")
Back to Journals » OncoTargets and Therapy » Volume 12
Shikonin-induced necroptosis in nasopharyngeal carcinoma cells via ROS overproduction and upregulation of RIPK1/RIPK3/MLKL expression
Received 7 January 2019
Accepted for publication 9 March 2019
Published 9 April 2019 Volume 2019:12 Pages 2605—2614
DOI https://doi.org/10.2147/OTT.S200740
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Tiancong Liu,1 Xun Sun,2 Zhiwei Cao1
1Department of Otorhinolaryngology – Head and Neck Surgery, Shengjing Hospital, China Medical University, Shenyang, Liaoning, People’s Republic of China; 2Department of Immunology, College of Basic Medicine, China Medical University, Shenyang, Liaoning, People’s Republic of China
Objective: Shikonin has inhibitory effects against nasopharyngeal carcinoma that are mediated through the apoptotic pathway. However, necroptosis signaling pathways may enable the elimination of apoptosis-resistant cancers when induced with targeted therapeutic agents. Thus, there is a need to clarify whether shikonin can cause necroptosis in nasopharyngeal carcinoma and to elucidate the underlying mechanisms.
Methods: In this study, we used the nasopharyngeal carcinoma cell line 5-8F and a 5-8F xenograft mouse model to evaluate the anticancer effects of shikonin. The viability and morphology of cells treated with shikonin were evaluated using CCK-8 assay and transmission electron microscopy, respectively. In addition, the expression levels of RIPK1, RIPK3, and MLKL were analyzed by western blotting, and the activities of caspase-3 and caspase-8 and levels of reactive oxygen species (ROS) were assessed.
Results: Shikonin exhibited a strong inhibitory effect on 5-8F cells in vitro and in vivo. The shikonin-treated 5-8F cells presented an electron-lucent cytoplasm, loss of plasma membrane integrity, and an intact nuclear membrane, indicating that shikonin induced necroptosis. Shikonin-induced cell death was inhibited by necrostatin-1. Moreover, RIPK1, RIPK3, and MLKL were upregulated by shikonin in a dose-dependent manner. Furthermore, shikonin significantly inhibited tumor growth in the 5-8F xenograft mouse model.
Conclusion: Shikonin induced 5-8F cell death via increased ROS production and the upregulation of RIPK1/RIPK3/MLKL expression, resulting in necroptosis. Thus, shikonin may represent a novel agent to treat nasopharyngeal carcinoma.
Keywords: nasopharyngeal carcinoma, necroptosis, reactive oxygen species, shikonin
Introduction
Nasopharyngeal carcinoma (NPC) is a common head and neck cancer with high morbidity among patients in Southeast Asia.1 Chemotherapy is used as an effective method to treat advanced-stage disease. Nonetheless, treatment failure largely due to local recurrence and distant metastasis remains a challenge.2,3 In addition, drug resistance is a prominent problem that can compromise the efficacy of chemotherapy.4
Cell death can occur through several pathways, including apoptosis, necroptosis, and autophagy. Necroptosis, a relatively newly discovered type of cell death, is morphologically similar to necrosis.5 With respect to morphology, necroptosis is characterized by a ruptured plasma membrane, gain in cell volume, swelling of organelles, and subsequent loss of intracellular contents. Necroptosis is a form of regulated necrosis that depends on receptor-interacting protein kinase 1 (RIPK1) and can be specifically inhibited by necrostatin-1 (Nec-1).6 Accumulating evidence has demonstrated that activated RIPK1 interacts with RIPK3 to facilitate RIPK1/RIPK3 necrosome formation.7,8 The necrosome recruits its downstream substrate, mixed lineage kinase domain-like protein (MLKL), which forms an oligomer, resulting in cellular membrane leakage and cell death.9 Necroptosis signaling pathways can be targeted by therapeutic agents to eliminate apoptosis/drug-resistant cancer cells, which is critical for the success of cancer chemotherapy.10
Shikonin (5,8-dihydroxy-2-[(1S)-1-hydroxy-4-methylpent-3-en-1-yl]naphthalene-1,4-dione) is a predominant type of naphthoquinone pigment that is extracted from the Chinese plant Lithospermum erythrorhizon, with a molecular weight of 288 kDa (Figure S1). L. erythrorhizon has been widely used for thousands of years in traditional Chinese medicine to treat burns, measles, carbuncles, and macular eruptions.11 In addition, shikonin has been reported to induce necroptosis in various tumors such as breast cancer, hepatocellular carcinoma, and osteosarcoma, which is accompanied by overproduction of reactive oxygen species (ROS) and mitochondrial injury.12–14 However, it remains unclear whether ROS participate in necroptosis by promoting RIPK1/RIPK3 necrosome formation.
Furthermore, previous research has shown that shikonin causes necroptosis in the CNE2Z cell line.15 Nonetheless, the underlying molecular mechanisms of shikonin-induced necroptosis are not fully understood. Herein, we extended this investigation to another type of NPC cell line, 5-8F cells, and a 5-8F xenograft mouse model. Clarifying the mechanisms of necroptosis by shikonin treatment may provide novel insight into the mechanism underlying the anticancer effects of shikonin.
