")
Back to Journals » Hepatic Medicine: Evidence and Research » Volume 7
Should NS5A inhibitors serve as the scaffold for all-oral anti-HCV combination therapies?
Authors Janardhan S, Reau N
Received 19 December 2014
Accepted for publication 11 February 2015
Published 16 April 2015 Volume 2015:7 Pages 11—20
DOI https://doi.org/10.2147/HMER.S79584
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Gerry Lake-Bakaar
Sujit V Janardhan, Nancy S Reau
Center for Liver Diseases, Section of Gastroenterology, Hepatology, and Nutrition, Department of Medicine, University of Chicago, Chicago, IL, USA
Abstract: Chronic hepatitis C virus (HCV) infection represents a global health problem that affects up to 130–150 million people worldwide. The HCV treatment landscape has been transformed recently by the introduction of direct-acting antiviral (DAA) agents that target viral proteins, including the NS3 protease, the NS5B polymerase, and the NS5A protein. Treatment with multiple DAAs in combination has been shown to result in high rates of sustained virologic response, without the need for pegylated interferon, and a shorter duration of therapy compared with interferon-based regimens; however, the optimal combination of DAAs has yet to be determined. The class of NS5A inhibitors has picomolar potency with pangenotypic activity, and recent clinical studies have shown these inhibitors to be an important component of DAA combination regimens. This review discusses the rational design of an optimal anti-HCV DAA cocktail, with a focus on the role of NS5A in the HCV life cycle, the attributes of the NS5A class of inhibitors, and the potential for NS5A inhibitors to act as a scaffold for DAA-only treatment regimens.
Keywords: hepatitis C virus, NS5A, therapy, direct-acting antiviral
Introduction
Chronic hepatitis C virus (HCV) infection continues to be a significant economic, humanistic, and social burden on the world’s population.1 Current estimates indicate that up to 130–150 million people worldwide are infected with HCV, resulting in up to 350,000–500,000 deaths per year, predominantly from cirrhosis and hepatocellular carcinoma.2,3 The incidence of acute HCV infections peaked between 1970 and 1990, and chronic HCV infection reached a peak in 2001. Despite this, HCV-related complications are increasing worldwide because of the aging HCV-infected population and the lag between initial HCV infection and disease-related complications. The highest prevalence of HCV-related cirrhosis is projected to be between 2010 and 2030.4 Along with contributions from other chronic liver diseases, increases in HCV-related cirrhosis have led to rising rates of cirrhosis-related mortality, which have gone from being the 14th leading cause of US mortality in 1990 to the 8th leading cause in 2011. This represents a 43% increase in deaths and a 39% increase in life-years lost.5
Complications related to hepatitis C have increased in part because of the limited efficacy and poor tolerability of previously available therapies.4,6 However, during the last 5 years, the HCV treatment landscape has been transformed by the introduction of direct-acting antiviral (DAA) agents targeting HCV. This has resulted in improved treatment efficacy, shortened treatment duration, and more tolerable therapies. Modeling data have indicated these therapies have the potential to significantly reduce the prevalence of cirrhosis and related complications.4 Available clinical trial data have suggested that treatment regimens targeting multiple components of the HCV life cycle are most adept at inducing high rates of sustained virologic response (SVR) without the need for interferon.7–11 However, the exact combination of DAAs that most effectively eliminates HCV while minimizing treatment-related adverse effects is not clear.
This review discusses the rational design of an optimal anti-HCV DAA cocktail. We begin by discussing currently available therapies and their limitations. We then provide an overview of the HCV life cycle and identify viral targets of DAAs. We focus primarily on the role of a particular viral protein, nonstructural (NS) protein 5A (NS5A), in viral replication and function. We discuss the critical elements of an ideal anti-HCV regimen, based in part on lessons learned from the evolution of treatments for other infectious diseases. We then review literature demonstrating the value of combination DAA therapy. Finally, we discuss how DAAs targeting NS5A may serve as the ideal scaffold for DAA combination HCV therapy.
Limitations of historic and current therapies: the need for an optimized combination regimen to treat HCV
The combination of peginterferon-alfa and ribavirin (PEG/RBV) was introduced as first-line therapy for HCV treatment after a series of landmark trials in 2001.12–14 The efficacy of this regimen was limited by a number of factors, including the genotype of the virus being treated. Seven distinct genotypes of HCV (GT1–GT7) have been identified.15 In addition to affecting the response to therapy, HCV genotype also influences disease progression and the development of hepatocellular carcinoma.16,17 The most prevalent genotype worldwide is GT1, although geographic variance is recognized.18 Unfortunately, PEG/RBV therapy was least effective against GT1 HCV. Its efficacy was even lower in patients with a variety of baseline host and viral characteristics that predicted treatment failure. These characteristics included patients with cirrhosis who stood to benefit the most from viral eradication. In addition, many patients were intolerant of treatment-related adverse effects, including psychiatric effects, severe fatigue, malaise, and blood dyscrasias. Despite this, PEG/RBV remained the standard-of-care HCV therapy for a decade.19,20
The introduction of DAA agents targeting specific HCV proteins represented a major milestone in improving anti-HCV therapy. The US Food and Drug Administration approved the protease inhibitors telaprevir and boceprevir in 2011. When added to PEG/RBV, these agents increased treatment effectiveness for GT1 HCV while decreasing treatment duration for some patients.21–24 However, several factors limited the clinical effectiveness of these regimens. These medications were frequently associated with increased drug-related adverse events (rash, anemia, ano-rectal discomfort, asthenia, etc), leading to poor tolerability and frequent discontinuation of therapy, especially when used in “real-life” environments outside of clinical trials.25,26 In addition, these therapies were highly selective for those infected with GT1 HCV and were less effective in patients with cirrhosis, those who had failed previous PEG/RBV therapy, and those who had unfavorable polymorphisms in a host interferon-responsive gene (IL28B).23,24,27–29 Finally, treatment failure in this regimen was often associated with the emergence of resistance-associated viral variants (RAVs) that were no longer sensitive to these DAA agents, as reviewed elsewhere.30
Two additional DAA agents, the protease inhibitor simeprevir and the nucleotide analog RNA-dependent RNA polymerase inhibitor sofosbuvir, were approved by the US Food and Drug Administration in 2013.31–36 In conjunction with PEG/RBV, both were associated with high rates of SVR and shortened treatment duration for GT1 HCV infection. Importantly, sofosbuvir was also approved in combination with RBV alone for treatment of GT2/GT3 HCV, representing the first all-oral (noninterferon-based) HCV regimens approved by the US Food and Drug Administration. Despite these recent advances, clinically significant RAVs were identified during clinical trials with simeprevir, and this agent has known drug–drug interactions that complicate its universal use.32,33,37 In addition, both of these new agents still require the addition of PEG/RBV for the treatment of GT1 HCV, which precludes treatment in a large portion of HCV-infected individuals who are either intolerant of treatment-related adverse effects or ineligible because of the risk for treatment-related decompensation.
