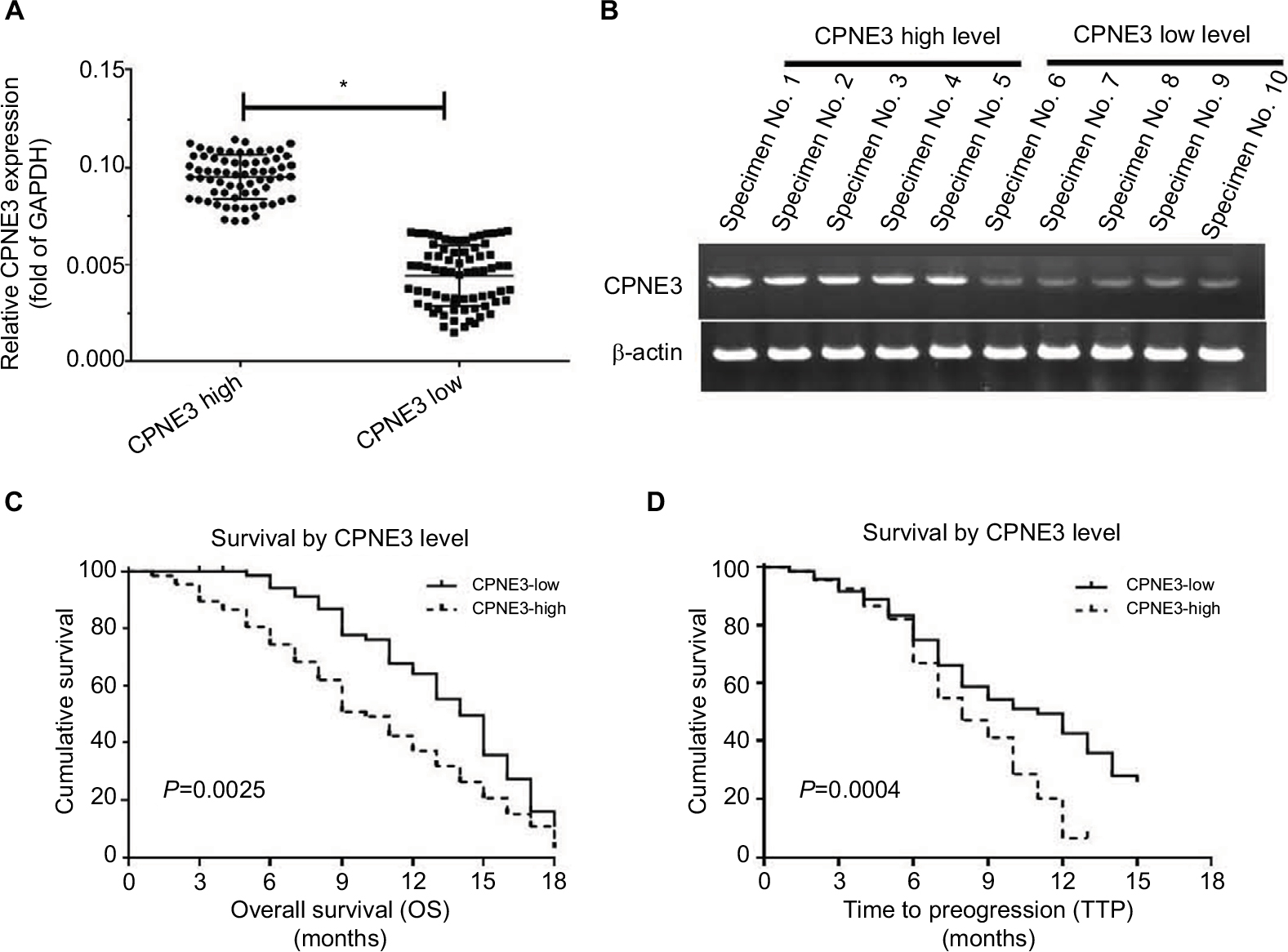 | Figure 1 Endogenous mRNA level of CPNE3 in advanced hepatocellular carcinoma (HCC) tissues related to clinical outcome of patients who received sorafenib treatment. Notes: (A and B) Endogenous mRNA level of CPNE3 was identified by quantitative polymerase chain reaction (qPCR) as relative RNA (A) or represented DNA electrophoresis bands from ten representative specimens (five high and five low) (B). (C) OS of patients who received sorafenib. (D) Time to progress of patients who received sorafenib. (C and D) Survival analysis was conducted using the Kaplan–Meier and log-rank test. (A) *P<0.05 versus CPNE3 high group or CPNE3 low group; (C) P=0.0025 versus CPNE3 high group or CPNE3 low group; (D) P=0.0004 versus CPNE3 high group or CPNE3 low group. |
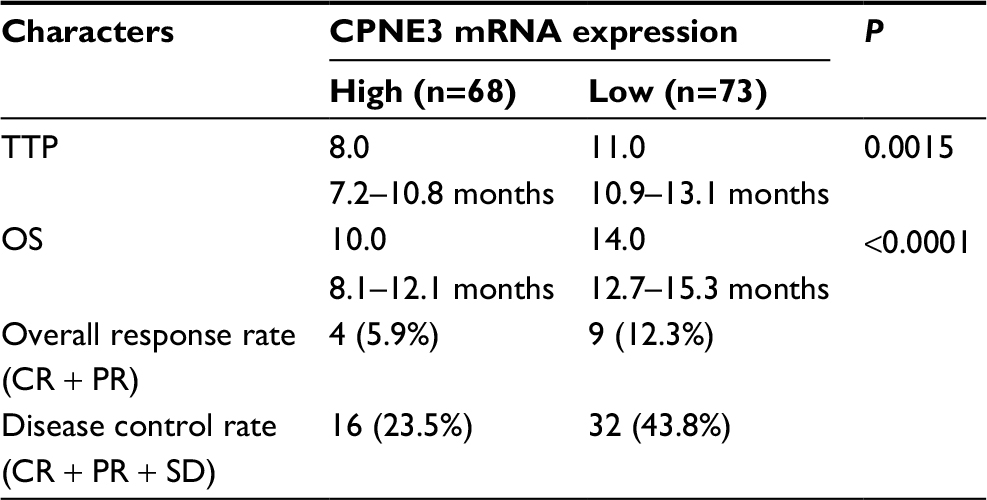 | Table 2 CPNE3 expression and clinical outcome of sorafenib treatment Abbreviations: TTP, time to progress; OS, overall survival; PR, partial remission; CR, complete remission; SD, stable disease. |
Moreover, the overall response rate (complete response [CR] + partial response [PR]) and disease control rate (CR + PR + stable disease [SD]) of patients who received sorafenib in the CPNE3-high group (CR + PR, 5.9%; CR + PR + SD, 23.5%) are much lower than those for patients in the CPNE3-low group (CR + PR, 12.3%; CR + PR + SD, 43.8%; Table 2). The results indicated that high level of endogenous CPNS3 expression is associated with poor prognosis with sorafenib therapy.
