")
Back to Journals » Cancer Management and Research » Volume 12
Significance of CXCL12/CXCR4 Ligand/Receptor Axis in Various Aspects of Acute Myeloid Leukemia
Authors Yazdani Z , Mousavi Z, Moradabadi A, Hassanshahi G
Received 20 October 2019
Accepted for publication 3 March 2020
Published 24 March 2020 Volume 2020:12 Pages 2155—2165
DOI https://doi.org/10.2147/CMAR.S234883
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Zinat Yazdani,1 Zahra Mousavi,2 Alireza Moradabadi,1 Gholamhossein Hassanshahi1,3
1Department of Hematology and Blood Banking, Kerman University of Medical Sciences, Kerman, Iran; 2Department of Hematology and Medical Laboratory Sciences, Iranshahr University of Medical Sciences, Iranshahr, Iran; 3Molecular Medicine Research Center, Institute of Basic Medical Sciences Research, Rafsanjan University of Medical Sciences, Rafsanjan, Iran
Correspondence: Gholamhossein Hassanshahi
Department of Hematology and Blood Banking, Kerman University of Medical Sciences, Kerman, Iran
Tel +98 391 5234 0035
Fax +98 391 522 5209
Email [email protected]
Abstract: Acute myeloid leukemia (AML) is defined as an aggressive disorder which is described by accumulation of immature malignant cells into the bone marrow. Chemokine-receptor axes are defined as factors involved in AML pathogenesis and prognosis. The chemokine receptor CXCR4 along with its ligand, CXCL12 fit in important players that are actively involved in the cross-talk between leukemia cells and bone marrow microenvironment. Therefore, according to the above introductory comments, in this review article, we have focused on delineating some parts played by CXCL12/CXCR4 axis in various aspects of AML malignancy. Targeting both leukemic and stromal cell interaction is nowadays accepted as a wide and attractive strategy for improving the outcome of treatment in AML in a non-cell autonomous manner. This strategy might be employed in a wide variety of AML patients regardless of their causative mutations. In addition to several potential targets involved in the disruption of malignant leukemic cells from their specific protective niches, compounds which interfere with CXCL12/CXCR4 axis have also been explored in multiple early-phase established clinical trials. Moreover, extensive research programs are exploring novel leading mechanisms for leukemia-stromal interactions that appear to find out novel therapeutic targets within the near future.
Keywords: SDF-1α, CXCL12, CXCR4, chemokine axis, AML
Introduction
Acute myeloid leukemia (AML) is defined as an aggressive disorder which is defined as assembling of the immature malignant cells within the bone marrow (BM). Most of the adult patients suffering from AML passed away by this disease, even upon further treatment with high-doses of multi-agent chemotherapy and allogeneic stem cell transplantation. The heterogeneous classification of AML is on the basis of cytogenetic mutations in parallel with molecular aberration findings.1,2 Factors involved in the prognosis of AML are heterogeneous and new parameters to be well clarified yet. Chemokines and their corresponding receptors are introduced as paramount factors involved in both pathogenesis and prognosis of AML. Concerning to the production, chemokines are also classified as “homeostatic” that control both leukocyte homing and lymphocyte recirculation at normal circumstances, and “inflammatory” which are generated in response to inflammatory as well as immune stimuli. It is also valuable to note that, however, the major activity of chemokines is the co-ordination of the leukocyte recruitment in both physiologic and pathologic circumstances; they are also able to mediate other biological functions, varying from regulation of cell differentiation and proliferation to cell survival and senescence. Hence, this is of particular relevance to consider that chemokine receptors are extensively expressed by several normal as well as malignant non-leukocyte cell types.3–8
Despite the sensitivity to chemotherapy, the long-term disease-free survival in AML sufferers remained low and it was reported that the majority of the patients most often enter the relapse phase from minimal residual disease (MRD).9 BM is one of the main locations for MRD where the adhesion of AML cells to the BM components may provide protection from the chemotherapy reagents.10 The CXCL12/CXCR4 chemokine-receptor is an important player that is actively involved in the cross-talk between leukemia cells and BM microenvironment. So the CXCL12/CXCR4 chemokine-receptor plays a role in MRD in AML patients.11,12
A large body of evidence has addressed the involvement of chemokine/receptor axes (specifically CXCL12/CXCR4) in the tropism of leukemic cells. BM stromal cells are the main sources of CXCL12 and BM-residing blasts express CXCR4 more intensively than those found within the circulation. The CXCL12/CXCR4 interaction axis appears to facilitate the retention of AML blasts within the BM.10 Consequently, elevated CXCR4 expression on AML blasts is considered as an independent risk factor for relapse and overall poor prognosis.13,14
CXCL12/CXCR4 polymorphisms played roles in AML patients. The genetic variation in CXCL12/CXCR4 was correlated with the clinical presentation and the complete remission of AML patients. Also some mutations affect CXCR4 expression. Disruption of the CXCL12/CXCR4 interaction axis by CXCR4 inhibitors represented a novel and promising strategy for the therapy of AML by targeting the BM microenvironment. A number of small antagonists have been designed to specially target CXCR4.15
Therefore, according to the above introductory comments, in the current review article, we focused on delineating crucial parts played by CXCL12/CXCR4 axis in various aspects of AML malignancy. By the fact to achieve this, we have searched multiple articles within different database literature using several motor engine searches and web pages including PubMed, Google scholar, Scopus, etc. Relatively, several keywords such as chemokine, SDF-1α, CXCL12, CXCR4, CXCR7, acute leukemia, and AML have also been searched.
Downstream Signaling Pathways Activated by CXCL12/CXCR4, 7 Interaction Axes in AML
The CXCL12 signaling pathway plays a key role within cross-talk of the leukemic cells and BM microenvironment interactions.7,16 To perform its activities, CXCL12 attaches to two namely receptors, CXCR4 and CXCR7 and further transduces on the mTOR pathway in pancreatic and gastric cancers, T cell leukemia, and the human renal cancer cells. Additionally, as described earlier in the text, chemokines are a superfamily of chemoattractant proteins that induce cytoskeletal rearrangement for firm adhesion to specific cells and directional migration. Further activation, chemokine/receptors axes trigger a cascade of multiple cellular events, varied from receptor dimerization, recruitment of heterotrimeric G proteins, and activation of the Janus kinase (JAK) and signal transducer and activator of transcription (STAT), PI3K (phosphatidylinositol-3-kinases), mitogen-activated protein kinases (MAPK) to extracellular signal-regulated kinases (ERK). Accumulating pieces of evidence has indicated that these axes in addition to CXCL12/CXCR4 are actively involved in the regulation of tumor development processes including tumor growth, progression, and metastasis17–20 (Figure 1).