Materials and methods
Reagents
Roswell Park Memorial Institute (RPMI)-1640 medium and fetal bovine serum (FBS) were obtained from Gibco (Grand Island, NY, USA). Shikonin, N-acetyl-L-cysteine (NAC), and necrostatin-1 (Nec-1) were procured from Sigma (St. Louis, MO, USA). Pan-caspase inhibitor z-VAD-fmk was obtained from Tocris (Bristol, UK). The Caspase-8 Fluorescence Metric Assay and Caspase-3 Fluorescence Metric Assay Kits were bought from KeyGEN BioTECH (Nanjing, China). The Reactive Oxygen Species Detection Kit and Protein Concentration Assay Kit were obtained from Beyotime Institute of Biotechnology (Wuhan, China). Annexin V Apoptosis Detection Kit I was purchased from BD Pharmingen (San Jose, CA, USA). Dimethyl sulfoxide (DMSO) was procured from Biosharp (Hefei, China). Radioimmunoprecipitation assay (RIPA) buffer, containing protease and phosphatase inhibitors, was obtained from Sigma (St. Louis, MO, USA). Anti-RIPK1, anti-RIPK3, anti-MLKL, anti-β-actin, and anti-GAPDH antibodies were obtained from Cell Signaling (Danvers, MA, USA) and diluted 1:1,000 in TBST. Anti-rabbit IgG HRP-linked antibody and anti-mouse IgG HRP-linked antibody were also obtained from Cell Signaling (Danvers, MA, USA) and diluted 1:5,000 in TBST. The enhanced chemiluminescence (ECL) western blotting detection reagent was purchased from Wanleibio (Shanghai, China). Cell Counting Kit-8 was purchased from Dojindo (Kumamoto, Japan)
Cell culture
The human 5-8F NPC cell line was obtained from Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). 5-8F cells were cultured in RPMI-1640 medium supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (100 U/mL) at 37 °C in a 5% CO2 humidified atmosphere. The medium was replaced every 2 d. The cells were harvested at 75% confluence with 0.25% trypsin containing 0.02% ethylenediaminetetraacetic acid (EDTA). Shikonin was dissolved in DMSO to a stock concentration of 20 mmol/L and stored in the dark at −20 °C. Nec-1 and z-VAD-fmk were dissolved in DMSO to a stock concentration of 20 mM and 2 mM, respectively. NAC was dissolved in phosphate-buffered saline (PBS) to a stock concentration of 400 mM. Different concentrations of shikonin, Nec-1 (50 μM), z-VAD-fmk (20 μM), and NAC (5 mM) were prepared in culture medium for cell treatments. The maximum final concentration of DMSO was less than 0.1% for each treatment.
Cell proliferation assay
The viability of 5-8F cells was measured using the CCK-8 assay kit, according to the manufacturer’s instructions. 5-8F cells were seeded at a density of 7.5×103 cells in each well (200 μL) of a 96-well plate and cultured for 24 h. The cells were then treated with different concentrations of shikonin (2.5, 5.0, 7.5, 10, and 15 μM) for 6, 12, 24, and 48 h. Each well was incubated with 10 μL of CCK-8 at 37 °C for 2 h. Subsequently, the absorbance (A) of the solution was measured using an automatic multi-well spectrophotometer (Bio-Rad, Hercules, CA, USA) at a wavelength of 540 nm. Treatment efficacy was determined by comparing the cell viability for the treated and untreated cells. The IC50, representing the shikonin concentration required to inhibit 50% of cell proliferation, was calculated using GraphPad Prism 6 (GraphPad Software, Inc. La Jolla, CA, USA). All experiments were repeated at least three times.
Detection of cell death by flow cytometry
Cell death modality was determined using the Annexin V Apoptosis Detection Kit I, according to the manufacturer’s instructions. Briefly, 5×105 cells were plated and pretreated with 50 μM Nec-1 for 3 h. The cells were then treated with 7.5 μM shikonin for 6 h. After treatment, the cells were washed twice with PBS and then resuspended in 200 μL binding buffer. The cells (100 μL) were transferred to a 5 mL culture tube and stained with 5 μL Annexin V-FITC and 5 μL propidium iodide (PI) for 15 min at room temperature in the dark. After the addition of 400 μL binding buffer into each tube, the apoptotic cells were quantified using a fluorescence-activated cell sorting (FACS) Calibur flow cytometer (BD Biosciences, San Jose, CA, USA). The data were acquired by collecting 2×104 cells per tube. Both early apoptotic (Annexin V-positive, PI-negative) and late apoptotic (double positive for Annexin V and PI) cells were detected; the percentage of stained cells was subsequently analyzed. The rate of cell death in each quadrant was analyzed using Flowing software version 2.4.1.