Copine-III expression higher in HCC cells than in hepatic non-tumor L-02 cells
Next, the expression of copine-III was evaluated in cell lines. As shown in Figure 2A, the HCC cell lines express a higher level of copine-III than that in hepatic non-tumor cells L-02. MHCC97-H, a highly aggressive HCC cell line, expresses the highest levels of copine-III among HCC cells. Thus, MHCC97-H cells were used to for knockdown of copine-III expression, whereas L-02 cells were used for overexpression of copine-III.
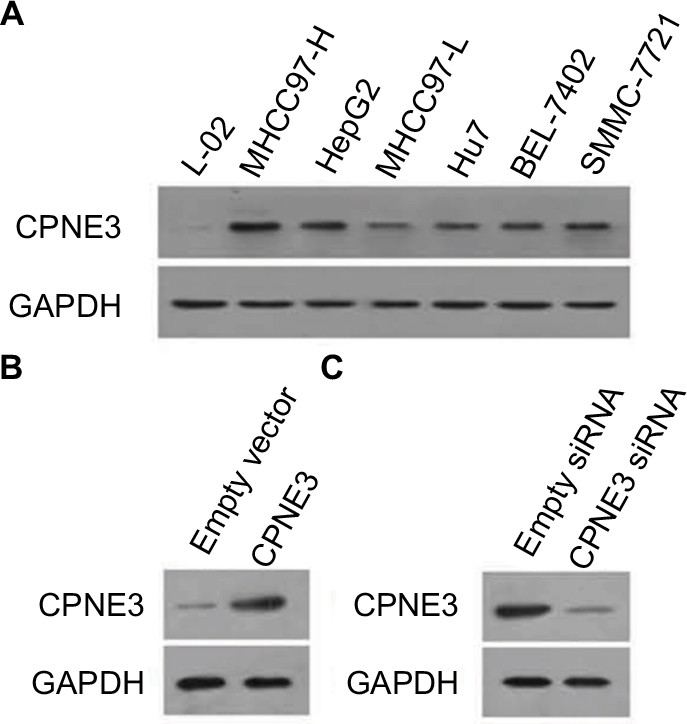 | Figure 2 Expression of copine-III in hepatic cell lines. Notes: (A) Endogenous protein level of copine-III was detected in L-02, MHCC97-H, MHCC97-L, Hu7, BEL-7402, SMMC-7721, and HepG2 cell lines by Western blotting. CPNE3 expression vectors boosted the expression of copine-III in L-02 cells (C), and infection of siCPNE3 significantly reduced the expression of copine-III in MHCC97-H cells (B). |
Then, we constructed the adenoviral particles of CPNE3 siRNA to knock down endogenous copine-III in MHCC97-H cells and the expressing viral vector to overexpress CPNE3 in L-02 cells. The results showed that Ad-siCPNE3 could decrease the protein level of copine-III in MHCC97-H cells as compared with parental cells or cells infected with Ad-control (control siRNA, Figure 2B). Meanwhile, infection with the CPNE3-expressing vector (Ad-CPNE3) increased the expression of copine-III in L-02 cells (Figure 2C).
Copine-III knockdown enhances the inhibitory effects of sorafenib on MHCC97-H cell survival
To discover the effect of copine-III in the sorafenib resistance of HCC, we compared the efficacy of sorafenib administration on MHCC97-H cells with and without copine-III. As shown in Figure 3A and B, silencing copine-III significantly enhanced the efficacy of sorafenib on MHCC97-H cells in a dose- or time-dependent manner. The IC50 value of sorafenib on MHCC97-H cells was decreased from 1.15±0.11 to 0.25±0.05 μmol/L.