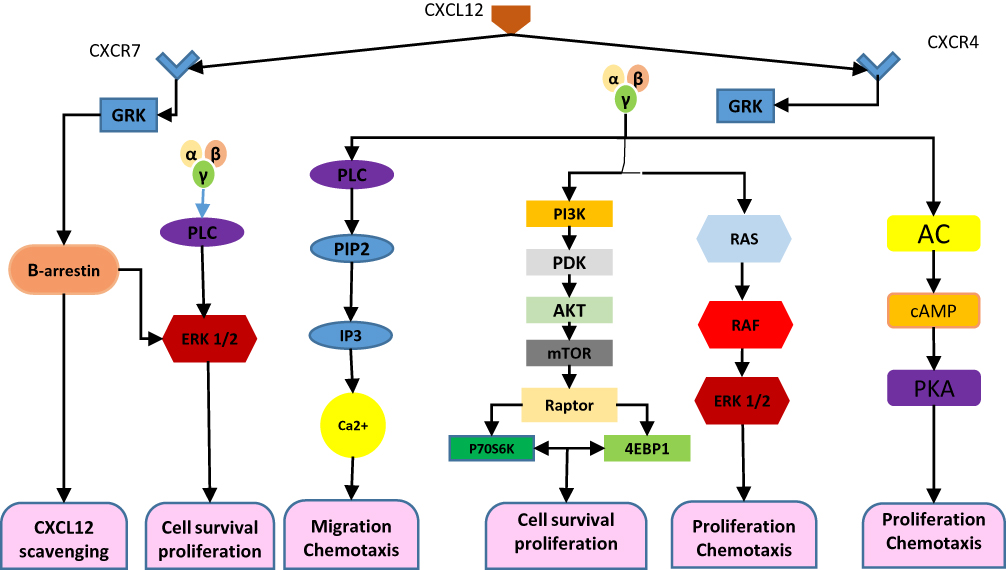 |
Figure 1 Exhibits the involvement of transduction pathway in AML. CXCL12 signal. Notes: CXCL12 acts by attaching to its cognate receptors CXCR4 and CXCR7, which can form homodimers or heterodimers. CXCL12–CXCR4 interaction activated by CXCL12 subsequently triggers GPCR signaling through PI3K/Akt, PLC/IP3, and ERK1/2 pathways, and mobilization of Ca2+ from endoplasmic reticulum by inhibition of adenyl cyclase mediated cAMP production, thus regulating cell survival, proliferation, and chemotaxis. While CXCL12 bands to CXCR7, activation of the β-arrestin may lead to scavenging of CXCL12. It can alternatively activate a MAP kinase (PLC/ERK1/2) pathway through β-arrestin and controls cell survival. Abbreviations: AC, adenylyl cyclase; PLC, phospholipase C; PIP2, phosphatidylinositol 4, 5-bisphosphate; IP3, inositol 1,4,5 trisphosphate; PI3K, phosphoinositide-3 kinase; ERK1/2, extracellular regulated kinase. 1/2; GRK, GPCR kinase; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; PDK1, pyruvate dehydrogenase kinase 1; mTOR, mechanistic target of rapamycin; 4EBP1, 4E-binding protein 1. |
CXCR4 Is Crucially Involved in Prognosis and Relapse of AML
Expression of CXCR4 by cancer cells is inversely related to the disease prognosis and serves as an independent factor from other prognostic parameters. Novel findings obtained by research teams have referred to the expression of CXCR4 by tumor-initiating cancer stem cells (CSCs) and this is in favor of the fact that CXCR4 is pivotally involved in therapy resistance, relapse, metastasis, and poor clinical outcomes.21
CXCR4 has been well indicated to be intensively expressed by a subset of myeloid cells of the AML patients and its expression is associated with poor prognosis in the patient. CXCR4 is likely to serve as an independent prognostic factor for the survival of leukemic cells in AML patients. In a related study, Ahn et al analyzed the expression of CXCR4 in AML patients and revealed the prognostic value of CXCR4 in 53 AML patients. Their data indicated that the expression of CXCR4 was remarkably induced in 26 patients. Researchers addressed that in older AML patients (with more than 60 years’ age) a complete remission was achieved following induction therapy and the expression of CXCR4 was not associated with a better progression-free survival (PFS). Whereby several studied proteins such as nucleophosmin1 (NPM1), CCAAT/enhancer-binding protein alpha (CEBP-α), FMS-like tyrosine kinase 3-Internal Tandem Duplication (FLT3 ITD), CXCR4, and FLT3 D835 were assessed, only CXCR4 was associated with PFS. Several studies indicated that the cytoplasmatic dislocation of NPM1 in AML is critical to its oncogenicity. Nucleophosmin contains amyloidogenic regions that are able to form toxic aggregates under physiological conditions.22,23 NPMc+ is an attractive therapeutic target, its similarity to the wt protein, that is strictly required for cell survival, has prevented the development of therapeutics directed against NPM1 itself. Very recently, it has been demonstrated that specific degradation of NPMc+ leads to the reduction of key features of the leukemic program definitively establishing that NPM1mutants act in a gain-of-function manner to maintain AML.1,4 Additionally, NPM1 mutants acquire new properties such as the ability to interact and inhibit the cell death activity of caspase-6 and caspase-8 in the cytoplasm.24
Its C-terminal domain (CTD) is endowed with a globular structure consisting of a three-helix bundle in the wild-type form that is disrupted by AML mutations.25
The polypeptide spanning 264–298 residues of NPM1 in type A mutation (H2mutA) NPM1 is an abundant multifunctional protein belonging to the nucleoplasmin family of nuclear chaperones. AML mutated protein is translocated into the cytoplasm (NPM1c+) retaining all functional domains except the loss of a unique NoLs (nucleolar localization signal) at the C-term domain (CTD) and the subsequent disruption of a three-helix bundle as tertiary structure. The oligomeric state of NPM1 is of utmost importance for its biological roles and an aggregation propensity of distinct regions of CTD to leukomogenic potentials of AML mutations.22 One study suggests that the direct interaction of several regions of NPM1CTD with cellular membranes could be implicated in diseases where NPM1 is mutated and/or where its overexpression is cytoxic.26
In another study, scientists investigated a polypeptide spanning the third and second helices of the bundle of type A mutated CTD. By a combination of several techniques, we ascertained the amyloid character of the aggregates and of fibrils resulting from a self-recognition mechanism. Further amyloid assemblies resulted cytoxic in MTT assay strengthening a new idea of a therapeutic strategy in AML consisting in the self-degradation of mutated NPM1.21
Applying multivariate analysis to explore PFS, it was demonstrated that CXCR4 serves as an independent prognostic factor for PFS, in addition to overall survival (OS). The expression of CXCR4 in these patients and has shown that 15 out of 22 sufferers had a normal karyotype. Overall these findings propose that overexpression of CXCR4 is related with poor prognosis in AML patients. Specifically, the overexpression of CXCR4 is considerably more frequent in AML patients exhibiting normal karyotype than others and is a marker of more aggressive disease in this population. Finally, based on literature data, CXCR4 expression could contribute to the risk assessment of patients with AML.26
Role of CXCL12/CXCR4 Axis in Extra Medullary AML
Extramedullary AML (EAML) was described as the presence of leukemic blasts in other organs such as skin, muscle, bone, gingival tissue, and brain, rather than BM-EAML, comprises 10–40% of pediatric AML. It has been documented that EAML is associated with poor prognosis in several, but not all studies.28,29
Faaij et al showed the involvement of CXCL12/CXCR4 axis in EAML of the skin in pediatric AML in 15 pediatric AML patients.13 The relatively downexpression of CXCR4 and CXCR7 by AML cells in PB and BM renders their role in skin-homing of leukemic blasts; however, both receptors have been shown to be involved in tumor cells survival. Notably, CXCL12/CXCR4 axis has been claimed that it is not directly involved in the survival of AML blasts; however, CXCL12/CXCR7 does play a role instead.