Evaluation of mode of cell death by transmission electron microscopy
5-8F NPC cells were cultured and treated with 7.5 μM shikonin for 6 h. The cells were harvested using 0.25% trypsin, washed with PBS, and fixed with 3% ice-cold glutaraldehyde and 2% paraformaldehyde in 0.1 M PBS (pH 7.4) at 4 °C. The 5-8F cells were post-fixed with 1% osmium tetroxide with 0.1% potassium ferricyanide, dehydrated through a graded series of ethanol, and embedded in Epon (EnergyBeam Sciences, Agawam, MA, USA). A Reichert ultramicrotome (Leica, Wetzlar, Germany) was used to cut sections of 65 nm thickness. The ultrathin sections were stained with 1% uranyl acetate and 0.1% lead citrate and examined using a transmission electron microscope (JEOL, Pleasanton, CA, USA).
Gel electrophoresis and western blotting
5-8F NPC cells were directly lysed with RIPA buffer for 30 min on ice. The lysates were centrifuged at 12,000×g for 10 min at 4 °C to obtain the supernatant. The protein content in the supernatant was determined using the Bio-Rad Protein Assay kit. Equal amounts of protein were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred onto polyvinylidene difluoride membranes. The membranes were blocked with 5% skim milk in PBS for 1 h and washed with Tris-buffered saline containing 0.1% Tween 20, three times. The membranes were incubated overnight at 4 °C with anti-RIPK1, anti-RIPK3, anti-MLKL antibodies, anti-GAPDH and anti-β-actin, followed by secondary antibodies. The blots were washed and the immunoreactive proteins were visualized using a gel imager (Bio-Rad) with ECL western blotting detection reagents. Densitometry was performed using Kodak ID image analysis software (Eastman Kodak Company, Rochester, NY, USA).
Evaluation of caspase-3 and -8 in shikonin-induced apoptosis or necroptosis
The activity of caspase-3 was measured using the Caspase-3 Fluorescence Metric Assay Kit. The cultured cells in 96-well plates were pretreated with 20 μM z-VAD-fmk or 50 μM Nec-1 for 3 h prior to treatment with 7.5 μM shikonin. The assay was conducted according to the instructions provided in the assay kit. The samples were read using a 485 nm excitation filter and 535 nm emission filter with a fluorescence microplate reader (BioTek Synergy HT, Winooski, VT, USA), and the results are expressed as the fold increase from the basal level (control cells). The activity of caspase-8 was measured similarly, with at least three repetitions.
Detection of intracellular ROS levels
The average level of intracellular ROS was evaluated in 5-8F cells loaded with the fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA). DCFH-DA was first diluted with serum-free medium to a concentration of 10 μM. The cells were cultured in a 96-well plate, following which the supernatant was removed. DCFH-DA was then added to each well. The substrate solution was discarded after incubation at 37 °C for 30 min, and the cells were washed thoroughly with PBS. Afterward, ROS were evaluated in cells incubated with 7.5 μM shikonin following pretreatment with 50 μΜ Nec-1 or 5 mM NAC. Untreated cells were used as the control. A multi-detection microplate reader was used to observe the DCFH-DA at an excitation wavelength of 485 nm and an emission wavelength of 528 nm. The ROS level is expressed as relative fluorescence units (RFU). 5-8F cells were treated either with 5 mM NAC alone at the indicated time points or with 5 mM NAC for 1 h followed by shikonin at the indicated concentrations. The cell viability was then determined by CCK-8 assay.
Antitumor effect of shikonin in vivo
Five-week-old female Balb/C nude mice were purchased from Beijing Vital River Laboratory Animal Technology Co. Ltd (Beijing, China). The mice were randomly divided into the control group and the experimental group. 5-8F cells (3×106 cells per animal) were injected into the right flank of the experimental nude mice. The mice that developed tumors were intraperitoneally injected with shikonin (0.5 mg/kg) every 3 d for 21 d.15 In contrast, the negative control group was injected with PBS every 3 d for 21 d. The nude mice were weighed before each injection. After the experiment, tumors were excised and the tumor weights were measured. The tumors were preserved in 4% formalin solution, sectioned into 6 μm thick slices and stained with hematoxylin and eosin (H&E).
Statistical analyses
The data are presented as mean ± SE. To compare the data, paired t-test and one-way analysis of variance (ANOVA) were used. All data represent at least three independent experiments and are expressed as mean ± SE. Results with a P-value of <0.05 were considered significant.
Results
Shikonin inhibited 5-8F cell proliferation in a dose- and time-dependent manner
We performed a CCK-8 assay to examine the effect of shikonin on the viability of 5-8F cells. As shown in Figure 1A, shikonin significantly inhibited 5-8F cell proliferation in a time- and dose-dependent manner. The cytotoxic effect was evident at 6 h in the cell line; after 6 h of incubation with shikonin at concentrations of 2.5, 5.0, 7.5, 10, and 15 μM, the average viability of 5-8F cells decreased to 92.3%, 77.5%, 53.2%, 40.3%, and 32.5% when compared with that of the control group, respectively. As a result, the IC50 of shikonin after 6 h of treatment was 7.5 μM. Thus, 7.5 μM shikonin was considered the optimum concentration and was used in subsequent experiments.