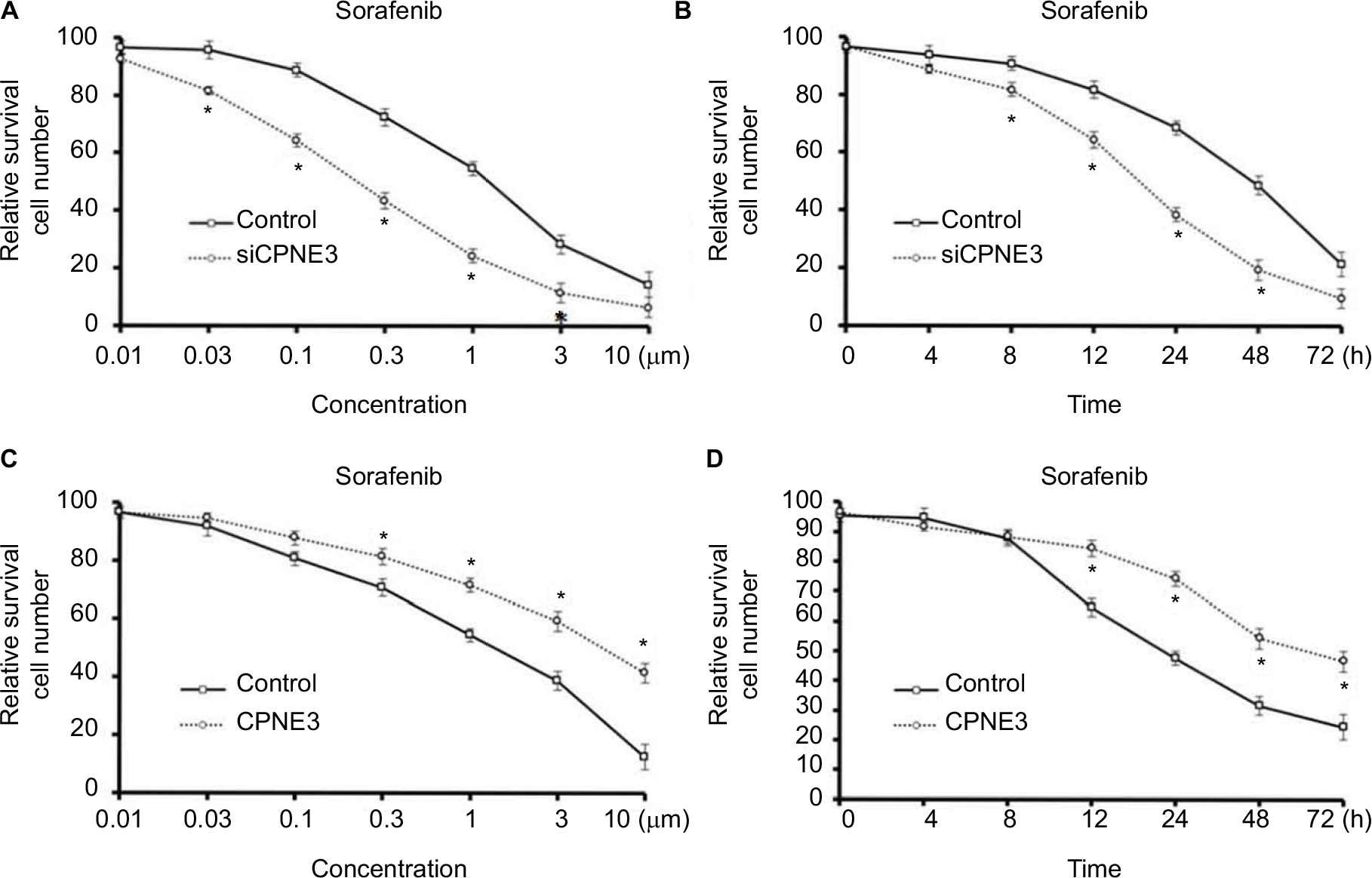 | Figure 3 Copine-III regulated the sensitivity of cells to sorafenib. Notes: (A) MHCC97-H cells, which were infected with control siRNA or siCPNE3, were treated with indicated concentrations of sorafenib and analyzed by MTT assays. (B) MHCC97-H cells, which were infected with control siRNA or siCPNE3, were treated with IC50 concentration of sorafenib and analyzed by MTT assays at indicated time points. (C) L-02 cells, which were infected with control or CPNE3, were treated with indicated concentration of sorafenib and analyzed by MTT-assays. (D) MHCC97-H cells, which were infected with control or CPNE3, were treated with the IC50 concentration of sorafenib and analyzed by MTT assays at indicated time points. *P<0.05 versus with control group or CPNE3 Group. |
To further examine the role of copine-III, MTT and colony-formation experiments were conducted in L-02 cells with or without copine-III overexpression. Sorafenib inhibited L-02 cells proliferation in a dose- (Figure 3C) and time-dependent (Figure 3D) manner. Overexpression of copine-III significantly attenuated the effect of sorafenib on L-02 cells (Figure 3C). The IC50 value of sorafenib on MHCC97-H cells was increased from 2.12±0.33 to 8.55±0.60 μmol/L. Therefore, copine-II would decrease the effect of sorafenib in HCC cells.
Silencing copine-III enhances the inhibitory activity of sorafenib on MHCC97-H cell in vitro metastasis
Metastasis is one of the foremost features of human malignancies. Sorafenib has been expected to reduce tumor metastasis. In line with this, we found that the IC50 concentration of sorafenib inhibited the in vitro invasion of MHCC97-H cells (Figure 4A). Silencing copine-III significantly enhanced the inhibitory activity of sorafenib on MHCC97-H cell in vitro invasion (Figure 4A). Similar results were obtained from in vitro migration examination (Figure 4B). These data show that copine-III knockdown upregulates the inhibitory activity of sorafenib on MHCC97-H cell in vitro invasion or migration.