According to the ex vivo and in situ observations, both CCL3/CCR5 and CXCL12/CXCR4 interaction axes facilitate the retention of AML cells within the skin, where the CXCL12/CXCR7 interaction further prolongs their survival.13
Roles Played by CXCL12/CXCR4 Polymorphisms in AML
SDF-1α-3´A
In AML, blasts attach to the bloodstream and may migrate to the extramedullary sites which are variative from one patient to another.29 Dommange et al examined the polymorphism in CXCL12 and malignant cells dissemination/tissue infiltration in AML patients and suggested that a polymorphism within the CXCL12 coding gene (which is known as the CXCL12 G801A or SDF-1α-3´A) is able to influence the blast dissemination and tissue infiltration in AML. This CXCL12 (SDF-1α-3´A) genetic variation was assessed in 86 adult patients alongside with 100 healthy subjects. The allelic status and CXCR4 expression on BM blasts were analyzed in relation to peripheral blood blast (PBB) count and frequency of extramedullary tumor sites of the SDF-1α-3´A carrier status (801G/A, 801A/A) was associated with a higher PBB count compared to 801G/G homozygous patients as well as higher frequency of extramedullary tumor sites. Furthermore, the PBB count was well associated with the expression of CXCR4 in 801A carriers. This genetic variation was also correlated with the clinical presentation of AML.32
Andersen et al explored the role of CXCL12-(SDF-1α-3´A) polymorphism in the complete remission (CR) phase in AML patients. Researchers recruited 214 adult type AML patients. One hundred and fourteen patients (53%) were females and 156 were males. In the study, M3 patients were excluded and M4 and M5 sufferers were considered isolated from the others (due to the presence of higher frequency of extramedullary disease). In order to examine the influence of CXCL12-(SDF-1α-3´A) polymorphism on complete remission, disease-free survival (DFS) and overall survival, they considered 156 patients (73% of total included patients) who required intensive treatment. Following diagnosis, investigators did not find a significant difference in hemoglobin, leukocyte or platelet count, percentage of either PB or BM blasts in relation to CXCL12-(SDF-1α-3´A) genetic variation. They found that among 156 patients who received a homogeneous treatment protocol, 88 were GG and 68 had one or both A alleles. In 114 patients (73%) entered a CR; 61 were GG and 53 had A allele, corresponding to 69% and 78% of the patients in each genotype group. Briefly, their data were not in favor of a role for CXCL12-(SDF-1α-3´A) polymorphism in prognosis of AML patients.31
El-Ghany et al have examined the CXCL12-(SDF-1α-3´A) gene variants and their influence on the malignant cell dissemination and tissue infiltration in AML patients. The CXCL12 genotyping was performed by polymerase chain reaction-restriction fragment length polymorphism (PCR_RFLP) for 48 AML: 38 de novo AML and 10 CML. They have also enrolled 50 age- and gender-matched control subjects. Authors showed that the frequency of wild genotype to the heterozygous genotype ratio was 1/2 in AML patients, while in CML patients, the frequency of wild genotype to the heterozygous genotype was 3/7. In the control subjects, 57.2% exhibited wild genotype, whereas 42.8% had heterozygous genotype and no significant difference was detected between AML patients and controls. An insignificant association was observed between wild and heterozygous genotypes regarding clinical, laboratory data and extramedullary dissemination; however, CXCL12-(SDF-1α-3´A) was not correlated with either increased risk of AML or extramedullary blast dissemination.32
rs2228014
Zheng et al have examined the role of CXCL12-(SDF-1α-3´A) genetic variation along with CXCR4 expression in AML and leukemia cell dissemination in 926 subjects, including 466 de novo AML patients and 460 healthy subjects. They have demonstrated that the distribution of CT and CT+TT for rs2228014 polymorphism in CXCR4 were significantly increased in AML patients, compared to healthy subjects. However, rs1801157 in CXCL12 has demonstrated no considerable difference in genotype distribution and allele frequency between AML patients and healthy subjects. For the two combined SNPs, they have not observed a marked difference between the wild-type GG-CC genotypes and non-GG-CC genotypes in AML patients and healthy subjects. Furthermore, the number of peripheral blood leukemia-cells (PBLCs) was not remarkably affected by rs1801157 and rs2228014. Overall, it appears that only CT genotype of rs2228014 was remarkably associated with the risk of AML, as they addressed, but it did not play a role in leukemia cells invading the bloodstream. However, rs1801157 and the two combined SNPs were not only associated with increased AML risk they were not also correlated with extramedullary leukemia-cell dissemination.34
Relationship Between CXCR4 Expression and Other Mutations
FLT3-ITD
A functional interaction has been proposed between FMS-like tyrosine kinase 3 (FLT3) receptor and CXCR4.34 The relapse phase of AML is associated with internal tandem duplication (ITD) mutations of the FLT3 receptor and the expression of CXCR4 in FLT3-ITD-bearing AML is associated with poor diagnosis whereby CXCR4 was inhibited, AML blasts were responsive to the chemotherapy.36
Jacobi et al have examined the effects of FLT3-ITD on cellular proliferation and CXCR4-mediated recruitment of human hematopoietic progenitor cells (HPPCs) alongside with their response to the CXCR4 inhibition. To achieve this, primary blasts obtained from the AML patients with FLT3-ITD or FLT3 wild-type were examined. In addition, human CD34+ hematopoietic progenitor cells were transduced with >70% human FLT3-ITD containing retroviral vectors and further STAT5a, STAT3, and ERK1/2 phosphorylation and CXCR4 expression were simultaneously analyzed by flow cytometry. Following analysis, it has been shown that FLT3-ITD transgene overexpressing human HPPCs migration toward CXCL12 was dramatically reduced along with significant reduction in BM homing in non-obese diabetic severe combined immune deficient (SCID) mice. Co-culture of FLT3-ITD–positive AML blasts or human HPPCs with BM stromal cells resulted in a strong proliferation advantage and increased early cobblestone area–forming cells compared to FLT3–wild-type AML blasts. In response to ABDS3100, as CXCR4 inhibitor both cobblestone area–forming cells and proliferation of FLT3-ITD–positive cells were remarkably decreased but has not influenced FLT3–wild-type cells, proposing the critical interaction between CXCR4 and FLT3-ITD. Based on Jacobia et al's report, CXCR4-mediated cell proliferation was pivotally reduced and control of the leukemic burden may provide a novel therapeutic strategy for treatment of FLT3-ITD–positive AML patients.35
BM-derived stromal cells secrete CXCL12, in a constitutive fashion and CXCR4 overexpression has been claimed to be associated with FLT3/ITD AML.34,36 Overexpression of FLT3/ITD mutants in Ba/F3 cells is correlated with activated CXCL12/CXCR4 axis.37