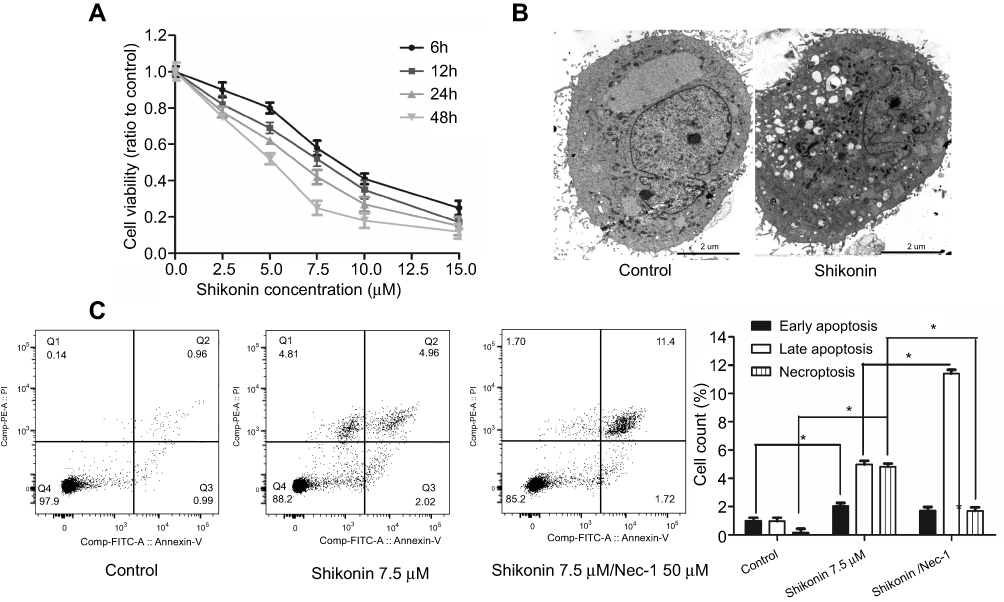 | Figure 1 Shikonin induced necroptosis in 5-8F cells. (A) 5-8F cells were treated with different concentrations of shikonin for 6, 12, 24, and 48 h. Cell viability was assessed by CCK-8 assay. (B) 5-8F cells were treated with or without 7.5 μΜ shikonin and morphological changes were examined by transmission electron microscopy. Scale bar =2 μm. (C) Shikonin-induced mode of death in the 5-8F cell line. The cells were pretreated with Nec-1 for 3 h prior to shikonin treatment. After 6 h of incubation with shikonin, the percentage of both necroptotic cells (stained with PI only) and late apoptotic cells (stained with both Annexin V and PI) were determined. The data are representative of three independent experiments. *P<0.05 versus untreated cells. |
Shikonin induced necroptosis in 5-8F cells
To clarify the mode of death in 5-8F cells treated with shikonin, we observed the cells under a transmission electron microscope. The shikonin-treated 5-8F cells showed typical nuclear fragmentation, a ruptured plasma membrane, gain in cell volume, swelling of organelles, and subsequent loss of intracellular contents, which were consistent with the characteristics of necroptosis (Figure 1B), compared with normal 5-8F cells.
Shikonin induced both apoptosis and necroptosis in 5-8F cells
To investigate the mode of 5-8F cell death caused by shikonin, flow cytometry with FITC-conjugated Annexin V and PI staining were performed. As shown in Figure 1C, the control cells were negative for both Annexin V-FITC and PI. After 6 h of incubation with 7.5 μM shikonin, 4.81% cells showed the induction of necroptosis (Annexin V−/PI+), 4.96% of the cells were in early apoptosis (Annexin V+/PI−) phase. To further clarify the mode of cell death by shikonin, 5-8F cells were treated with shikonin in combination with Nec-1. Pretreatment with Nec-1 reduced cell necroptosis caused by shikonin to 1.7%. In contrast, the rate of early apoptosis of shikonin/Nec-1-treated cells increased significantly compared with that of the control cells.
Involvement of caspase-3 and caspase-8 in shikonin-induced apoptosis or necroptosis
Treatment of 5-8F cells with 7.5 μΜ shikonin significantly increased the activity of caspase-8 (Figure 2A) and caspase-3 (Figure 2B) compared with those of the control (P<0.05). Pretreatment of cells with z-VAD-fmk (20 μM) significantly inhibited shikonin-induced apoptosis in 5-8F cells compared with that in the control (P<0.05). In contrast, pretreatment with Nec-1 (50 μM) significantly increased the activity of caspase-8 and caspase-3.