These limitations have led investigators to explore the effectiveness of antiviral therapy using multiple DAA agents. The combination of the NS5A inhibitor daclatasvir with sofosbuvir established the ability to achieve high rates of SVR in difficult-to-treat populations (GT1, previous PEG/RBV failures).7 Studies such as this have provided proof of concept that combination DAA therapy may indeed be the key to generating an all-oral, highly effective, and durable therapy against hepatitis C. In fact, the potential of highly effective and more tolerable DAA regimens that do not require interferon has led many physicians to “warehouse” patients until these regimens become available.38–40 However, the question of which DAA agents or classes to include in an ideal anti-HCV DAA cocktail remains unanswered.
An overview of HCV replication
A basic understanding of the HCV life cycle and the viral elements targeted by DAA agents is needed to identify which agents should be combined into an optimized anti-HCV regimen. HCV is a member of the Flaviviridae family in the genus Hepacivirus.41 Similar to other members of this genus, it is a single-stranded RNA virus with positive polarity. Its genetic makeup consists of a 9.6 kbp RNA genome with a single open reading frame flanked by 5′- and 3′-untranslated regions.
The details of HCV life cycle events have been extensively reviewed.42–46 Briefly, HCV travels through the bloodstream associated with apolipoproteins and adheres to hepatocytes via interaction with the low-density lipoprotein receptor. HCV interacts with other host proteins to facilitate its entry into the cell and its release of the viral RNA genome into the cytoplasm. RNA translation occurs in the endoplasmic reticulum (ER) via an internal ribosome entry site in the 5′-untranslated region to produce a single 3,000 amino acid viral polyprotein. The polyprotein is cleaved by both cellular signal peptidases and viral proteases into 10 viral proteins (a detailed structure/function discussion of each protein was previously reviewed).43,45 The polyprotein contains structural elements, including the core capsid protein and the envelope glycoproteins E1 and E2, as well as a number of NS proteins. The putative ion channel/viroporin p7 is involved in viral assembly and release of infectious particles. NS protein 2a (NS2A) is a cysteine protease that facilitates cleavage between itself and NS3. NS3 primarily acts as a protease that facilitates processing of the remainder of the NS proteins (NS4A, NS4B, NS5A, NS5B). NS3 also has helicase activity that facilitates replication and inactivates factors involved in the host innate immune response. NS4A acts as a multifunctional cofactor to NS3: stabilizing the protein’s position in cellular membranes, preventing its degradation, and increasing the catalytic activity of its protease domain. HCV replication occurs in the cytosol in a membranous compartment known as the membranous web, which is closely associated with the ER. Densely populated with replication complexes consisting of viral NS proteins and host machinery, this compartment essentially acts as a virion-producing “factory.” The main function of NS4B is to function in concert with other “replicase factors,” such as NS3, NS4A, NS5A, and NS5B, in developing this compartment. NS5A is a multifunctional phosphoprotein that has no enzymatic activity, but is required for RNA replication, membranous web formation, and viral particle formation. Further discussion of NS5A structure and function can be found in subsequent sections of this paper. NS5B is an RNA-dependent RNA polymerase that replicates the HCV genome. HCV replication in the membranous web and budding of new virions from the ER occur in close proximity to lipid droplets and cellular compartments involved in the synthesis of very low density lipoprotein. This allows virions to associate with lipoprotein molecules to form lipoviroparticles and be secreted via the host endogenous secretory pathway. Although reinfection can occur via an extracellular route, it is believed that the major route of transmission in vivo occurs via cell-to-cell transmission, thereby avoiding the effects of neutralizing antibodies.
Role of NS5A in the HCV life cycle
NS5A structure and functional interactions
NS5A is a multifunctional phosphoprotein approximately 447 amino acids in length (in GT1 HCV). It exists in a basal and hyperphosphorylated state (p56 and p58, respectively). Hyperphosphorylation appears to act as a switch that regulates different NS5A functions, specifically, replication and viral particle formation,47,48 and is reviewed elsewhere.49 The structure of NS5A consists of several distinct subdomains, including an amino-terminal 31 amino acid amphipathic alpha helix, followed by three structural domains (D1–D3) that are separated by two low-complexity sequences (LCS1 and LCS2) (Figure 1).