The FLT3 inhibitors have been applied for the improvement of the dismal prognosis of AML patients who exhibit FLT3 mutations. Clinical findings in AML patients with FLT3 inhibitor monotherapy indicated that peripheral blood responses are more pronounced than the BM ones.39 Kensuke Kojima et al have examined the involvement of p53 in BM stromal cells in stromal cell-mediated resistance to FLT3 inhibition in FLT3 mutant AML and indicated that the namely FLT3 inhibitor (eg, FI-700) induced apoptosis in FLT3 mutant AML cells. This induced apoptosis was further abolished under stromal co-culture circumstances which appear to be mediated, at least in part, by CXCL12/CXCR4 axis, whereby the stromal cells proliferation was inhibited, it has significantly reduced by pre-treatment with the HDM2 (human ortholog of murine double minute 2). Activation of P53 signaling by Nutlin-3a was not cytotoxic to stromal cells; however, it has reduced the CXCL12 mRNA levels and secretion of CXCL12 partially via p53-mediated decreasing hypoxia-induced factor-1 (HIF-1). These authors have also demonstrated that activation of P53 in stromal cells has blunted the stromal cell-mediated resistance to FLT3 inhibition, along with CXCL12 down-regulation. This, in fact, proposes that the combination of HDM2 antagonists and FLT3 inhibitors is probably effective in clinical trials that are targeting mutant FLT3 leukemia.38
NPM
Zhang et al claimed that further NPM (nucleophosmin) suppression, the chemotactic responses to CXCL12 were elevated, while inversely the overexpression of a cytosolic NPM mutant has reduced CXCL12-induced chemotaxis. Also, CXCR4 mutants coupled to G proteins and phosphorylated (Zhang et al, 2002) were more associated with NPM than the wild-type receptors. Reversal of this conformation with T140 as a potent inverse agonist which switches off the receptor architectural equilibrium has decreased the NPM interaction with CXCR4 in an immune precipitation assay. In contrast, activation of wild-type CXCR4 by CXCL12 has drastically affected NPM with cytoplasmic loops of the receptor, raising the possibility that the interaction may be indirect and involves additional subunits present in the milieu of chronic desensitization.40
Chou et al established a mouse knocked-down model for NPM1 mutation inserting TCTG after nucleotide c.857 and showed that the expression of CXCL12/CXCR4-related genes was significantly suppressed in mutant myeloid precursors, compared to wild-type NPM1 myeloid precursors. In a similar fashion, the suppressed CXCL12/CXCR4 was detected by genome-wide expression microarray analysis in BM mononuclear cells in NPM1-mutated AML patients, compared to NPM1-wild AML.40
The expression of CXCR4 and pCXCR4 along with NPM1 mutations and their correlation with prognosis in adult AML patients were examined by Konoplev et al. In 117 untreated adult AML patients, they used immunohistochemistry techniques to detect the expression of CXCR4 within the BM tissue as well as phosphorylated CXCR4. All of the studied subjects were also examined for NPM1 mutations by PCR-based techniques and the following results were obtained: CXCR4 expression was detected in 75 (64%) while pCXCR4 was presented in 31 (26%) of patients. Mutations of NPM1 were detected in 63 (54%) patients and was not correlated with either CXCR4 or pCXCR4 expression. The median survival period was 5 years with a median follow-up of 8 months. Lack of association was observed between CXCR4 and pCXCR4 and NPM1 mutations which may propose that the CXCR4 pathway is critically involved in AML pathogenesis regardless of NPM pathways.41
Inhibitors of CXCR4
On the basis of tumor type, stage of cancer, and immunological contexture, the inhibition of chemokines or their receptors may aid positive or deleterious effects on disease development.42
More recently investigations evidenced that CXCL12/CXCR4 axis plays remarkable parts in leukemic cell resistance to the signal transduction inhibitor as well as chemotherapy-induced apoptosis. Disruption of the CXCL12/CXCR4 interaction axis by CXCR4 inhibitors represented a novel and promising strategy for the therapy of AML by targeting the BM microenvironment. Several research teams sought to examine whether the CXCR4 antagonists have beneficial effects on both survival and oriented locomotion of AML cells in vivo and in vitro. A number of small antagonists have been designed to specially target CXCR4, such as AMD3100. Some other analogues and peptides were also designed on the amino-terminal region of the chemokines such as T22, TN14003, and CTCE-9908. In experimental models of tumor, TN14003 has remarkably reduced pulmonary metastasis,43,44 and CTCE-9908 displayed inhibitory effects against primary tumors as well as anti-metastatic in animal models of several tumors, including melanoma, osteosarcoma, breast, and prostate.45,46 In case of prostate, the intraperitoneal delivery of CTCE-9908 was also accompanied by inhibition of Vascular endothelial growth factor (VEGF) and angiogenesis, and attenuated recruitment of myeloid host cells.47 In contrast, CTCE-9908 has not inhibited metastasis (caused by the intra-cardiac inoculation of breast cancer cells); however, it has reduced the size of metastasis in all examined organs.47 Recent pieces of evidence demonstrated that treatment of tumor cells with CTCE-9908 in vitro has led to multinucleation, G2/M arrest, and further intensive mitosis.48
AMD3100
AMD3100 as a CXCR4 antagonist has elevated the peripheral mobilization of hematopoietic stem cells. The clinical application of AMD3100 has recently been approved.29
4F-Benzoyl-TN14003
The CXCR4 antagonist, 4F-benzoyl-TN14003 which is also called (BKT140) was shown to exhibit preferential cytotoxicity against malignant cells with hematopoietic origin such as AML cells.49
Ulocuplumab
Ulocuplumab (BMD-936564/MDX-1338), as a fully humanized type of immunoglobulin G4 (IgG4) monoclonal antibody (MAb) which selectively recognizes the human CXCR4 has also been introduced by Kuhne et al. It has been reported that ulocuplumab has antitumor properties against several tumors such as subcutaneous xenograft models of APL (acute promyelocytic leukemia) and facilitates apoptosis of a profile of cell lines including AML generated types. More importantly, they have explained that the apoptosis in an antibody-induced manner is one of the most related mechanisms by which tumor cells proliferation as well as tumor progression is inhibited.50
LY2624587
LY2624587 is introduced as another humanized anti-CXCR4 IgG4 MAb which also represents a promising potential for induction of apoptosis in human lymphoma and leukemia both in vivo and in vitro.51 Pre-clinical data, obtained from another newly designed anti-CXCR4 IgG1 MAb, PF-06747143, was also recently introduced during annual meeting of the American Society of Hematology and antibody designers proposed that cholesterol-dependent cytolysin (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC) are amongst related mechanisms responsible for its anti-leukemia effect in AML cell lines.52 PF-06747143 exerts its anti-leukemic impact similarly as monotherapy in primary models of AML xenograft.53 These pre-clinical data, alongside with assumed mechanisms for AML, propose that anti-CXCR4 MAb may have promising effects in clinical applications; however, their toxic adverse effects on the processes of normal hematopoiesis deserved to be well elucidated in future studies.