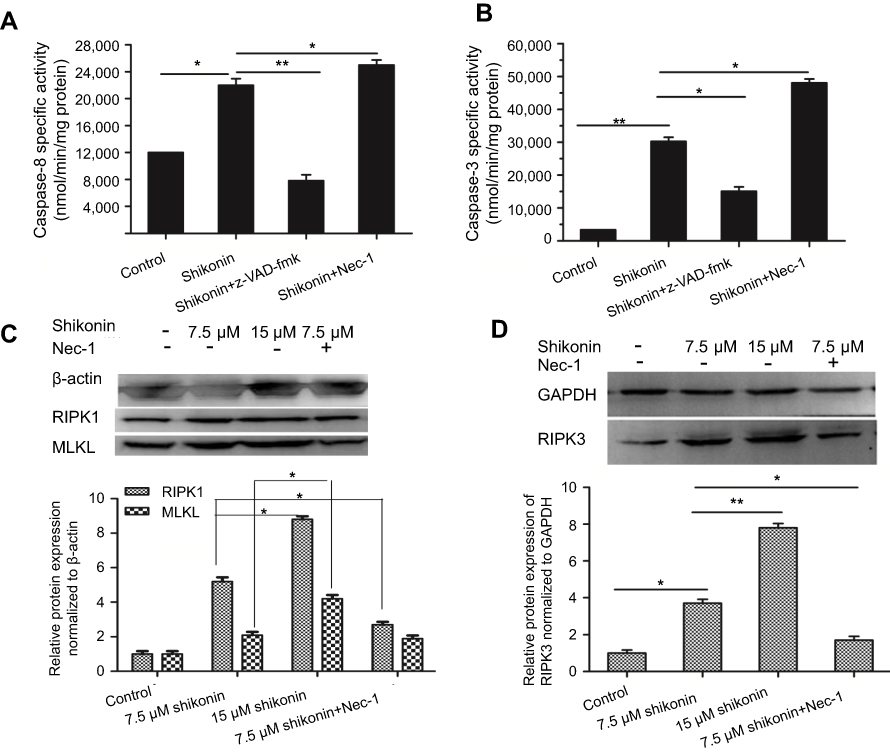 | Figure 2 (A and B) Activity of caspase-8 and caspase-3 in the presence of shikonin in 5-8F cells. 5-8F cells were treated with 7.5 μM shikonin in the absence or presence of 50 μM Nec-1 and 20 μM z-VAD-fmk for 6 h, whereas untreated cells were used as a control. Pretreatment with Nec-1 and z-VAD-fmk was performed for 3 h before shikonin treatment. (C and D) Western blotting analysis of the expression of RIPK1, RIPK3, and MLKL. *P<0.05 versus untreated cells; **P<0.01 versus the control group. |
Shikonin upregulated the expression of necroptosis-associated proteins RIPK1, RIPK3, and MLKL
To elucidate the molecular mechanisms underlying the inhibitory effects of shikonin on 5-8F cell viability, we examined the expression of RIPK1, RIPK3, and MLKL in 5-8F cells treated with different concentrations of shikonin. RIPK1, RIPK3, and MLKL were significantly upregulated in 5-8F cells treated with higher concentrations of shikonin (Figure 2C and D). To clarify the relationship between Nec-1 and the key proteins of necroptosis, we performed western blotting to examine the effects of Nec-1 on the expression of RIPK1, RIPK3, and MLKL. We found that pretreatment with 50 μM Nec-1 markedly decreased the levels of RIPK1, RIPK3, and MLKL when compared with those of the control group. This indicated that the expression of RIPK1, RIPK3, and MLKL in 5-8F cells was upregulated significantly by 7.5 μM shikonin and suppressed by pretreatment with Nec-1.
Involvement of ROS in shikonin-induced necroptosis
Compared to the control cells, cells treated with 7.5 μΜ shikonin showed significantly higher levels of ROS (Figure 3A). In addition, treatment with 50 μM Nec-1 and 5 mM NAC considerably inhibited the elevation of ROS levels. Furthermore, the fluorescence intensity of cells treated with shikonin alone was stronger than that of cells pretreated with antioxidant NAC. As shown in Figure 3B, pretreatment with NAC protected the cells from shikonin mediated cell death. We performed western blotting to examine the effects of NAC on the expression of proteins associated with necroptosis, RIPK1, RIPK3, and MLKL. We found that pretreatment with NAC attenuated the increase in RIPK1, RIPK3, and MLKL induced by shikonin treatment (Figure 3C and D). Taken together, our data suggest that treatment with shikonin resulted in the generation of large amounts of ROS, which led to necroptosis.