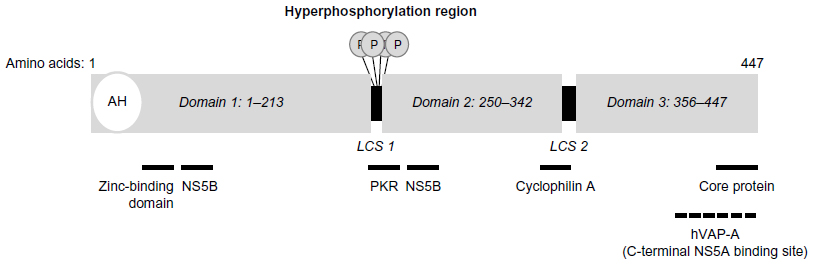 | Figure 1 NS5A structure and function; proposed interaction sites with other cellular and viral proteins are summarized below the schematic diagram. |
The amino-terminal alpha helix domain anchors the protein to cellular membranes and facilitates its recruitment to lipid droplets.50,51 The D1 domain contains a zinc-binding domain and facilitates formation of NS5A homodimers. Interestingly, the crystal structures of purified NS5A dimers suggest the existence of two distinct configurations (claw-like and back-to-back) that may have different functions during replication.52–55 Studies have also suggested that NS5A monomers and dimers may form large multimeric complexes.52 D2 and D3 are unfolded domains that are able to interact with both viral and cellular proteins.45,56–58 NS5A interactions with NS4B, NS5B, and the core protein facilitate membranous web formation, RNA replication, and viral particle formation, respectively.59–61 Interaction of NS5A with cyclophilin A is required for HCV replication.62 NS5A interaction with human vesicle-associated membrane protein-associated protein A facilitates localization of HCV replication complex components to lipid rafts.63 Modulation of the NS5A phosphorylation state is mediated by casein kinases 1 and 2, as well as a variety of other cellular kinases, including phosphatidylinositole-4-phosphate kinase III-α.64,65 NS5A is also an RNA-binding protein, as domains D1–D3 have all been shown to have RNA-binding activity.66,67 D1 and D2 play a critical role in RNA replication. In contrast, much of the D3 domain is not required for replication but appears to be critical for viral particle formation through its interaction with core protein.59,68,69
NS5A in viral replication and beyond
Although the exact role of NS5A in the viral life cycle remains to be fully characterized, NS5A appears to play a role at multiple points in this process, including membranous web formation, replication complex formation and function, and viral particle formation. Mutational analysis identifying three distinct NS5A regions required for RNA replication suggest a clear role in the viral replication complex.70 The ability of NS5A to dimerize/multimerize, bind RNA, and interact with other cellular and viral proteins involved in RNA replication has led to the formation of models depicting NS5A as a critical component in building a network of viral replication complexes within membranous vesicles (Figure 2).71 NS5A also appears to play an important role in modulating the cellular environment to favor HCV replication by interacting with a variety of cellular proteins and signaling pathways, as reviewed elsewhere.57 It has been postulated to downregulate the host interferon response by binding and inactivating the interferon-responsive, ds-RNA-activated protein kinase R.72 It has also been shown to upregulate transcription of the cytokine interleukin 8, which is known to attenuate the interferon response.73–75 Interestingly, interleukin 8 is upregulated in HCV-infected individuals relative to noninfected controls and is further upregulated in patients who are nonresponsive to interferon-based therapy.76 NS5A also appears to interfere with the ability of host cells to undergo apoptosis in response to viral infection. Transgenic mice expressing NS5A in hepatocytes were resistant to TNF-induced hepatic apoptosis.77 In addition, NS5A inhibits apoptotic stimuli by sequestering proapoptotic proteins such as p53 and the bcl-2 family member bax.78,79 NS5A has also been shown to modulate several signaling pathways within hepatocytes, including mitogenic, cell survival, calcium, and reactive oxygen signaling pathways, as reviewed elsewhere.57
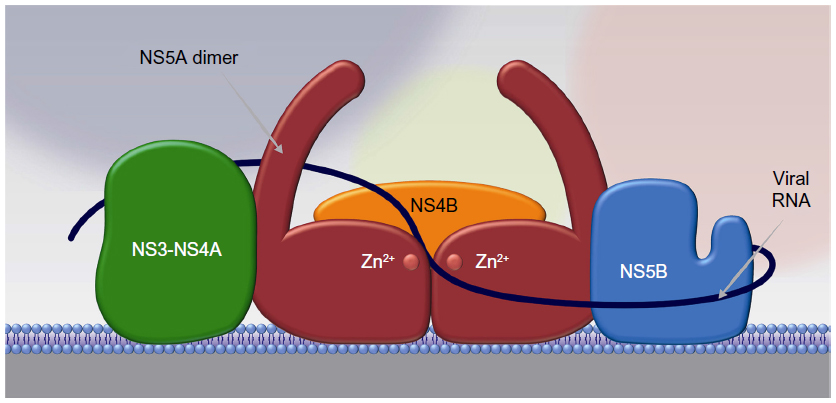 | Figure 2 NS5A acts as a scaffold for other proteins in the viral replication complex. |
How to build an optimal anti-HCV regimen
Examination of the HCV life cycle reveals several candidate targets for antiviral therapy. Accordingly, several molecular inhibitors of these targets (DAA agents) have been developed and have demonstrated remarkable antiviral activity in vitro and in vivo. What remains unclear is which classes of DAA agents to use alone or in combination to generate an optimal anti-HCV regimen. To create an ideal regimen, one must first clearly define the desired characteristics of such a regimen. Examining the evolution of therapies for other infectious diseases, such as the Human Immunodeficiency Virus (HIV) and Mycobacterium tuberculosis (TB), can help identify these characteristics. Similar to HCV, HIV and TB express error-prone polymerases that predispose these organisms to the development of mutations that confer resistance to therapeutic agents. In HCV and HIV, this is exacerbated by high rates of replication.80,81 Therapies for HIV and TB were revolutionized with the discovery that therapeutic regimens that target multiple components of the infectious life cycle synergistically increased the potency of the individual agents and decreased the development of organisms resistant to the combination regimen.82,83 Further optimization of these therapies required investigation into drug combinations that would minimize the adverse events and drug–drug interactions associated with these therapies.