LY2510924
In the most recent investigations, Cho BS and his team demonstrated a markedly anti-leukemic activity for LY2510924 as peptidic CXCR4 inhibitor as well as a monotherapy, and in combination with anti-AML chemotherapy. The in vitro obtained data showed that LY2510924 has drastically disrupted CXCL12/CXCR4 interaction axis in AML cells at nano-molar concentrations and has further inhibited the proliferation of AML cells rather than apoptosis (in contrast to BKT140). They also demonstrated that in AML xenograft models, LY2510924 caused mobilization of leukemic cells into the periphery and inhibited several survival factors which were produced by activation of the CXCL12/CXCR4 axis, and induces myeloid differentiation; thereby, producing anti-leukemic effects as monotherapy. These anti-leukemia functions of the compound are firmly in synergy with chemotherapeutic compounds such as cytarabine and doxorubicin in xenograft models, resembling standard inductive chemotherapy in human trials.54
Clinical Applications of CXCR4 Inhibitors
BL-8040
BL-8040 (BKT140) as a peptidic CXCR4 inhibitor that has pro-apoptotic activity directly against AML cells.49,55 A Phase 1/2 trial was performed on relapsed/refractory AML patients. In this trial participated patients received BL-8040 for two days as monotherapy and the therapy protocol was again followed by BL-8040 and cytarabine for five upcoming days. Preliminary documented findings of the study revealed that all examined doses of BL-8040 were safely tolerated. The couple of days BL-8040 treatment has also mobilized approximately 40.2-fold increase in immature AML progenitors from the BM along with decreased number of leukemia progenitor cells (by 58%) in the BM at the third day. The apoptosis of AML blasts was observed in five out of nine samples and was well correlated with a 3.1-fold elevation in differentiated monocytes and granulocytes. During the period of dose escalation, a CR/CRi of 38% was achieved by 22 AML patients. These achievements propose that the induction of apoptosis and differentiation, in parallel with mobilization of AML blasts from the protective marrow, may act as mechanistic pathway for CXCR4 inhibition.56
Ibrutinib
The effect of ibrutinib on CXCL12/CXCR4-mediated migration in AML was also examined by Zaitseva et al and the CXCR4 expression was analyzed by flow cytometry and followed by chemotactic analysis using transwell plates. The AML blasts were treated with CXCL12 and cellular protein extracts were subjected to Western blotting for pBTK, BTK, pMAPK, MAPK, and β-actin as the housekeeping protein. The AML blasts were also pretreated with the escalation measures of ibrutinib and as similar to CXCL12. The effects of ibrutinib on the migration of AML cell lines U937, MV4-11, HL60, and THP-1 in response to CXCL12. Interestingly researchers found that ibrutinib was able to actively block the migration of all AML cell lines. Finally, they tested the activity of ibrutinib on CXCL12-induced migration on a wide spectrum of primary the AML blasts in vitro from a broad range of ages in adult patients and across a range of World Health Organization (WHO) AML subclasses. Ibrutinib has inhibited primary AML blast migration. Taken into account these data may explain that ibrutinib has limited the CXCL12/CXCR4-based migration of human AML cells.57
Synthetic Peptide E5
Li et al demonstrated that synthetic peptide E5 has the capacity to elevate the therapeutic efficiency of various chemotherapeutics on AML both in vitro and in vivo. They have claimed that E5 was able to abolish BM stromal cell-provided protection to leukemia cells, as well as enhance the occurrence of apoptosis which was induced by various chemotherapeutics in several AML cell lines, E5 has also increased the number of periphery AML cells out of stromal niches, in a rodent xenograft AML model. Whereby E5 was added to vincristine and cyclophosphamide, it had also the ability to inhibit the infiltration of AML cells into several organs, including BM, liver, and spleen, and extended the lifespan of AML mice compared with mice treated with chemotherapy alone. Moreover, cytotoxicity was not observed for E5 when the histological analysis was performed and clinical parameters of serum for toxicity were determined in vivo.58
siRNA
Although siRNA carrier technology and siRNA-mediated silencing method as a therapy are developed for various leukemia, its application in AML has yet to be explored in detail at the present time. Consistently, CXCL12/CXCR4 antagonists have been employed against a wide spectrum of leukemic cells in both in vitro and clinical trials and very few studies applied the siRNA technology as a therapeutic test for leukemia cell lines. Although only few studies utilized siRNA technology, siRNA therapy has been progressing into clinical trials as a cancer therapy.59,60 Beneficial advantages of CXCR4 as a target for siRNA therapies have also been established for treatment of solid tumors.61,62
In a more recent attempt, Landry et al targeted the CXCL12/CXCR4 axis by lipopolymer contained complexes of siRNA in AML and found an advantageous therapeutic potential for most promising lipopolymer in siRNA-mediated silencing of the CXCL12/CXCR4 axis in AML. They have also revealed that CXCR4 protein expression (by immune staining) was effectively attenuated when siRNA delivery was undertaken to THP-1 cells with the polymer PEI2-CA. The down-expression of CXCR4, in turn, has also led to a decreased cell number possibly, due to inhibiting proliferation as they have demonstrated by the dye dilution technique; however, CXCL12 was predominantly generated by bone marrow stromal cell (BMSC). These researchers have indicated that reduced cell number is mediated by CXCL12 siRNA silencing with lipopolymer/siRNA complexes; nonetheless, no enhanced effect was found when CXCL12 and CXCR4 were simultaneously silenced. The CXCR4 pathway could also mediate partial chemo-resistance to cytarabine exposure where silencing CXCR4 would then reduce cellular resistance to the drug. Investigators have reported an elevated CXCR4 resistance to cytarabine when AML cells were grown with homing bone marrow stromal cell (hBMSC), proposing that other BMSC secreted factor(s) might not be significant in their culture system. They have also claimed that CXCR4 silencing has considerably decreased the THP-1 cell attachment to hBMSC. Therefore, to completely dislodge leukemic cells from the BM, several above mentioned adhesion molecules deserved to be targeted. In spite of the detachment of leukemic cells from the BM microenvironment, it is considered as a major goal for targeting theCXCR4. Prevention of adhesion may not possibly be required for disruption of activating signaling via CXCR4 pathway which in turn aids the activation of survival pathways.63