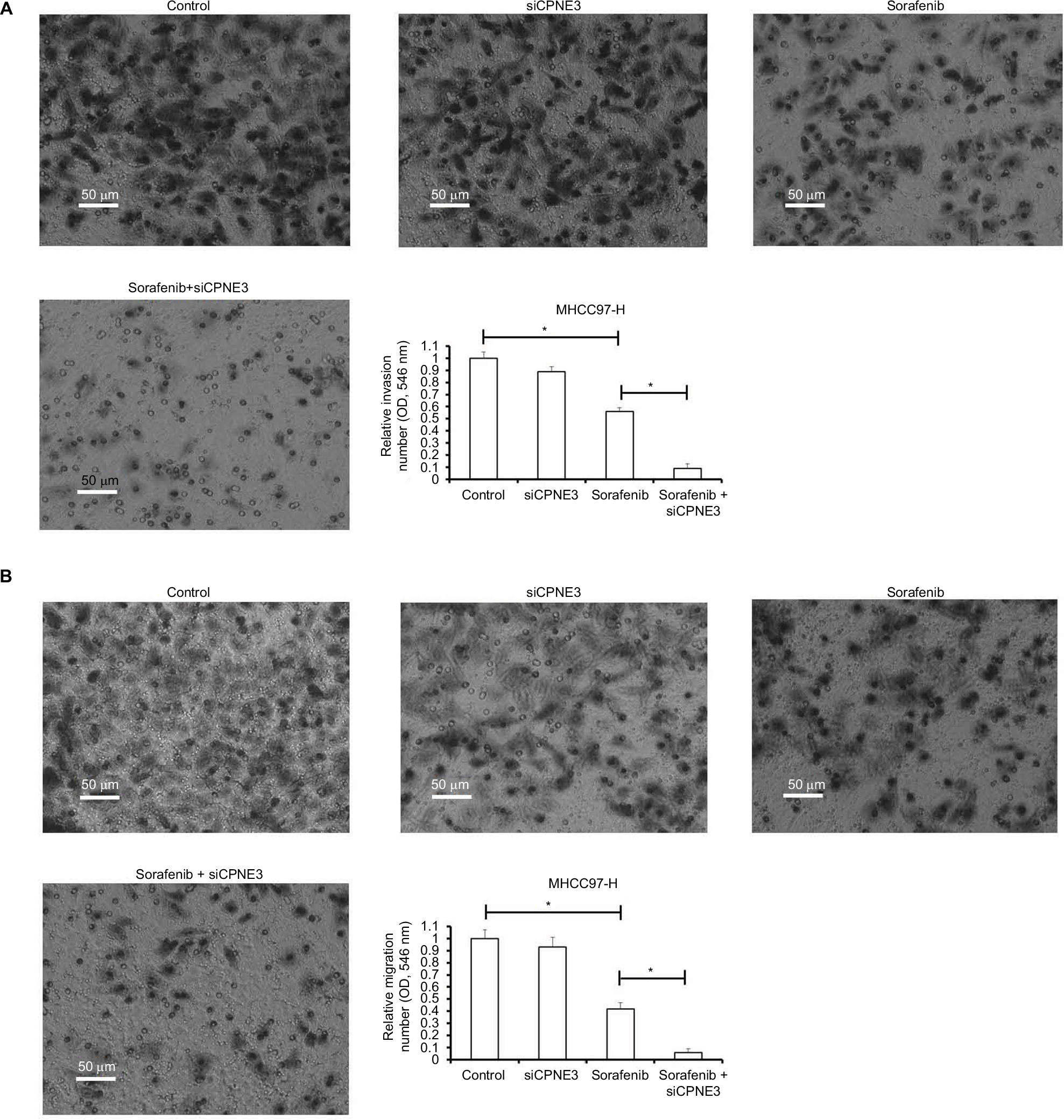 | Figure 4 Silencing copine-III enhances the effect of sorafenib on MHCC97-H cell in vitro invasion and migration. Notes: MHCC97-H cells, which were infected with control siRNA or siCPNE3, were treated with indicated concentrations of sorafenib and analyzed by Transwell analysis. (A) In vitro invasion of MHCC-97-H cells; (B) the in vitro migration of MHCC97-H cells. *P<0.05. |
Copine-III knockdown enhances the inhibitory effects of sorafenib on MHCC97-H cell survival
Further, the role of copine-III in sorafenib-induced apoptosis was determined. As shown in Figure 5, sorafenib induced the apoptosis of MHCC97-H cells (Figure 5A and C). Apoptotic cells increased from 1.12% to 22.5% (Figure 5E). Knockdown of copine-III enhanced the inhibitory activity of sorafenib on MHCC97-H (Figure 5A, and C–E). Apoptotic cells increased from 22.5% to 36.6%. The results confirmed that CPNE3 knockdown upregulates the effect of sorafenib on MHCC97-H cell apoptosis.
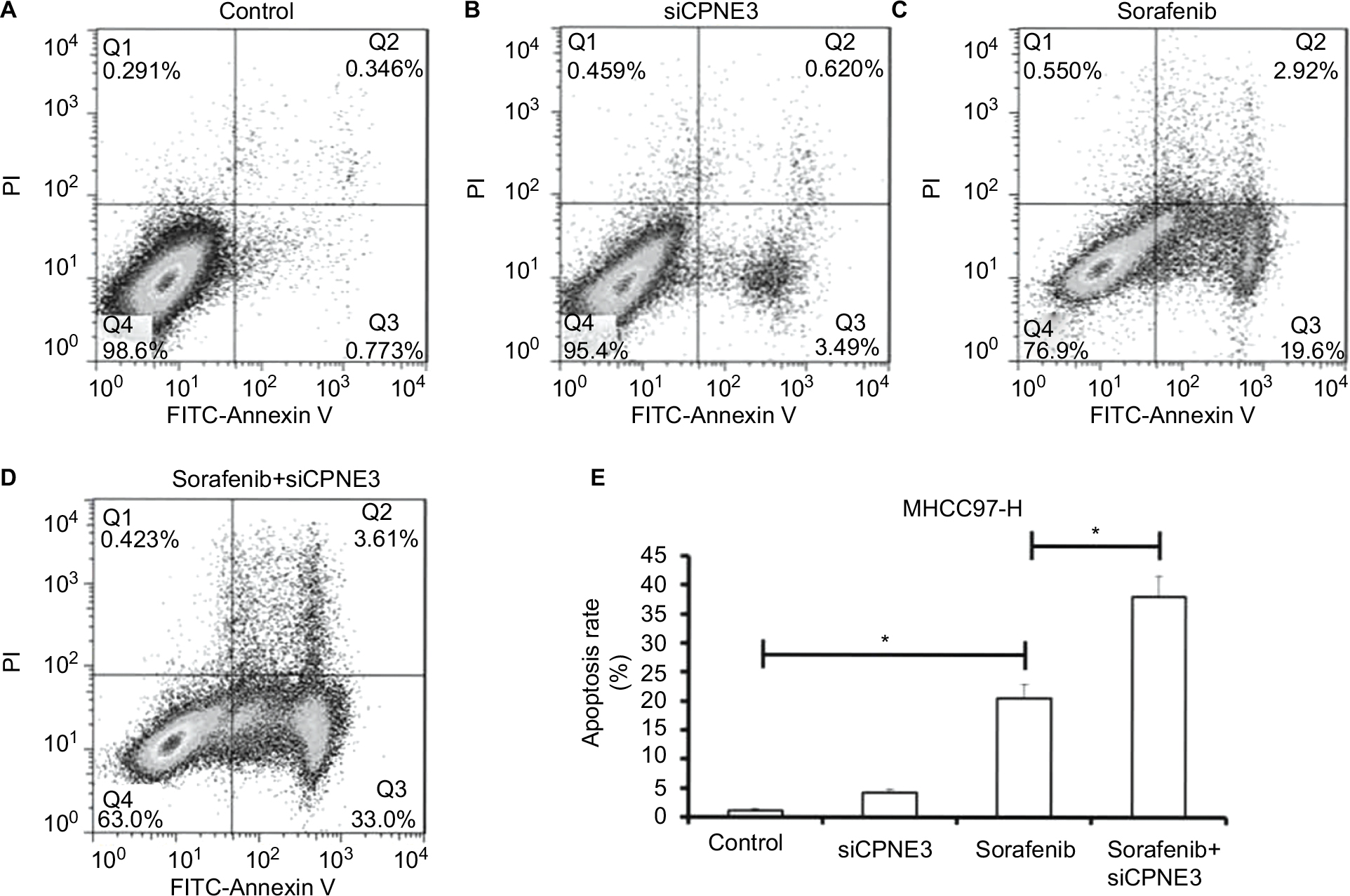 | Figure 5 Silencing copine-III enhances sorafenib-induced MHCC97-H cell apoptosis. Notes: (A–E) MHCC97-H cells, which were infected with control siRNA or siCPNE3, were treated with indicated concentrations of sorafenib and analyzed by flow cytometer. *P<0.05. Abbrevaiation: FITC, fluorescein isothiocyanate. |
CPNE3 knockdown enhanced the inhibitory effect of sorafenib on HepG2 cells in vivo growth
We next established a tumor-bearing nude mice model via MHCC97-H cell injection for further in vivo study. Treatment with sorafenib could significantly reduce the tumor volume (Figure 6A and B) or tumor weight (Figure 6C) of subcutaneous tumors formed by MHCC97-H cells, whereas copine-III knockdown further increased this effect, significantly (Figure 6).