These observations and the limitations of historic and current HCV therapies help define the characteristics of an optimal anti-HCV regimen. Such a regimen would be all-oral (interferon-free), be well tolerated with little to no treatment-related monitoring required (anemia, leukopenia, etc), and have very few drug–drug interactions. An ideal regimen would also provide pangenotypic activity, have a high barrier to the development of clinically significant RAVs, and target multiple elements of the viral life cycle. Finally, to optimize patient compliance and increase the accessibility of the therapy to more providers, it would be a potent regimen that allows for short duration of therapy, abrogates the need for response-guided therapy, and allows for once-daily dosing. Identifying the characteristics of individual DAA agents that meet these criteria either alone or in combination with other agents will allow for rational design of an ideal anti-HCV therapy.
The benefits of combination therapy
Studies using DAA combinations such as daclatasvir and sofosbuvir established the proof of concept that this approach can result in effective all-oral therapies against HCV.7 Although clinical trial data have not clearly defined which classes of agents should be represented in an ideal anti-HCV therapy, they have provided insight into the value of DAA combinations. Studies combining DAAs of different classes have demonstrated the potential to overcome several baseline characteristics previously predictive of treatment failure, optimize treatment duration, and minimize adverse effects. A regimen consisting of the NS5B inhibitor sofosbuvir and RBV has been shown to have poor efficacy in patients with GT1 HCV who had previously failed PEG/RBV therapy.84 However, addition of the NS5A inhibitor ledipasvir to sofosbuvir (even in the absence of RBV) allowed for high levels of SVR in this patient population.11 In addition, treatment-naïve, noncirrhotic patients with GT1 HCV were able to achieve high levels of SVR after only 8 weeks of this regimen.10 The combination of the potent NS3/4A inhibitor ABT-450 boosted with ritonavir (ABT-450/r), the NS5A inhibitor ombitasvir (ABT-267), the nonnucleoside polymerase inhibitor dasabuvir (ABT-333), and RBV demonstrated high levels of SVR in previously difficult-to-treat cirrhotic patients.8 A recently published Phase II study demonstrated that the addition of sofosbuvir to simeprevir can overcome the effects of an NS3 mutation that predicts simeprevir treatment failure (Q80K).9,37 Overall, these studies demonstrate how combining different classes of DAA agents can greatly improve the efficacy, tolerability, and efficiency of anti-HCV therapy.
Why NS5A inhibitors may be a good scaffold for anti-HCV therapy
One difficulty in determining the components of an optimal anti-HCV regimen is identifying the DAA agents/classes that are most critical to the overall effectiveness of the combination regimen and can act as a scaffold to which other DAA classes are added. It stands to reason that such a scaffold would possess many, if not all, the characteristics desired in the overall combination regimen and would minimize the shortcomings of other components contained in the regimen.
Very few studies have examined the relative contributions of various DAA classes to the effectiveness of a combination regimen. One study examined the effect of removing an NS5A inhibitor, a nonnucleoside NS5B inhibitor, or RBV from a 12-week regimen consisting of ABT-450/r (NS3/4A), ombitasvir (NS5A), dasabuvir (NN NS5B), and RBV in treatment-naïve patients with GT1.85 The largest decline in SVR 24 weeks post-therapy was observed when the NS5A component was removed from the regimen (83% without NS5A versus 96% with). Although this was not a primary endpoint of the study and the difference trended toward, but did not achieve, statistical significance (P=0.06), these findings suggest the relative importance of the NS5A class to this combination DAA regimen. Consistent with this, every all-oral DAA combination regimen shown to be successful in published Phase III studies to date has contained an NS5A component.
NS5A inhibitors possess several characteristics that make them attractive candidate molecules to use as a scaffold for an anti-HCV regimen. Unlike first-generation protease inhibitors and nonnucleoside NS5B inhibitors, NS5A inhibitors have pangenotypic activity resulting from an NS5A inhibitor-binding site that is conserved among all HCV genotypes.86 In addition, certain NS5A inhibitors have been shown to have unprecedented antiviral activity and a long duration of action.87 The potency of these agents is likely related to their ability to disrupt a multitude of NS5A functions, including membranous web formation, RNA replication, and viral particle formation.88 Consistent with this, the NS5B inhibitor NM107 and the NS5A inhibitor daclatasvir had similar inhibitory effects on viral replication, but daclatasvir was far superior at reducing viral titers, suggesting a role for daclatasvir outside of viral replication and likely involving viral particle formation and release.89
NS5A inhibitors have no known cross-resistance with other DAA classes, making them ideal agents to use in patients who have failed previous therapies, including those containing DAA agents. It should be noted that several NS5A mutations have been identified that significantly reduce the antiviral activity of NS5A inhibitors when these agents are used alone. However, these baseline polymorphisms do not necessarily predict failure in DAA combination regimens. Treatment failure, when it did occur, was often associated with resistance mutations against multiple components of the regimen.7,8,10,11,88,90–92 In addition, recent data have shown that resistance to NS5A inhibitors can be overcome with the use of synergistic NS5A inhibitors. Gao et al demonstrated that treatment of viral variants expressing NS5A resistance mutations with one NS5A inhibitor (BMS-128) resensitized these variants to the antiviral action of another (daclatasvir), both in vitro and in vivo.93 The underlying mechanism is thought to be a conformational change in NS5A that is induced by binding of the first inhibitor. This conformational change is transmitted to adjacent NS5A molecules and resensitizes them to the action of the second “synergistic” NS5A inhibitor. This synergistic activity was shown to increase potency and resistance barriers across multiple resistance variants and genotypes. This raises the possibility that clinically significant post-treatment NS5A resistance variants could be retreated with a synergistic combination of NS5A inhibitors. The conformational change induced by an NS5A inhibitor may contribute to the agent’s potency in additional ways. One hypothesized mechanism by which NS5A inhibitors can exert profound antiviral effects in low concentration is by fragmenting NS5A multimers that act as RNA-binding “railroads” for replication.45 Whether the conformational change induced by NS5A inhibitor binding impairs NS5A multimerization (and, in turn, viral replication) remains to be determined, but this may represent an additional indirect mechanism through which NS5A inhibitors exert their effect.