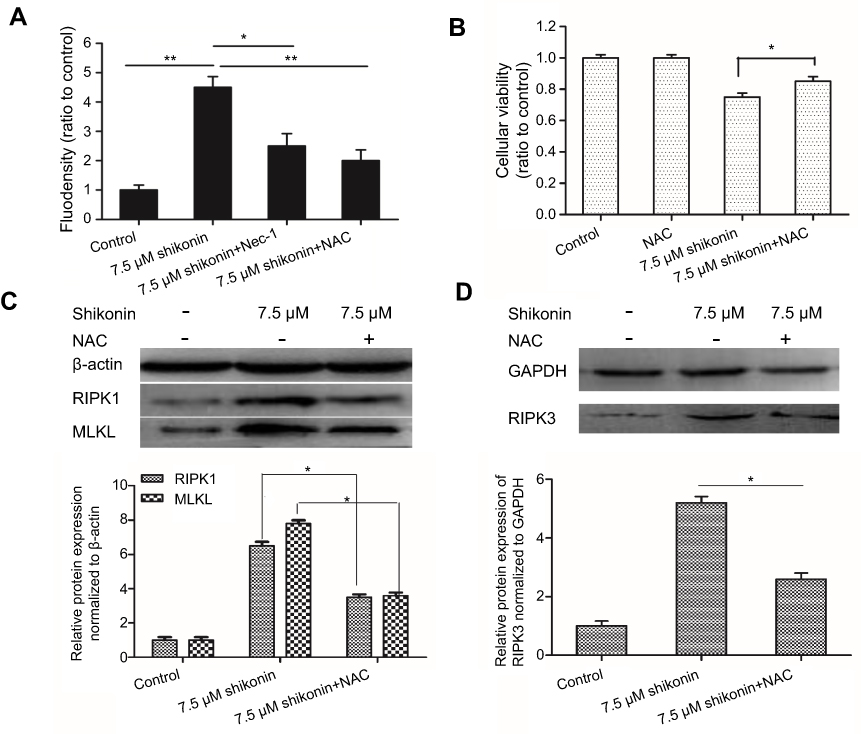 | Figure 3 (A) Shikonin mediated ROS production in 5-8F cells. (B) CCK-8 assay showed that the inhibitory effects of shikonin on the viability of 5-8F cells was suppressed significantly by pretreatment with N-acetyl-L-cysteine (NAC). (C and D) Pretreatment with NAC attenuated the shikonin-induced increase in RIPK1, RIPK3, and MLKL. *P<0.05 versus untreated cells; **P<0.01 versus the control group. |
In vivo antitumor effect of shikonin
In order to evaluate the antitumor effect of shikonin in vivo, we transplanted 5-8F cells into nude mice. Shikonin significantly inhibited the growth of the NPC tumors (Figure 4A and B) and markedly decreased the whole body weights of the treated nude mice (Figure 4C). In comparison with those of the negative control group, the tumor weights in the shikonin-treated group were significantly lower (Figure 4D). H&E staining of tumor tissue demonstrated that the degree of tumor necrosis in the treatment group (Figure 4F) was significantly higher than that in the control group (Figure 4E). Collectively, these results indicated that shikonin inhibited the growth of NPC in the 5-8F xenograft mouse model.
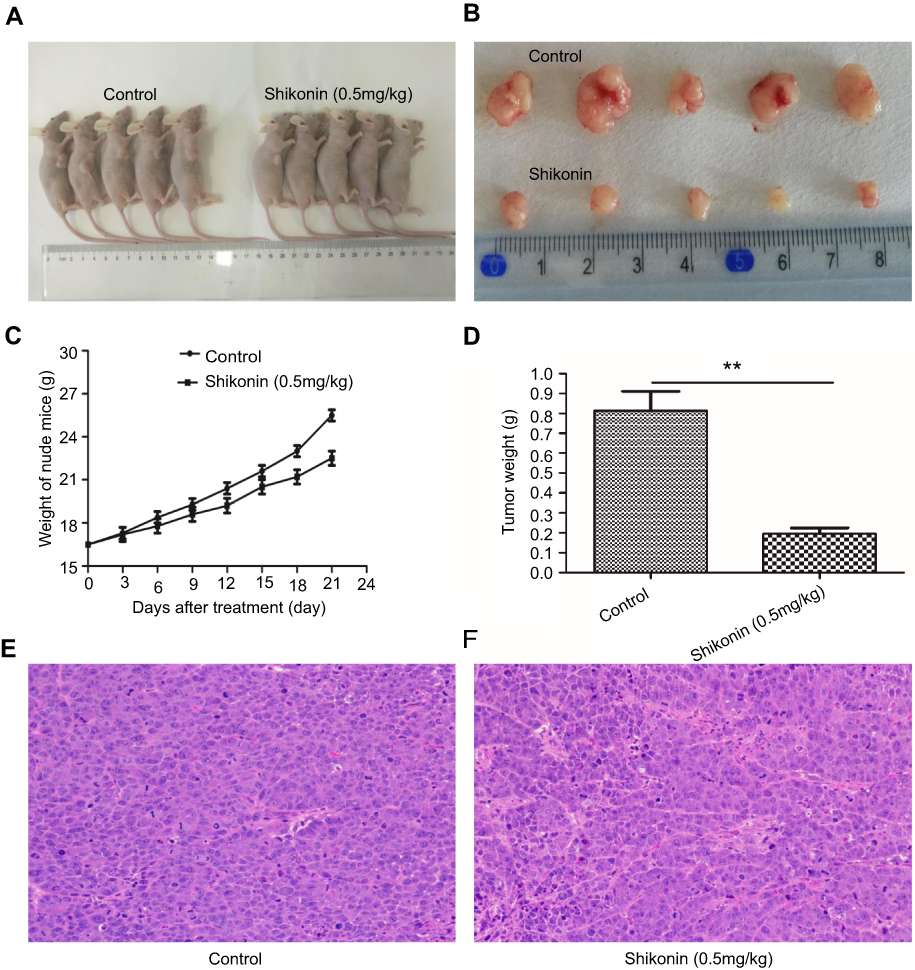 | Figure 4 Shikonin significantly inhibited tumor growth. (A) Photographs of tumor-bearing mice receiving different treatments. (B) Photographs of tumors under different treatments. (C) Plot showing the change in body weight of nude mice during treatment. (D) Weights of tumors after treatment. (E) Hematoxylin and eosin (H&E) staining of the control group (×200). (F) H&E staining of the test group (×200). *P<0.05 versus untreated group. |
Discussion
Accumulating evidence has demonstrated that complete treatment of advanced NPC is very challenging as a large group of NPC patients suffer from preexisting resistance to common chemotherapeutic agents and radiotherapeutic regimes. Fortunately, necroptosis induction effectively eliminates cancer cells that are resistant to apoptosis.16 Thus, necroptosis-based therapy is a promising option for NPC.