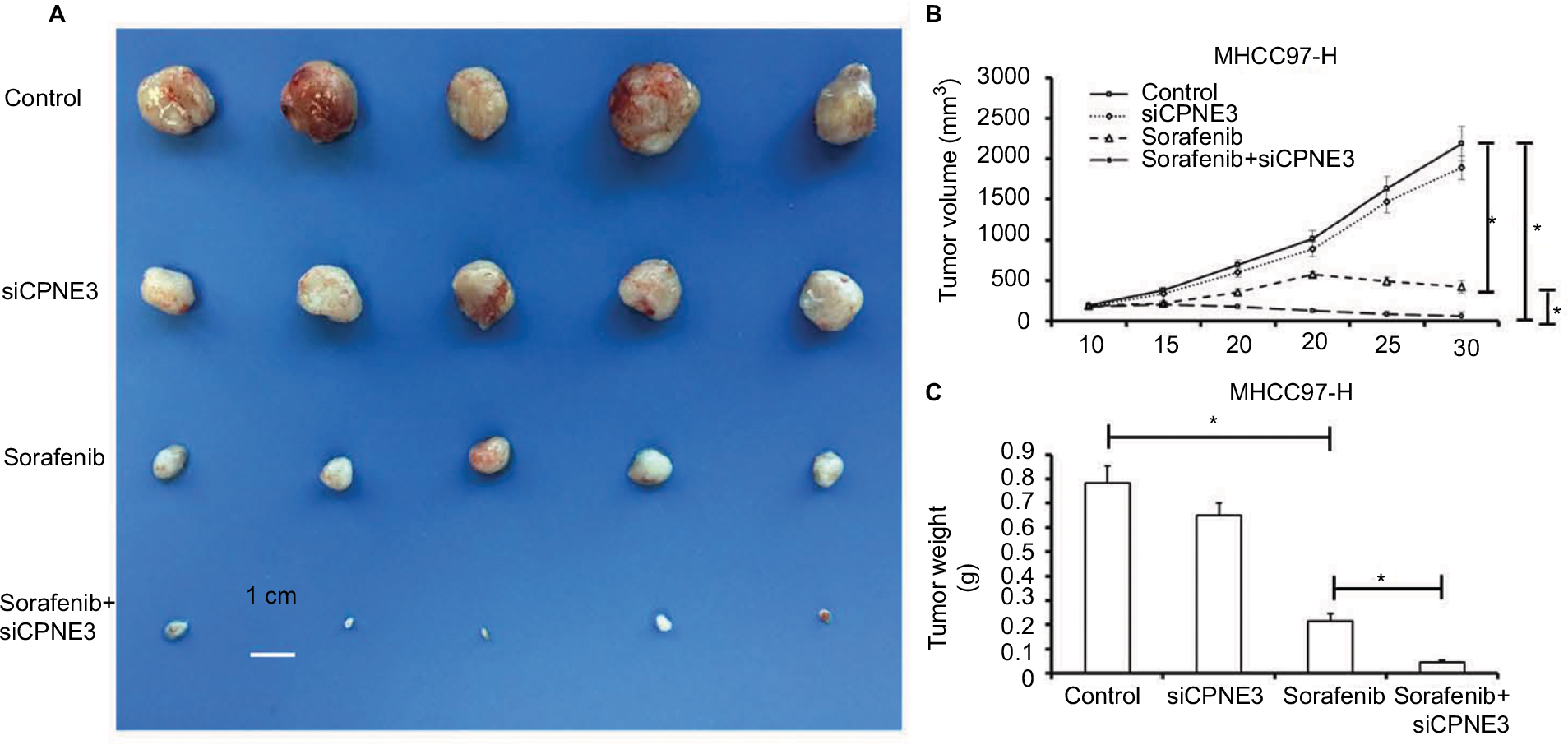 | Figure 6 Silencing copine-III enhances the effect of sorafenib on MHCC97-H cell subcutaneous growth. Notes: MHCC97-H cells, which were infected with control siRNA or siCPNE3, were treated with indicated concentration of sorafenib and analyzed in a nude mice mode. The size of tumors was shown as photographs (A), tumor growth curve by mean ± standard deviation (B), or tumor weight by mean ± standard deviation (C). *P<0.05. |
Next, the intrahepatic growth of MHCC97-H cells was determined. As shown in Figure 6, intrahepatic growth of MHCC97-H cells in mice liver was identified by positron emission tomography (PET)/CT screening. Sorafenib decreased the PET intensity in the mice’s liver location as compared with control, whereas knockdown of copine-III enhanced the effect of sorafenib (Figure 7A). PET images were confirmed by a liver-to-blood radioactivation examination (Figure 7A). Moreover, mice were harvested, and liver specimens were collected to identify the nodules formed by MHCC97-H cells. As shown in Figure 7B, sorafenib treatment inhibited the intrahepatic growth of MHCC987-H cells, and the nodules in mice’s liver formed by HCC cells were shrinking. Silencing of copine-III enhanced the antitumor effect of sorafenib in intrahepatic growth of HCC cells. The PET imaging of resected livers was also obtained (Figure 7B). Thus, the in vivo model confirms that silencing copine-III enhances the effect of sorafenib in inhibiting tumor growth.