Clinical data have shown the NS5A-inhibitor class to be well tolerated with a favorable adverse effect profile and low potential for meaningful drug–drug interactions. The low rate of adverse effects observed with NS5A inhibitors was likely a result of the high specificity of the inhibitors for the NS5A protein, which in turn has no direct enzymatic activity and lacks significant homology to any human protein. Furthermore, the potency of the NS5A inhibitors means they are able to exert maximal antiviral effects with low plasma concentrations. Importantly, clinical trial data from studies using NS5A-containing DAA combination regimens in cirrhotic patients have not reported an increased risk for adverse events in this group.8,92,94
The need for salvage therapies
It is important to recognize that widespread use of combination DAA therapy in the general population could result in individuals who develop RAVs against multiple components of an NS5A-based regimen. Similar phenomena have been seen in the treatment of other infectious diseases with the emergence of multidrug-resistant strains of TB and Clostridium difficile.80,95,96 Within HCV therapy, it should be noted that although baseline NS5A RAVs have been overcome in combination regimens, the emergence of treatment-associated NS5A RAVs has been observed in patients who experience relapse after treatment with an NS5A-containing combination regimen.7,8,10,11,88,90–92 Although the full clinical effect of treatment-associated NS5A RAVs needs to be investigated in larger studies of this relatively rare, relapsing population, the data highlight the importance of identifying additional agents or DAA classes that can serve as salvage therapy for patients who fail NS5A-based combination regimens. Of course, the exact composition of the regimen used for retreatment will be informed by the specific treatment-associated RAVs observed.
Although DAA agents against several potential targets in the HCV life cycle have been generated, only protease inhibitors and nucleotide inhibitors demonstrate properties that make them reasonable alternatives. Because most (first-generation) protease inhibitors are not pangenotypic, nucleotide inhibitors may be the best alternative class of DAA agents (available at this time) to act as a universal scaffold. In addition to pangenotypic activity, this class of agents is associated with low rates of resistance and a favorable adverse effect profile.97 Limitations of this class of agents include that they only target one aspect of the viral life cycle, and most need to be delivered as nucleoside prodrugs to allow membrane penetration. On entry into the cell, the nucleoside needs to be converted to a 5′-monophosphate in a rate-limiting step with eventual conversion to the triphosphate form.98 This rate-limiting step can be bypassed by designing membrane-permeable 5′-monophosphate nucleotide analogs such as sofosbuvir. However, to be membrane-permeable, this molecule requires several chemical modifications that need to be removed by cellular machinery before it can exert its antiviral effect.97 Whereas sofosbuvir has been most effective when used in combination with NS5A inhibitors such as ledipasvir and daclatasvir, recent phase 2 data have demonstrated its efficacy in achieving SVR in small subsets of difficult-to-treat patients when used as part of an all-oral combination DAA regimen, even in the absence of an NS5A inhibitor.7,9–11 Nucleotide analogs such as sofosbuvir may therefore be an important component of DAA regimens that target HCV variants that are resistant to combination NS5A inhibitor-based therapies. This highlights the need for further evaluation of this agent in patients who have failed all-oral regimens because of the development of treatment-related RAVs. Ultimately, the most effective regimen will be one that combines multiple pangenotypic agents with a high barrier to resistance (multiple “scaffolds”). The benefit of such an approach was investigated in a recently published trial sponsored by the National Institutes of Health, in which both an NS5A inhibitor (ledipasvir) and a nucleotide analog NS5B inhibitor (sofosbuvir) were combined with either a nonnucleoside NS5B inhibitor (GS-9669) or a protease inhibitor (GS-9451). Both therapies demonstrated the ability to achieve SVR after only 6 weeks of therapy.99 These data suggest that combining the most effective therapeutic scaffolds may further raise the standard of care for HCV therapy: minimizing treatment duration, adverse effects, and resistance while maintaining a regimen that works for all patients, regardless of clinical or virologic characteristics. Specifically, these regimens may allow for effective treatment of HCV genotypes that continue to demonstrate low SVR rates even in the all-oral DAA combination era (GT3) and avoidance of components that increase regimen-related adverse effects or drug–drug interactions (eg, ribavirin and ritonavir, respectively).100 Importantly, the ongoing development of second-generation DAA agents will allow clinicians to treat HCV with confidence today, knowing that relatively rare therapeutic failures caused by treatment-associated RAVs will likely be eligible for salvage therapies with future combination regimens.