Conclusion
Due to the considerable heterogeneity, in addition to the multi-line nature of AML, it was demonstrated by employing DNA sequencing studies, that there exist limitations for improving the therapy outcomes of medications that appropriately target deregulated signaling pathways. Targeting both leukemic and stromal cell interaction is nowadays accepted as a wide and attractive strategy for improving the outcome of treatment in AML in a non-cell autonomous manner. This strategy might be employed in a wide variety of AML patients regardless of their causative mutations. The lack of relationship between a particular mutation and along with broad applicability to a wide spectrum of disorders makes the microenvironment targeting a worthy endeavor. In addition to several potential targets involved in the disruption of malignant leukemic cells from their specific protective niches, compounds that interfere with the CXCL12/CXCR4 axis have also been explored in multiple early-phase established clinical trials. Overall, findings obtained from these types of trials and particularly with inhibitors of CXCR4 antagonists, found as promising and worth pursuing, if tolerated and efficient at an adequate level, CXCR4 inhibitors could be considered as a proper adjunct therapy accompanied with drugs which are directly targeting related mutations as for curative tools. Both present and future investigations may reveal the advantages of targeting the CXCL12/CXCR4 axis in AML. But, these inhibitors and antagonists in patients with CXCL12/CXCR4 mutated forms will have the same effect as in patients with wild-type forms and it will also help the treatment of this group of patients.
Also, in patients with other mutations, such as FLT3-ITD, inhibitory compounds should be administered at different doses than patients who do not have these mutations. Which of the inhibitors in these patients will be more effective?
Moreover, extensive research programs are exploring novel leading mechanisms for leukemia stromal interactions that appear to find out novel therapeutic targets within the near future.
Prospects
In the new year’s, the advent of targeted molecular therapies, new monoclonal antibodies, and strong inhibitors of small molecules suggests that we can ultimately have tools to improve the outcome in AML. Inhibiting the CXCL12/CXCR4 axis lonely or in combination with chemotherapy can help the treatment and preventing the recurrence of the disease. The recent surge in inhibitory molecules and antagonists research in a clinical trial is likely to lead to considerable advances in our understanding that which one will have an excellent effect on the treatment of AML in the short term and with the least dose. Also, we will gain knowledge about which inhibitors will reduce the use of chemotherapy drugs. We will understand that a combination of inhibitors and antagonists to be used or only one of them will lead us to the target. We also have to understand the side effects of targeted molecular therapies, new monoclonal antibodies, and strong inhibitors of small molecules. The role of the other chemokines and their receptors in pathogenesis and relapse of the disease in addition to the signals controlling the expression of chemokines and chemokine receptors will be examined; determining what patients will benefit from chemokine-axis inhibitors.
Data Sharing Statement
Please contact the corresponding author for data requests.
Ethics
The study was approved by the ethics committee at the Kerman University of Medical Science.
Acknowledgments
This project was financially supported by the Kerman University of Medical Sciences.
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. van Gils N, Verhagen H, Smit L. Reprogramming acute myeloid leukemia into sensitivity for retinoic acid-driven differentiation. Exp Hematol. 2017;52:12–23. doi:10.1016/j.exphem.2017.04.007
2. Yazdani Z, Mousavi Z, Ghasemimehr N, et al. Differential regulatory effects of chemotherapeutic protocol on CCL3_CCL4_CCL5/CCR5 axes in acute myeloid leukemia patients with monocytic lineage. Life Sci. 2019;240:117071.
3. Mantovani A, Savino B, Locati M, Zammataro L, Allavena P, Bonecchi R. The chemokine system in cancer biology and therapy. Cytokine Growth Factor Rev. 2010;21(1):27–39. doi:10.1016/j.cytogfr.2009.11.007
4. Khorramdelazad H, Bagheri V, Hassanshahi G, Zeinali M, Vakilian A. New insights into the role of stromal cell-derived factor 1 (SDF-1/CXCL12) in the pathophysiology of multiple sclerosis. J Neuroimmunol. 2016;290:70–75. doi:10.1016/j.jneuroim.2015.11.021
5. Karimabad MN, Hassanshahi G. Significance of CXCL12 in type 2 diabetes mellitus and its associated complications. Inflammation. 2015;38(2):710–717. doi:10.1007/s10753-014-9981-3
6. Tavakolian Ferdousie V, Mohammadi M, Hassanshahi G, et al. Serum CXCL10 and CXCL12 chemokine levels are associated with the severity of coronary artery disease and coronary artery occlusion. Int J Cardiol. 2017;233:23–28. doi:10.1016/j.ijcard.2017.02.011
7. Mousavi Z, Yazdani Z, Moradabadi A, Hoseinpourkasgari F, Hassanshahi G. Role of some members of chemokine/cytokine network in the pathogenesis of thalassemia and sickle cell hemoglobinopathies: a mini review. Exp Hematol Oncol. 2019;8(1):1–6. doi:10.1186/s40164-019-0145-x
8. Taherahmadi H, Moradabadi AR, Arjomand Shabestari A, Nazari J, Kahbazi MK. Antibiotic induced hemolytic anemia and thrombocytopenia among pediatric patients admitted to intensive care unit. Iran J Pediatr Hematol Oncol. 2019;9(1):9–16.