Morphology is an important factor in the evaluation of the mode of cell death. Transmission electron microscopy revealed that necroptosis is characterized by cell swelling, mitochondrial dysfunction, plasma membrane permeabilization, and the release of cytoplasmic content to the extracellular space.17 In light of these findings, the present investigation was conducted to evaluate the effects of shikonin on the induction of necroptosis. It is noteworthy that shikonin may provide a novel approach to overcome apoptosis-mediated drug resistance by inducing necroptosis.18
To determine the mode of 5-8F cell death induced by shikonin, we used the pan-caspase inhibitor z-VAD-fmk and Nec-1. The morphological findings suggested that shikonin induced dominant necroptotic death in 5-8F cells. Furthermore, when the apoptotic pathway was blocked by z-VAD-fmk, the necroptotic pathway became more dominant.13 On the contrary, when the necroptotic pathway was inhibited by Nec-1, apoptosis became the major route of cell death.13 Of note, shikonin exerted extremely low cytotoxicity in normal nasal epithelial cells in our previous study. Therefore, shikonin was considered an excellent candidate drug for the treatment of NPC. Targeting the weakness of cancer cells via the induction of necroptosis may accelerate cancer cell death or enhance the sensitivity of tumor cells to chemotherapeutic agents and radiotherapeutic regimes.
Our results support results from previous studies that have shown that RIPK1, RIPK3, and MLKL are crucial mediators of necroptosis.19,20 As expected, we found that the levels of RIPK1, RIPK3, and MLKL increased in 5-8F cells after treatment with shikonin. Overall, shikonin can induce necroptosis by regulating RIPK1/RIPK3/MLKL expression. In addition, Nec-1 inhibited the necroptosis of 5-8F cells by attenuating the increased expression of RIPK1/RIPK3/MLKL. Therefore, our results indicated that shikonin-induced necroptosis of NPC cells is associated with the upregulated expression of RIPK1/RIPK3/MLKL.
However, it remains unknown whether ROS participate in the regulation of signals leading to necroptosis mediated by RIPK1/RIPK3 necrosome formation. An elevation in intracellular ROS level and the destruction of mitochondria might result in the suppression of cancer cell migration, oxidative DNA damage, and arrest of the cell cycle.21,22 We have shown that shikonin induces the production of ROS in large quantities, which is followed by the disruption of mitochondrial transmembrane potential.23,24 Notably, we found that inhibition of ROS with the antioxidant NAC attenuated the protein levels of RIPK1, RIPK3, and MLKL. Our results are consistent with Huang et al who reported that the inhibition of ROS with NAC attenuated shikonin-induced necroptosis in glioma cells.4 Since scavenging ROS using NAC decreased the expression of RIPK1 and RIPK3, ROS might be upstream signals of RIPK1 and RIPK3. It is reasonable to shown that ROS not only are responsible for shikonin-induced NPC cell necroptosis but can also regulate the expression of RIPK1/RIPK3/MLKL. Lu reported that shikonin induced glioma cell necroptosis in vitro by ROS overproduction and ROS was the trigger that induces the activation of RIPK1/RIPK3 necrosome formation.23 Taken together,these findings suggests that ROS play a crucial role in the regulation of shikonin-induced necroptosis via the formation of positive feedback with RIPK1 and RIPK3.
Conclusion
The results revealed that shikonin can induce necroptosis by ROS overproduction and regulating the expression of RIPK1/RIPK3/MLKL. Thus, shikonin has the potential to be developed as a novel therapeutic agent to treat NPC. However, in-depth studies are required to determine its efficacy.
Ethics statement
All experiments were performed following the management system and operating rule for experimental animals at the China Medical University. All experiments were performed with the approval of the Ethics Committee of Shengjing Hospital of China Medical University (Shenyang, China).