Conclusion
Therapy against hepatitis C has been revolutionized in the past several years because of the tireless efforts of investigators in academics and the pharmaceutical industry to develop highly efficacious DAA agents while sharing a unified vision of discovery, cooperation, and cure. The previous “pipe dream” of all-oral, potent, tolerable, pangenotypic, and highly efficacious therapies for even difficult-to-treat HCV-infected populations is now a real possibility. However, to enter the final chapter in the battle against chronic HCV infection, these new therapies must be used judiciously in combinations that are most effective in viral eradication while being mindful of issues related to patient access to care, cost, and development of resistance. Although the future of HCV-related care remains uncertain, the outlook is certainly improved, and NS5A inhibitor-based DAA combination regimens may represent a rational, data-driven strategy for targeting chronic HCV infection moving forward.
Disclosure
Dr Janardhan reports no conflicts of interest in this work. Dr Reau would like to disclose both advisory and research support relationships with the following entities: AbbVie, Inc.; Gilead Sciences, Inc.; Merck and Co, Inc.; Bristol-Myers Squibb Company.
References
Dibonaventura MD, Yuan Y, Lescrauwaet B, et al. Multicountry burden of chronic hepatitis C viral infection among those aware of their diagnosis: a patient survey. PLoS One. 2014;9(1):e86070. | |
World Health Organization. Hepatitis C Key Facts. WHO Fact sheet No 164. Available from: http://www.who.int/mediacentre/factsheets/fs164/en/. Accessed May 14, 2014. | |
Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57(4):1333–1342. | |
Davis GL, Alter MJ, El-Serag H, Poynard T, Jennings LW. Aging of hepatitis C virus (HCV)-infected persons in the United States: a multiple cohort model of HCV prevalence and disease progression. Gastroenterology. 2010;138(2):513–521. | |
Murray CJL; US Burden of Disease Collaborators. The state of US health, 1990-2010: burden of diseases, injuries, and risk factors. JAMA. 2013;310(6):591–608. | |
Morrill JA, Shrestha M, Grant RW. Barriers to the treatment of hepatitis C. Patient, provider, and system factors. J Gen Intern Med. 2005;20(8):754–758. | |
Sulkowski MS, Gardiner DF, Rodriguez-Torres M, et al; AI444040 Study Group. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med. 2014;370(3):211–221. | |
Poordad F, Hezode C, Trinh R, et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med. 2014;370(21):1973–1982. | |
Lawitz E, Sulkowski MS, Ghalib R, et al. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: the COSMOS randomised study. Lancet. 2014;384(9956):1756–1765. | |
Kowdley KV, Gordon SC, Reddy KR, et al; ION-3 Investigators. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med. 2014;370(20):1879–1888. | |
Afdhal N, Reddy KR, Nelson DR, et al; ION-2 Investigators. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med. 2014;370(16):1483–1493. | |
Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347(13):975–982. | |
Hadziyannis SJ, Sette H Jr, Morgan TR, et al; PEGASYS International Study Group. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med. 2004;140(5):346–355. | |
Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358(9286):958–965. | |
Smith DB, Bukh J, Kuiken C, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59(1):318–327. | |
Raimondi S, Bruno S, Mondelli MU, Maisonneuve P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: a meta-analysis. J Hepatol. 2009;50(6):1142–1154. | |
Kanwal F, Kramer JR, Ilyas J, Duan Z, El-Serag HB. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of US Veterans with HCV. Hepatology. 2014;60(1):98–105. | |
Messina JP, Humphreys I, Flaxman A, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015;61(1):77–87. | |
Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54(4):1433–1444. | |
Ghany MG, Strader DB, Thomas DL, Seeff LB; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49(4):1335–1374. | |
Victrelis™ (boceprevir) [prescribing information]. Kenilworth, NJ: Merck Sharp and Dohme Corp; 2012. | |
Incivek™ (telaprevir) [prescribing information]. South Boston, MA: Vertex Pharmaceuticals; 2012. | |
Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364(25):2405–2416. | |
Poordad F, McCone J Jr, Bacon BR, et al; SPRINT-2 Investigators. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364(13):1195–1206. | |
Price JC, Murphy RC, Shvachko VA, Pauly MP, Manos MM. Effectiveness of telaprevir and boceprevir triple therapy for patients with hepatitis C virus infection in a large integrated care setting. Dig Dis Sci. 2014;59(12):3043–3052. | |
Belperio PS, Hwang EW, Thomas IC, Mole LA, Cheung RC, Backus LI. Early virologic responses and hematologic safety of direct-acting antiviral therapies in veterans with chronic hepatitis C. Clin Gastroenterol Hepatol. 2013;11(8):1021–1027. | |
Bacon BR, Gordon SC, Lawitz E, et al; HCV RESPOND-2 Investigators. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med. 2011;364(13):1207–1217. | |
McHutchison JG, Manns MP, Muir AJ, et al; PROVE3 Study Team. Telaprevir for previously treated chronic HCV infection. N Engl J Med. 2010;362(14):1292–1303. | |
Zeuzem S, Andreone P, Pol S, et al; REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med. 2011;364(25):2417–2428. | |
Gambarin-Gelwan M, Jacobson IM. Resistance-associated variants in chronic hepatitis C patients treated with protease inhibitors. Curr Gastroenterol Rep. 2012;14(1):47–54. | |