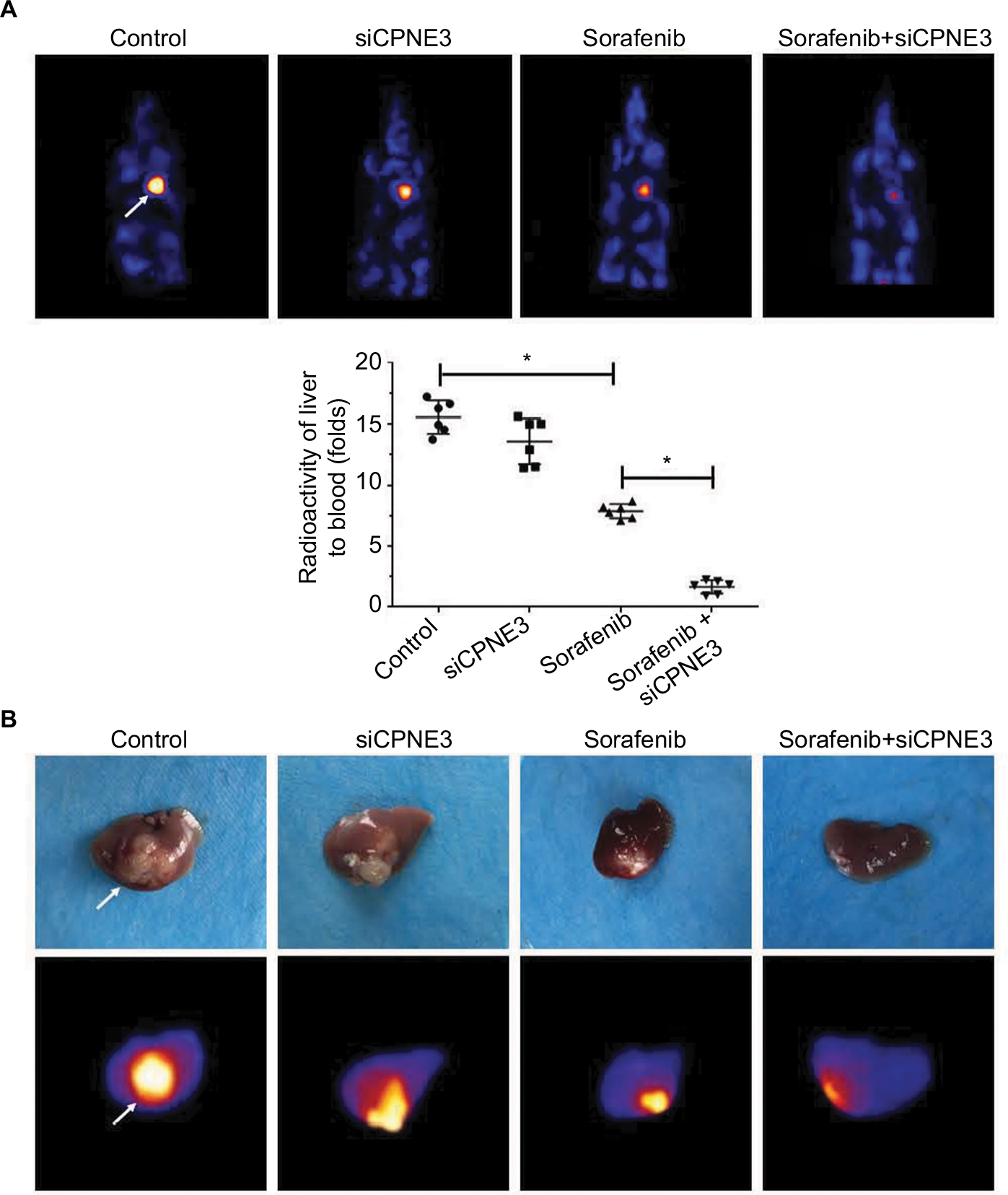 | Figure 7 Silencing copine-III enhances the effect of sorafenib on MHCC97-H cell intrahepatic growth. Notes: MHCC97-H cells, which were injected with control siRNA or siCPNE3, were injected into liver lobes to form intrahepatic nodules. Next, nude mice were treated with a solvent control or sorafenib. After 4–6 weeks of growth, tumor nodules formed by MHCC97-H cells in liver organs were examined by positron emission tomography (PET)/CT scanning. (A) Results showed the PET/CT images of whole animals or radioactivation of liver to blood; (B) images of tumor modules or PET/CT results from the liver. *P<0.05. |
Knocking down copine-III modulates the epithelial–mesenchymal transition (EMT) indicators
To examine potential copine-III function, the role of copine-III in indicators of epithelial–mesenchymal transition (EMT) was determined. Our results showed that silencing copine-III enhanced the expression of E-cadherin (an epithelial indicator) and decreased the expression of N-cadherin or vimentin (two mesenchymal markers; Figure 8). Therefore, copine-III may participate in MHCC97-H regulation via modulating its EMT process.
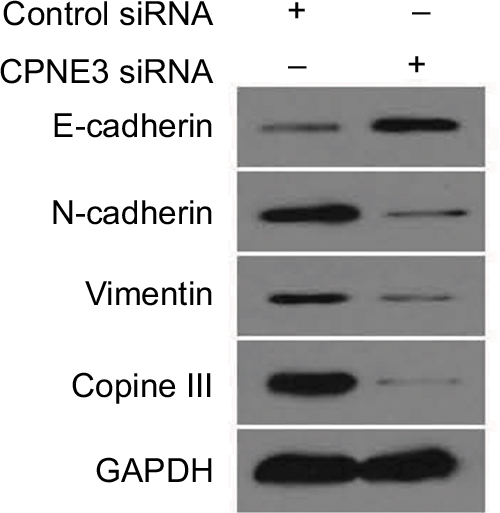 | Figure 8 Silencing copine-III modulates the expression of epithelial–mesenchymal transition (EMT) indicators. Notes: MHCC97-H cells, infected with control siRNA or siCPNE3, were harvested and analyzed by Western blotting assays. The expression of copine-III, E-cadherin, N-cadherin, or vimentin was detected via their antibodies. GAPDH was chosen as a loading control. |
Discussion
In the present work, our results show that copine-III participates in the sorafenib-resistance process of HCC cells. High levels of copine-III expression in clinical specimens were related to poor prognosis of sorafenib-treated patients with advanced HCC. Copine-III is highly expressed in HCC cells than in L-02 cells – a hepatic non-tumor cell line. MHCC97-H cells express the highest level of copine-III among HCC cell lines. Silencing copine-III significantly enhanced the sensitivity of MHCC97-H cells to sorafenib. On the other hand, overexpression of copine-III protected L-02 cells against sorafenib. Infection of copine-III siRNA enhances the antitumor effect of sorafenib by inducing MHCC97-H cell apoptosis and their in vitro invasion/migration. Moreover, silencing copine-III enhances the efficacy of sorafenib on HepG2 cells in vivo. These results all suggested that copine-III might be involved in the sorafenib resistance of HCC.