Acknowledgments
This research was supported by the National Natural Science Foundation of China (grant no. 81200730). Thanks are due to X Wang and FX Meng for their technical assistance and to DB Zhou and DQ Jin for their valuable discussion.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chua ML, Wee JT, Hui EP, Chan ATC. Nasopharyngeal carcinoma. Lancet. 2016;387(10022):1012–1024. doi:10.1016/S0140-6736(15)00055-0
2. Peng H, Chen L, Zhang Y, et al. The tumour response to induction chemotherapy has prognostic value for long-term survival outcomes after intensity-modulated radiation therapy in nasopharyngeal carcinoma. Sci Rep. 2016;6:24835. doi:10.1038/srep24835
3. Tang X-R, Li Y-Q, Liang S-B, et al. Development and validation of a gene expression-based signature to predict distant metastasis in locoregionally advanced nasopharyngeal carcinoma: a retrospective, multicentre, cohort study. Lancet Oncol. 2018;19(3):382–393. doi:10.1016/S1470-2045(18)30080-9
4. Huang C, Luo Y, Zhao J, et al. Shikonin kills glioma cells through necroptosis mediated by RIP-1. PLoS One. 2013;8(6):e66326. doi:10.1371/journal.pone.0066326
5. Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370(5):455–465. doi:10.1056/NEJMra1310050
6. Humphries F, Yang S, Wang B, Moynagh PN. RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ. 2015;22(2):225–236. doi:10.1038/cdd.2014.126
7. Li J, McQuade T, Siemer AB, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150(2):339–350. doi:10.1016/j.cell.2012.06.019
8. Shahsavari Z, Karami-Tehrani F, Salami S. Targeting cell necroptosis and apoptosis induced by shikonin via receptor interacting protein kinases in estrogen receptor positive breast cancer cell line, MCF-7. Anticancer Agents Med Chem. 2018;18(2):245–254. doi:10.2174/1871520617666170919164055
9. Wang H, Sun L, Su L, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133–146. doi:10.1016/j.molcel.2014.03.003
10. He G-W, Günther C, Thonn V, et al. Regression of apoptosis-resistant colorectal tumors by induction of necroptosis in mice. J Exp Med. 2017;214(6):1655–1662. doi:10.1084/jem.20160442
11. Chen C, Xiao W, Huang L, et al. Shikonin induces apoptosis and necroptosis in pancreatic cancer via regulating the expression of RIP1/RIP3 and synergizes the activity of gemcitabine. Am J Transl Res. 2017;9(12):5507–5517.
12. Zhang X, Cui J-H, Meng Q-Q, Li S-S, Zhou W, Xiao S. Advance in anti-tumor mechanisms of shikonin, alkannin and their derivatives. Mini Rev Med Chem. 2018;18(2):164–172. doi:10.2174/1389557517666170228114809
13. Shahsavari Z, Karami-Tehrani F, Salami S. Shikonin induced necroptosis via reactive oxygen species in the T-47D breast cancer cell line. Asian Pac J Cancer Prev. 2015;16(16):7261–7266. doi:10.7314/APJCP.2015.16.16.7261
14. Tang X, Zhang C, Wei J, Fang Y, Zhao R, Yu J. Apoptosis is induced by shikonin through the mitochondrial signaling pathway. Mol Med Rep. 2016;13(4):3668–3674. doi:10.3892/mmr.2016.4967
15. Zhang Z, Zhang Z, Li Q, et al. Shikonin induces necroptosis by reactive oxygen species activation in nasopharyngeal carcinoma cell line CNE-2Z. J Bioenerg Biomembr. 2017;49(3):265–272. doi:10.1007/s10863-017-9714-z
16. Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016;23(2):253–260. doi:10.1038/cdd.2015.92
17. Vanden Berghe T, Kaiser WJ, Bertrand MJ, Vandenabeele P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol Cell Oncol. 2015;2(4):e975093. doi:10.4161/23723556.2014.975093
18. Wang Y, Hao F, Nan Y, et al. PKM2 inhibitor shikonin overcomes the cisplatin resistance in bladder cancer by inducing necroptosis. Int J Biol Sci. 2018;14(13):1883–1891. doi:10.7150/ijbs.27854
19. Rodriguez DA, Weinlich R, Brown S, et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016;23(1):76–88. doi:10.1038/cdd.2015.70
20. Wang H-Y, Zhang B. Cobalt chloride induces necroptosis in human colon cancer HT-29 cells. Asian Pac J Cancer Prev. 2015;16(6):2569–2574.
21. Zhou Z, Lu B, Wang C, et al. RIP1 and RIP3 contribute to shikonin-induced DNA double-strand breaks in glioma cells via increase of intracellular reactive oxygen species. Cancer Lett. 2017;390:77–90. doi:10.1016/j.canlet.2017.01.004
22. Shahsavari Z, Karami-Tehrani F, Salami S, Ghasemzadeh M. RIP1K and RIP3K provoked by shikonin induce cell cycle arrest in the triple negative breast cancer cell line, MDA-MB-468: necroptosis as a desperate programmed suicide pathway. Tumour Biol. 2016;37(4):4479–4491. doi:10.1007/s13277-015-4258-5
23. Lu B, Gong X, Wang Z-Q, et al. Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation. Acta Pharmacol Sin. 2017;38(11):1543–1553. doi:10.1038/aps.2017.112
24. Liang W, Cui J, Zhang K, et al. Shikonin induces ROS-based mitochondria-mediated apoptosis in colon cancer. Oncotarget. 2017;8(65):109094–109106. doi:10.18632/oncotarget.22618
Supplementary material
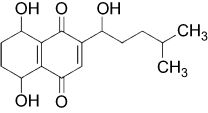 | Figure S1 Structure of shikonin. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.