Olysio™ (simeprevir) [prescribing information]. Titusville, NJ: Janssen Therapeutics; 2014. | |
Manns M, Marcellin P, Poordad F, et al. Simeprevir with pegylated interferon alfa 2a or 2b plus ribavirin in treatment-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-2): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2014; 384(9941):414–426. | |
Jacobson IM, Dore GJ, Foster GR, et al. Simeprevir with pegylated interferon alfa 2a plus ribavirin in treatment-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-1): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet. 2014;384(9941):403–413. | |
Jacobson IM, Gordon SC, Kowdley KV, et al; POSITRON Study; FUSION Study. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med. 2013;368(20):1867–1877. | |
Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368(20):1878–1887. | |
Sovaldi™ (sofosbuvir) [prescribing information]. Foster City, CA: Gilead Sciences, Inc.; 2014. | |
Jacobson GJ, Foster GR, Fried M, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in treatment-naïve patients: efficacy in difficult-to-treat patient sub-populations in the QUEST-1 and 2 Phase III trials. HCV Therapeutics: New Agents. Hepatology. 2013;58(S1):730A–760A. | |
Chen EY, Sclair SN, Czul F, et al. A small percentage of patients with hepatitis C receive triple therapy with boceprevir or telaprevir. Clin Gastroenterol Hepatol. 2013;11(8):1014–1020. | |
Tovo CV, de Mattos AA, de Almeida PR. Chronic hepatitis C genotype 1 virus: who should wait for treatment? World J Gastroenterol. 2014;20(11):2867–2875. | |
Shiffman ML, Benhamou Y. HCV F1/F2 patients: treat now or continue to wait. Liver Int. 2014;34 Suppl 1:79–84. | |
Simmonds P. The origin of hepatitis C virus. Curr Top Microbiol Immunol. 2013;369:1–15. | |
Bartenschlager R, Penin F, Lohmann V, André P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011;19(2):95–103. | |
Moradpour D, Penin F. Hepatitis C virus proteins: from structure to function. Curr Top Microbiol Immunol. 2013;369:113–142. | |
Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5(6):453–463. | |
Rupp D, Bartenschlager R. Targets for antiviral therapy of hepatitis C. Semin Liver Dis. 2014;34(1):9–21. | |
Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19(7):837–849. | |
Appel N, Pietschmann T, Bartenschlager R. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J Virol. 2005;79(5):3187–3194. | |
Tellinghuisen TL, Foss KL, Treadaway J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 2008;4(3):e1000032. | |
Huang Y, Staschke K, De Francesco R, Tan SL. Phosphorylation of hepatitis C virus NS5A nonstructural protein: a new paradigm for phosphorylation-dependent viral RNA replication? Virology. 2007;364(1):1–9. | |
Miyanari Y, Atsuzawa K, Usuda N, et al. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9(9):1089–1097. | |
Penin F, Brass V, Appel N, et al. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J Biol Chem. 2004;279(39):40835–40843. | |
Lim PJ, Chatterji U, Cordek D, et al. Correlation between NS5A dimerization and hepatitis C virus replication. J Biol Chem. 2012; 287(36):30861–30873. | |
Love RA, Brodsky O, Hickey MJ, Wells PA, Cronin CN. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J Virol. 2009;83(9):4395–4403. | |
Tellinghuisen TL, Marcotrigiano J, Gorbalenya AE, Rice CM. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J Biol Chem. 2004;279(47):48576–48587. | |
Tellinghuisen TL, Marcotrigiano J, Rice CM. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature. 2005;435(7040):374–379. | |
Hanoulle X, Verdegem D, Badillo A, Wieruszeski JM, Penin F, Lippens G. Domain 3 of non-structural protein 5A from hepatitis C virus is natively unfolded. Biochem Biophys Res Commun. 2009;381(4):634–638. | |
Macdonald A, Harris M. Hepatitis C virus NS5A: tales of a promiscuous protein. J Gen Virol. 2004;85(Pt 9):2485–2502. | |
Verdegem D, Badillo A, Wieruszeski JM, et al. Domain 3 of NS5A protein from the hepatitis C virus has intrinsic alpha-helical propensity and is a substrate of cyclophilin A. J Biol Chem. 2011;286(23):20441–20454. | |
Masaki T, Suzuki R, Murakami K, et al. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J Virol. 2008;82(16):7964–7976. | |
Romero-Brey I, Merz A, Chiramel A, et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012;8(12):e1003056. | |
Shimakami T, Hijikata M, Luo H, et al. Effect of interaction between hepatitis C virus NS5A and NS5B on hepatitis C virus RNA replication with the hepatitis C virus replicon. J Virol. 2004;78(6):2738–2748. | |
Grisé H, Frausto S, Logan T, Tang H. A conserved tandem cyclophilin-binding site in hepatitis C virus nonstructural protein 5A regulates Alisporivir susceptibility. J Virol. 2012;86(9):4811–4822. | |
Gao L, Aizaki H, He JW, Lai MM. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J Virol. 2004;78(7):3480–3488. | |
Quintavalle M, Sambucini S, Di Pietro C, De Francesco R, Neddermann P. The alpha isoform of protein kinase CKI is responsible for hepatitis C virus NS5A hyperphosphorylation. J Virol. 2006;80(22):11305–11312. | |
Reiss S, Harak C, Romero-Brey I, et al. The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS Pathog. 2013;9(5):e1003359. | |
Huang L, Hwang J, Sharma SD, et al. Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J Biol Chem. 2005;280(43):36417–36428. | |