Copine-III has been identified as a regulator of human cancer. Raufi and Tirona showed that copine-III can interact with ErbB2 (HER2) and promote migration of breast cancer cells.8 The receptor of activated C kinase 1 (i.e., RACK1) and Jun activation domain-binding protein 1 (Jab1) were also found later to participate in copine-III–ErbB2 interaction.9 Mo et al revealed the potential roles of copine-III in prostate cancer.25 Lin et al found the metastasis-promoting roles of copine-III in non-small cell lung cancer (NSCLC) via a quantitative proteomic analysis.26 Recently, Fu et al reported that high expression of CPNE3 predicts adverse prognosis in AML.27 Our results showed that copine-III would participate in sorafenib-resistance in advanced HCC treatment. These data expand our understanding of copine-III in human cancers. Interestingly, silencing endogenous copine-III did not significantly disrupt the in vitro tumor cell proliferation, invasion, or migration nor in vivo growth of MHCC97-H cells; furthermore, silencing copine-III did not induce the apoptosis of MHCC97-H cells. These may be due to the elevated expression of some types of receptor tyrosine kinases in HCC or other types of human cancer that compensates for the effects: a high level of EGFR has been identified in NSCLC28,29 and a high level of HER2 has been detected in some types of breast cancer.30,31 Clinical investigations showed that patients with advanced-stage HCC could not gain benefits from gefitinib (targeting EGFR) and trastuzumab (targeting ErbB2/HER2).32–35 Therefore, copine-III may have functions in the sorafenib resistance process in HCC which is independent of the copine-III–ErbB2 axis. Our results showed that silencing copine-III enhanced the expression of E-cadherin36 (an epithelial indicator) and decreased the expression of N-cadherin37 or vimentin38 (two mesenchymal markers). Therefore, inhibiting the EMT process of HCC cells would be a potential mechanism of copine-III function.
Moreover, Jung et al provided evidence that high expression of the CPNE3 genes would be a key cause of CSC process in AML pathogenesis.10 Moreover, the concept of CSCs is used to explain solid proliferation hierarchy and heterogeneity among cancer cells in solid tumors.39 In solid tumor tissues, CSCs would be an affiliated group of cancer cells that are resistant to chemotherapy.40 Attenuating the stemness of cancer cells would also be a useful strategy to enhance their susceptibility to antitumor agents.41 Among the identified surface markers of CSCs in human cancers, EpCAM,42 CD133,43 or CD9044 may be associated with HCC.45,46 Therefore, it is valuable to examine whether copine-III would modulate the expression of markers of CSCs associated with HCC and the roles of copine-III regulation of HCC CSCs.
Conclusion
We conclude that copine-III would enhance the resistance of HCC cells to sorafenib and be a novel prognosis indicator for patients who receive sorafenib as a therapeutic target for advanced HCC treatment.
Acknowledgment
The authors would like to thank Dr Tao Wang of the Beijing Institute of Pharmacology and Toxicology, Beijing, People’s Republic of China, for his help and advice.
Disclosure
The authors report no conflicts of interest in this work.
References
Chen J, Zaidi S, Rao S, et al. Analysis of genomes and transcriptomes of hepatocellular carcinomas identifies mutations and gene expression changes in the transforming growth factor-β pathway. Gastroenterology. 2018;154(1):195–210. | ||
Guo H, Wu T, Lu Q, et al. Surgical resection improves long-term survival of patients with hepatocellular carcinoma across different Barcelona Clinic Liver Cancer stages. Cancer Manag Res. 2018;10:361–369. | ||
Sherman M. Regorafenib for treatment of hepatocellular carcinoma. Hepatology. 2018;67(3):1162–1165. | ||
Reiss KA, Yu S, Mamtani R, et al. Starting dose of sorafenib for the treatment of hepatocellular carcinoma: a retrospective, multi-institutional study. J Clin Oncol. 2017;35(31):3575–3581. | ||
Kim DW, Talati C, Kim R. Hepatocellular carcinoma (HCC): beyond sorafenib-chemotherapy. J Gastrointest Oncol. 2017;8(2):256–265. | ||
Zhu YJ, Zheng B, Wang HY, Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38(5):614–622. | ||
Li J, Wu PW, Zhou Y, et al. Rage induces hepatocellular carcinoma proliferation and sorafenib resistance by modulating autophagy. Cell Death Dis. 2018;9(2):225. | ||
Raufi A, Tirona MT. Prospect of the use of checkpoint inhibitors in hepatocellular cancer treatments. Cancer Manag Res. 2017;9:19–27. | ||
Choi HY, Park N, Na JB, Ko ES, Park JY, Yoo JC. Direct binding of Copine3 with Jab1 activates downstream ErbB2 signaling and motility in SKBr3 breast cancer cells. Oncol Rep. 2016;35(2):1147–1152. | ||
Jung N, Dai B, Gentles AJ, Majeti R, Feinberg AP. An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nat Commun. 2015;6:8489. | ||
Wei S, Zhong L, Wang X, Zhang W. Low expression of GATA3 promotes cell proliferation and metastasis in gastric cancer. Cancer Manag Res. 2017;9:769–780. | ||
Ying H, Xu Z, Chen M, Zhou S, Liang X, Cai X. Overexpression of Zwint predicts poor prognosis and promotes the proliferation of hepatocellular carcinoma by regulating cell-cycle-related proteins. Onco Targets Ther. 2018;11:689–702. | ||
Zhao J, Bai Z, Feng F, et al. Cross-talk between EPAS-1/HIF-2α and PXR signaling pathway regulates multi-drug resistance of stomach cancer cell. Int J Biochem Cell Biol. 2016;72:73–88. | ||
Hou J, Hong Z, Feng F, et al. A novel chemotherapeutic sensitivity-testing system based on collagen gel droplet embedded 3D-culture methods for hepatocellular carcinoma. BMC Cancer. 2017;17(1):729. | ||
Feng F, Lu YY, Zhang F, et al. Long interspersed nuclear element ORF-1 protein promotes proliferation and resistance to chemotherapy in hepatocellular carcinoma. World J Gastroenterol. 2013;19(7):1068–1078. | ||