9. Matsunaga T, Takemoto N, Sato T, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9(9):1158–1165. doi:10.1038/nm909
10. Estey E, Dohner H. Acute myeloid leukaemia. Lancet (London, England). 2006;368(9550):1894–1907. doi:10.1016/S0140-6736(06)69780-8
11. Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23(1):43–52. doi:10.1038/leu.2008.299
12. Azin H, Vazirinejad R, Ahmadabadi BN, et al. The SDF-1 3ʹa genetic variation of the chemokine SDF-1alpha (CXCL12) in parallel with its increased circulating levels is associated with susceptibility to MS: a study on Iranian multiple sclerosis patients. J Mol Neurosci. 2012;47(3):431–436. doi:10.1007/s12031-011-9672-6
13. Faaij CM, Willemze AJ, Revesz T, et al. Chemokine/chemokine receptor interactions in extramedullary leukaemia of the skin in childhood AML: differential roles for CCR2, CCR5, CXCR4 and CXCR7. Pediatr Blood Cancer. 2010;55(2):344–348. doi:10.1002/pbc.22500
14. Eckert F, Schilbach K, Klumpp L, et al. Potential role of CXCR4 targeting in the context of radiotherapy and immunotherapy of cancer. Front Immunol. 2018;9:3018. doi:10.3389/fimmu.2018.03018
15. Yu Z, Han-bo C, Wen-jun L, Li Z. The CXCL12 (SDF-1)/CXCR4 chemokine axis: oncogenic properties, molecular targeting, and synthetic and natural product CXCR4 inhibitors for cancer therapy. Chin J Nat Med. 2018;16(11):801–810. doi:10.1016/S1875-5364(18)30122-5
16. Circelli L, Sciammarella C, Guadagno E, et al. CXCR4/CXCL12/CXCR7 axis is functional in neuroendocrine tumors and signals on mTOR. Oncotarget. 2016;7(14):18865–18875. doi:10.18632/oncotarget.v7i14
17. Nazari A, Khorramdelazad H, Hassanshahi G. Biological/pathological functions of the CXCL12/CXCR4/CXCR7 axes in the pathogenesis of bladder cancer. Int J Clin Oncol. 2017;22(6):991–1000. doi:10.1007/s10147-017-1187-x
18. Moosavi SR, Khorramdelazad H, Amin M, et al. The SDF-1 3ʹA genetic variation is correlated with elevated intra-tumor tissue and circulating concentration of CXCL12 in glial tumors: a study on Iranian anaplastic astrocytoma and glioblastoma multiforme patients. J Mol Neurosci. 2013;50(2):298–304. doi:10.1007/s12031-013-9954-2
19. Moradabadi A, Farsinejad A, Khansarinejad B, Fatemi A. Development of a high resolution melting analysis assay for rapid identification of JAK2 V617F missense mutation and its validation. Exp Hematol Oncol. 2019;8(1):10. doi:10.1186/s40164-019-0134-0
20. Peitzsch C, Cojoc M, Kurth I, Dubrovska A. Implications of CXCR4/CXCL12 Interaction for Cancer Stem Cell Maintenance and Cancer Progression. In Babashah S, editor. Cancer Stem Cells: Emerging Concepts and Future Perspectives in Translational Oncology. Berlin: Springer; 2015: 89-130.
21. Di Natale C, La Manna S, Malfitano AM, et al. Structural insights into amyloid structures of the C-terminal region of nucleophosmin 1 in type A mutation of acute myeloid leukemia. Biochim Biophys Acta Proteins Proteomics. 2019;1867(6):637–644. doi:10.1016/j.bbapap.2019.01.010
22. Di Natale C, Scognamiglio PL, Cascella R, et al. Nucleophosmin contains amyloidogenic regions that are able to form toxic aggregates under physiological conditions. FASEB J. 2015;29(9):3689–3701. doi:10.1096/fj.14-269522
23. Scognamiglio PL, Di Natale C, Leone M, et al. Destabilisation, aggregation, toxicity and cytosolic mislocalisation of nucleophosmin regions associated with acute myeloid leukemia. Oncotarget. 2016;7(37):59129–59143. doi:10.18632/oncotarget.v7i37
24. La Manna S, Roviello V, Scognamiglio PL, et al. Amyloid fibers deriving from the aromatic core of C-terminal domain of nucleophosmin 1. Int J Biol Macromol. 2019;122:517–525. doi:10.1016/j.ijbiomac.2018.10.210
25. De Santis A, La Manna S, Krauss IR, et al. Nucleophosmin-1 regions associated with acute myeloid leukemia interact differently with lipid membranes. Biochim Biophys Acta. 2018;1862(4):967–978. doi:10.1016/j.bbagen.2018.01.005
26. Ahn JY, Seo K, Weinberg OK, Arber DA. The prognostic value of CXCR4 in acute myeloid leukemia. Appl Immunohistochem Mol Morphol. 2013;21(1):79–84.
27. Bisschop MM, Revesz T, Bierings M, et al. Extramedullary infiltrates at diagnosis have no prognostic significance in children with acute myeloid leukaemia. Leukemia. 2001;15(1):46–49. doi:10.1038/sj.leu.2401971
28. Kobayashi R, Tawa A, Hanada R, Horibe K, Tsuchida M, Tsukimoto I. Extramedullary infiltration at diagnosis and prognosis in children with acute myelogenous leukemia. Pediatr Blood Cancer. 2007;48(4):393–398. doi:10.1002/pbc.v48:4
29. Peled A, Tavor S. Role of CXCR4 in the pathogenesis of acute myeloid leukemia. Theranostics. 2013;3(1):34–39. doi:10.7150/thno.5150
30. Dommange F, Cartron G, Espanel C, et al. CXCL12 polymorphism and malignant cell dissemination/tissue infiltration in acute myeloid leukemia. FASEB J. 2006;20(11):1913–1915. doi:10.1096/fj.05-5667fje
31. Ponziani V, Mannelli F, Bartalucci N, et al. No role for CXCL12-G801A polymorphism in the development of extramedullary disease in acute myeloid leukemia. Leukemia. 2008;22(3):669–671. doi:10.1038/sj.leu.2404938
32. El-ghany HM, El-saadany ZA, Bahaa NM, Ibrahim NY, Hussien SM. Stromal cell derived factor-1 (CXCL12) chemokine gene variant in myeloid leukemias. Clin Lab. 2014;60(5):735–741. doi:10.7754/Clin.Lab.2013.130445
33. Zheng Q, Shuai X, Ye Y, et al. The role of polymorphisms of stromal-derived factor-1 and CXC receptor 4 in acute myeloid leukemia and leukemia cell dissemination. Gene. 2016;588(2):103–108. doi:10.1016/j.gene.2016.04.059
34. Rombouts EJ, Pavic B, Lowenberg B, Ploemacher RE. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood. 2004;104(2):550–557. doi:10.1182/blood-2004-02-0566
35. Jacobi A, Thieme S, Lehmann R, et al. Impact of CXCR4 inhibition on FLT3-ITD-positive human AML blasts. Exp Hematol. 2010;38(3):180–190. doi:10.1016/j.exphem.2009.12.003