Hwang J, Huang L, Cordek DG, et al. Hepatitis C virus nonstructural protein 5A: biochemical characterization of a novel structural class of RNA-binding proteins. J Virol. 2010;84(24):12480–12491. | |
Appel N, Zayas M, Miller S, et al. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008;4(3):e1000035. | |
Hughes M, Griffin S, Harris M. Domain III of NS5A contributes to both RNA replication and assembly of hepatitis C virus particles. J Gen Virol. 2009;90(Pt 6):1329–1334. | |
Fridell RA, Valera L, Qiu D, Kirk MJ, Wang C, Gao M. Intragenic complementation of hepatitis C virus NS5A RNA replication-defective alleles. J Virol. 2013;87(4):2320–2329. | |
Gu M, Rice CM. Structures of hepatitis C virus nonstructural proteins required for replicase assembly and function. Curr Opin Virol. 2013; 3(2):129–136. | |
Gale MJ Jr, Korth MJ, Tang NM, et al. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997; 230(2):217–227. | |
Girard S, Shalhoub P, Lescure P, et al. An altered cellular response to interferon and up-regulation of interleukin-8 induced by the hepatitis C viral protein NS5A uncovered by microarray analysis. Virology. 2002;295(2):272–283. | |
Khabar KS, Al-Zoghaibi F, Al-Ahdal MN, et al. The alpha chemokine, interleukin 8, inhibits the antiviral action of interferon alpha. J Exp Med. 1997;186(7):1077–1085. | |
Polyak SJ, Khabar KS, Paschal DM, et al. Hepatitis C virus nonstructural 5A protein induces interleukin-8, leading to partial inhibition of the interferon-induced antiviral response. J Virol. 2001;75(13):6095–6106. | |
Polyak SJ, Khabar KS, Rezeiq M, Gretch DR. Elevated levels of interleukin-8 in serum are associated with hepatitis C virus infection and resistance to interferon therapy. J Virol. 2001;75(13):6209–6211. | |
Majumder M, Ghosh AK, Steele R, et al. Hepatitis C virus NS5A protein impairs TNF-mediated hepatic apoptosis, but not by an anti-FAS antibody, in transgenic mice. Virology. 2002;294(1):94–105. | |
Chung YL, Sheu ML, Yen SH. Hepatitis C virus NS5A as a potential viral Bcl-2 homologue interacts with Bax and inhibits apoptosis in hepatocellular carcinoma. Int J Cancer. 2003;107(1):65–73. | |
Lan KH, Sheu ML, Hwang SJ, et al. HCV NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene. 2002;21(31):4801–4811. | |
Gillespie SH. Evolution of drug resistance in Mycobacterium tuberculosis: clinical and molecular perspective. Antimicrob Agents Chemother. 2002;46(2):267–274. | |
Soriano V, Perelson AS, Zoulim F. Why are there different dynamics in the selection of drug resistance in HIV and hepatitis B and C viruses? J Antimicrob Chemother. 2008;62(1):1–4. | |
Gulick RM, Mellors JW, Havlir D, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med. 1997;337(11):734–739. | |
Hammer SM, Squires KE, Hughes MD, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N Engl J Med. 1997;337(11):725–733. | |
Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med. 2013;368(1):34–44. | |
Kowdley KV, Lawitz E, Poordad F, et al. Phase 2b trial of interferon-free therapy for hepatitis C virus genotype 1. N Engl J Med. 2014; 370(3):222–232. | |
Belda O, Targett-Adams P. Small molecule inhibitors of the hepatitis C virus-encoded NS5A protein. Virus Res. 2012;170(1–2):1–14. | |
Gao M, Nettles RE, Belema M, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465(7294):96–100. | |
Gao M. Antiviral activity and resistance of HCV NS5A replication complex inhibitors. Curr Opin Virol. 2013;3(5):514–520. | |
Guedj J, Dahari H, Rong L, et al. Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Proc Natl Acad Sci U S A. 2013;110(10):3991–3996. | |
Karino Y, Toyota J, Ikeda K, et al. Characterization of virologic escape in hepatitis C virus genotype 1b patients treated with the direct-acting antivirals daclatasvir and asunaprevir. J Hepatol. 2013;58(4):646–654. | |
Suzuki Y, Ikeda K, Suzuki F, et al. Dual oral therapy with daclatasvir and asunaprevir for patients with HCV genotype 1b infection and limited treatment options. J Hepatol. 2013;58(4):655–662. | |
Manns M, Pol S, Jacobson IM, et al; HALLMARK-DUAL Study Team. All-oral daclatasvir plus asunaprevir for hepatitis C virus genotype 1b: a multinational, phase 3, multicohort study. Lancet. 2014;384(9954):1597–1605. | |
Gao M, O’Boyle DR II, Lemm JA, et al. Synergistic Interactions Of Hcv Ns5A Replication Complex Inhibitors Sensitize Resistant Variants And Enhance The Efficacy Of Daclatasvir (Dcv, Bms-790052) In Vitro And In Vivo. ABSTR 1424. J Hepatol. 2013;58:S573–S574. | |
Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet. 2014;383(9916):515–523. | |
Huang H, Weintraub A, Fang H, Nord CE. Antimicrobial resistance in Clostridium difficile. Int J Antimicrob Agents. 2009;34(6):516–522. | |
Shah D, Dang MD, Hasbun R, et al. Clostridium difficile infection: update on emerging antibiotic treatment options and antibiotic resistance. Expert Rev Anti Infect Ther. 2010;8(5):555–564. | |
Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother. 2013;14(9):1161–1170. | |
Coats SJ, Garnier-Amblard EC, Amblard F, et al. Chutes and ladders in hepatitis C nucleoside drug development. Antiviral Res. 2014;102:119–147. | |
Kohli A, Osinusi A, Sims Z, et al. Virological response after 6 week triple-drug regimens for hepatitis C: a proof-of-concept phase 2A cohort study. Lancet. 2015;385(9973):1107–1113. | |
Goossens N, Negro F. Is genotype 3 of the hepatitis C virus the new villain? Hepatology. 2014;59(6):2403–2412. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.