Chen Y, Feng F, Gao X, et al. miRNA153 reduces effects of chemotherapeutic agents or small molecular kinase inhibitor in HCC cells. Curr Cancer Drug Targets. 2015;15(3):176–187. | ||
Li L, Kang L, Zhao W, et al. miR-30a-5p suppresses breast tumor growth and metastasis through inhibition of LDHA-mediated Warburg effect. Cancer Lett. 2017;400:89–98. | ||
Feng Y, Xu X, Zhang Y, et al. HPIP is upregulated in colorectal cancer and regulates colorectal cancer cell proliferation, apoptosis and invasion. Sci Rep. 2015;5:9429. | ||
Ma D, Jia H, Qin M, et al. miR-122 induces radiosensitization in non-small cell lung cancer cell line. Int J Mol Sci. 2015;16(9):22137–22150. | ||
Costa GA, de Souza SB, da Silva Teixeira LR, et al. Tumor cell cholesterol depletion and V-ATPase inhibition as an inhibitory mechanism to prevent cell migration and invasiveness in melanoma. Biochim Biophys Acta. 2018;1862(3):684–691. | ||
Jia H, Yang Q, Wang T, et al. Rhamnetin induces sensitization of hepatocellular carcinoma cells to a small molecular kinase inhibitor or chemotherapeutic agents. Biochim Biophys Acta. 2016;1860(7):1417–1430. | ||
An L, Li DD, Chu HX, et al. Terfenadine combined with epirubicin impedes the chemo-resistant human non-small cell lung cancer both in vitro and in vivo through EMT and Notch reversal. Pharmacol Res. 2017;124:105–115. | ||
Feng F, Jiang Q, Cao S, et al. Pregnane X receptor mediates sorafenib resistance in advanced hepatocellular carcinoma. Biochim Biophys Acta. 2018;1862(4):1017–1030. | ||
Xu X, Fan Z, Liang C, et al. A signature motif in LIM proteins mediates binding to checkpoint proteins and increases tumour radiosensitivity. Nat Commun. 2017;8:14059. | ||
Mo W, Zhang J, Li X, et al. Identification of novel AR-targeted microRNAs mediating androgen signalling through critical pathways to regulate cell viability in prostate cancer. PLoS One. 2013;8(2):e56592. | ||
Lin HC, Zhang FL, Geng Q, et al. Quantitative proteomic analysis identifies CPNE3 as a novel metastasis-promoting gene in NSCLC. J Proteome Res. 2013;12(7):3423–3433. | ||
Fu L, Fu H, Qiao J, et al. High expression of CPNE3 predicts adverse prognosis in acute myeloid leukemia. Cancer Sci. 2017;108(9):1850–1857. | ||
Vega JF, Ramos J, Cruz VL, et al. Molecular and hydrodynamic properties of human epidermal growth factor receptor HER2 extracellular domain and its homodimer: experiments and multi-scale simulations. Biochim Biophys Acta. 2017;1861(9):2406–2416. | ||
Hoesl C, Röhrl JM, Schneider MR, Dahlhoff M. The receptor tyrosine kinase ERBB4 is expressed in skin keratinocytes and influences epidermal proliferation. Biochim Biophys Acta. 2018;1862(4):958–966. | ||
Oh S, Kim H, Nam K, Shin I. Egr-1 is required for neu/HER2-induced mammary tumors. Cell Signal. 2018;45:102–109. | ||
Ezzoukhry Z, Louandre C, Trécherel E, et al. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int J Cancer. 2012;131(12):2961–2969. | ||
Blivet-Van Eggelpoël MJ, Chettouh H, Fartoux L, et al. Epidermal growth factor receptor and HER-3 restrict cell response to sorafenib in hepatocellular carcinoma cells. J Hepatol. 2012;57(1):108–115. | ||
Bodzin AS, Wei Z, Hurtt R, Gu T, Doria C. Gefitinib resistance in HCC mahlavu cells: upregulation of CD133 expression, activation of IGF-1R signaling pathway, and enhancement of IGF-1R nuclear translocation. J Cell Physiol. 2012;227(7):2947–2952. | ||
Xian ZH, Zhang SH, Cong WM, Wu WQ, Wu MC. Overexpression/amplification of HER-2/neu is uncommon in hepatocellular carcinoma. J Clin Pathol. 2005;58(5):500–503. | ||
Hsu C, Huang CL, Hsu HC, Lee PH, Wang SJ, Cheng AL. HER-2/neu overexpression is rare in hepatocellular carcinoma and not predictive of anti-HER-2/neu regulation of cell growth and chemosensitivity. Cancer. 2002;94(2):415–420. | ||
Zheng L, Xu M, Xu J, et al. ELF3 promotes epithelial-mesenchymal transition by protecting ZEB1 from miR-141-3p-mediated silencing in hepatocellular carcinoma. Cell Death Dis. 2018;9(3):387. | ||
Hou J, Ge C, Cui M, et al. Pigment epithelium-derived factor promotes tumor metastasis through an interaction with laminin receptor in hepatocellular carcinomas. Cell Death Dis. 2017;8(8):e2969. | ||
Hammouda MB, Riahi-Chebbi I, Souid S, et al. Macrovipecetin, a C-type lectin from Macrovipera lebetina venom, inhibits proliferation migration and invasion of SK-MEL-28 human melanoma cells and enhances their sensitivity to cisplatin. Biochim Biophys Acta. 2018;1862(3):600–614. | ||
Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13(10):727–738. | ||
Zeuner A, De Maria R. Not so lonely at the top for cancer stem cells. Cell Stem Cell. 2011;9(4):289–290. | ||
Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–768. | ||
Kumar R, Yu F, Zhen YH, et al. PD-1 blockade restores impaired function of ex vivo expanded CD8+ T cells and enhances apoptosis in mismatch repair deficient EpCAM+ PD-L1+ cancer cells. Onco Targets Ther. 2017;10:3453–3465. | ||
Mitra A, Yan J, Xia X, et al. IL6-mediated inflammatory loop reprograms normal to epithelial-mesenchymal transition+ metastatic cancer stem cells in preneoplastic liver of transforming growth factor beta-deficient β2-spectrin+/- mice. Hepatology. 2017;65(4):1222–1236. | ||
Yamashita T, Honda M, Nakamoto Y, et al. Discrete nature of EpCAM+ and CD90+ cancer stem cells in human hepatocellular carcinoma. Hepatology. 2013;57(4):1484–1497. | ||
Wang Y, Xu M, Ke ZJ, Luo J. Cellular and molecular mechanisms underlying alcohol-induced aggressiveness of breast cancer. Pharmacol Res. 2017;115:299–308. | ||
Marchetti P, Trinh A, Khamari R, Kluza J. Melanoma metabolism contributes to the cellular responses to MAPK/ERK pathway inhibitors. Biochim Biophys Acta. 2018;1862(4):999–1005. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.