36. Grundler R, Brault L, Gasser C, et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J Exp Med. 2009;206(9):1957–1970. doi:10.1084/jem.20082074
37. Fukuda S, Broxmeyer HE, Pelus LM. Flt3 ligand and the Flt3 receptor regulate hematopoietic cell migration by modulating the SDF-1alpha(CXCL12)/CXCR4 axis. Blood. 2005;105(8):3117–3126. doi:10.1182/blood-2004-04-1440
38. Kojima K, McQueen T, Chen Y, et al. p53 activation of mesenchymal stromal cells partially abrogates microenvironment-mediated resistance to FLT3 inhibition in AML through HIF-1alpha-mediated down-regulation of CXCL12. Blood. 2011;118(16):4431–4439. doi:10.1182/blood-2011-02-334136
39. Zhang W, Navenot JM, Frilot NM, Fujii N, Peiper SC. Association of nucleophosmin negatively regulates CXCR4-mediated G protein activation and chemotaxis. Mol Pharmacol. 2007;72(5):1310–1321. doi:10.1124/mol.107.037119
40. Chou SH, Ko BS, Chiou JS, et al. A knock-in Npm1 mutation in mice results in myeloproliferation and implies a perturbation in hematopoietic microenvironment. PLoS One. 2012;7(11):e49769. doi:10.1371/journal.pone.0049769
41. Chen Y, Jacamo R, Konopleva M, Garzon R, Croce C, Andreeff M. CXCR4 downregulation of let-7a drives chemoresistance in acute myeloid leukemia. J Clin Invest. 2013;123(6):2395–2407. doi:10.1172/JCI66553
42. Ma Y, Adjemian S, Galluzzi L, Zitvogel L, Kroemer G. Chemokines and chemokine receptors required for optimal responses to anticancer chemotherapy. Oncoimmunology. 2014;3(1):e27663. doi:10.4161/onci.27663
43. Takenaga M, Tamamura H, Hiramatsu K, et al. A single treatment with microcapsules containing a CXCR4 antagonist suppresses pulmonary metastasis of murine melanoma. Biochem Biophys Res Commun. 2004;320(1):226–232. doi:10.1016/j.bbrc.2004.05.155
44. Tamamura H, Hori A, Kanzaki N, et al. T140 analogs as CXCR4 antagonists identified as anti-metastatic agents in the treatment of breast cancer. FEBS Lett. 2003;550(1–3):79–83. doi:10.1016/S0014-5793(03)00824-X
45. Kim SY, Lee CH, Midura BV, et al. Inhibition of the CXCR4/CXCL12 chemokine pathway reduces the development of murine pulmonary metastases. Clin Exp Metastasis. 2008;25(3):201–211. doi:10.1007/s10585-007-9133-3
46. Richert MM, Vaidya KS, Mills CN, et al. Inhibition of CXCR4 by CTCE-9908 inhibits breast cancer metastasis to lung and bone. Oncol Rep. 2009;21(3):761–767.
47. Porvasnik S, Sakamoto N, Kusmartsev S, et al. Effects of CXCR4 antagonist CTCE-9908 on prostate tumor growth. Prostate. 2009;69(13):1460–1469. doi:10.1002/pros.21008
48. Kwong J, Kulbe H, Wong D, Chakravarty P, Balkwill F. An antagonist of the chemokine receptor CXCR4 induces mitotic catastrophe in ovarian cancer cells. Mol Cancer Ther. 2009;8(7):1893–1905. doi:10.1158/1535-7163.MCT-08-0966
49. Beider K, Begin M, Abraham M, et al. CXCR4 antagonist 4F-benzoyl-TN14003 inhibits leukemia and multiple myeloma tumor growth. Exp Hematol. 2011;39(3):282–292. doi:10.1016/j.exphem.2010.11.010
50. Kuhne MR, Mulvey T, Belanger B, et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res. 2013;19(2):357–366. doi:10.1158/1078-0432.CCR-12-2333
51. Peng S-B, Zhang X, Paul D, et al. Inhibition of CXCR4 by LY2624587, a fully humanized anti-CXCR4 antibody induces apoptosis of hematologic malignancies. PLoS One. 2016;11(3):e0150585. doi:10.1371/journal.pone.0150585
52. Pernasetti F, Liu S-H, Hallin M, et al. A novel CXCR4 antagonist IgG1 antibody (PF-06747143) for the treatment of hematological malignancies. Blood. 2014;124(21):2311. doi:10.1182/blood.V124.21.2311.2311
53. Zhang Y, Saavedra E, Tang R, et al. Targeting acute myeloid leukemia with a new CXCR4 antagonist IgG1 antibody (PF-06747143) in NOD/SCID mice. Blood. 2015;126(23):1362. doi:10.1182/blood.V126.23.1362.1362
54. Cho BS, Zeng Z, Mu H, et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood. 2015;126(2):222–232. doi:10.1182/blood-2015-02-628677
55. Zhang Y, Patel S, Abdelouahab H, et al. CXCR4 inhibitors selectively eliminate CXCR4-expressing human acute myeloid leukemia cells in NOG mouse model. Cell Death Dis. 2012;3(10):e396. doi:10.1038/cddis.2012.137
56. Borthakur G, Ofran Y, Nagler A, et al. The peptidic CXCR4 antagonist, BL-8040, significantly reduces bone marrow immature leukemia progenitors by inducing differentiation, apoptosis and mobilization: results of the dose escalation clinical trial in acute myeloid leukemia. Blood. 2015;126(23):2546. doi:10.1182/blood.V126.23.2546.2546
57. Zaitseva L, Murray MY, Shafat MS, et al. Ibrutinib inhibits SDF1/CXCR4 mediated migration in AML. Oncotarget. 2014;5(20):9930–9938. doi:10.18632/oncotarget.v5i20
58. Li X, Guo H, Duan H, et al. Improving chemotherapeutic efficiency in acute myeloid leukemia treatments by chemically synthesized peptide interfering with CXCR4/CXCL12 axis. Sci Rep. 2015;5:16228. doi:10.1038/srep16228
59. Burnett JC, Rossi JJ, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. 2011;6(9):1130–1146. doi:10.1002/biot.201100054
60. Tabernero J, Shapiro GI, LoRusso PM, et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013. doi:10.1158/2159-8290.CD-12-0429
61. Abedini F, Ismail M, Hosseinkhani H, et al. Effects of CXCR4 siRNA/dextran-spermine nanoparticles on CXCR4 expression and serum LDH levels in a mouse model of colorectal cancer metastasis to the liver. Cancer Manag Res. 2011;3:301–309. doi:10.2147/CMR.S11678
62. Liang Z, Yoon Y, Votaw J, Goodman MM, Williams L, Shim H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res. 2005;65(3):967–971.
63. Landry B, Gul-uludag H, Plianwong S, et al. Targeting CXCR4/SDF-1 axis by lipopolymer complexes of siRNA in acute myeloid leukemia. J Control Release. 2016;224:8–21. doi:10.1016/j.jconrel.2015.12.052
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.