")
Back to Journals » Drug Design, Development and Therapy » Volume 14
SGLT2 Inhibitors: A Novel Player in the Treatment and Prevention of Diabetic Cardiomyopathy
Received 26 June 2020
Accepted for publication 23 September 2020
Published 6 November 2020 Volume 2020:14 Pages 4775—4788
DOI https://doi.org/10.2147/DDDT.S269514
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Na Li, Hong Zhou
Department of Endocrinology, Second Hospital of Hebei Medical University, Shijiazhuang, People’s Republic of China
Correspondence: Hong Zhou
Department of Endocrinology, Second Hospital of Hebei Medical University, Shijiazhuang, People’s Republic of China
Tel +86-151-3011-9625
Email [email protected]
Abstract: Diabetic cardiomyopathy (DCM) characterized by diastolic and systolic dysfunction independently of hypertension and coronary heart disease, eventually develops into heart failure, which is strongly linked to a high prevalence of mortality in people with diabetes mellitus (DM). Sodium–glucose cotransporter type2 inhibitors (SGLT2Is) are a novel type of hypoglycemic agent in increasing urinary glucose and sodium excretion. Excitingly, the EMPA-REG clinical trial proved that empagliflozin significantly reduced the relative risk of cardiovascular (CV) death and hospitalization for heart failure (HHF) in patients with type 2 DM (T2DM) plus CV disease (CVD). The EMPRISE trial showed that empagliflozin decreased the risk of HHF in T2DM patients with and without a CVD history in routine care. These beneficial effects of SGLT2Is could not be entirely attributed to glucose-lowering or natriuretic action. There could be potential direct mechanisms of SGLT2Is in cardioprotection. Recent studies have shown the effects of SGLT2Is on cardiac iron homeostasis, mitochondrial function, anti-inflammation, anti-fibrosis, antioxidative stress, and renin–angiotensin–aldosterone system activity, as well as GlcNAcylation in the heart. This article reviews the current literature on the effects of SGLT2Is on DCM in preclinical studies. Possible molecular mechanisms regarding potential benefits of SGLT2Is for DCM are highlighted, with the purpose of providing a novel strategy for preventing DCM.
Keywords: diabetes, diabetic cardiomyopathy, SGLT2 inhibitor, cardioprotection, molecular mechanism
Introduction
Diabetes mellitus (DM) and related cardiovascular (CV) diseases are growing at an astonishing speed. The Framingham Heart Study suggested that the risk of heart failure (HF) independently increases up to twofold in men and fivefold in women compared with age-matched control groups in DM.1 Even after adjusting for other risk factors, such as age, hypertension, hypercholesterolemia, and coronary heart disease, there is still an increase in the incidence of HF in patients with DM. Diabetic cardiomyopathy (DCM) is defined as myocardial structural and functional abnormalities in the absence of coronary artery disease, hypertension, and other cardiac valvular disease in DM.2 The main basic pathological changes of DCM include cardiomyocyte apoptosis, hypertrophy, fibrosis, and microvascular lesions combined with oxidative stress and inflammation. Progression of DCM can be divided into three stages. Initially, it manifests as a subclinical status characterized by cardiac structural abnormalities without functional changes. Then, these structural abnormalities of myocardial fibrosis and related diastolic dysfunction gradually appear in DM, which often progresses to HF with preserved ejection fraction. Finally, aggravated structural changes in the heart ultimately lead to systolic dysfunction and HF with decreased ejection fraction.3 Sodium–glucose cotransporter type 2 inhibitors (SGLT2Is) have a glucose-lowering effect via a specific renal action by increasing glucosuria independently of insulin. Therefore, they have extra benefits of weight loss and a low risk of hypoglycemia.4
Recent CV-outcomes trials have revealed an unexpectedly strong cardioprotective role of SGLT2Is in patients with type 2 DM (T2DM), who are at high risk of CV diseases (CVDs).5–7 Empagliflozin (EMPA-REG OUTCOME, canagliflozin (CANVAS Program), and dapagliflozin (DECLARE-TIMI 58) trials all showed a reduction in CV mortality and hospitalization for HF (HHF) in patients with T2DM within months of taking SGLT2Is.6 The EMPRISE trial also demonstrated that empagliflozin decreased risk of HHF in T2DM patients treated in routine care with or without CVD history.8 As such, SGLT2Is represent the first class of therapeutic hypoglycemic agents that could decrease the risk of HHF in patients with T2DM. The beneficial cardiac effects of SGLT2Is are partly due to metabolic improvement, such as reduction in glycated hemoglobin (HbA1c), body weight, uric acid, and visceral fat, or to hemodynamic improvement, including lowering blood pressure, osmotic diuresis, and reduced cardiac load.9,10 However, the huge cardiac benefits of SGLT2Is cannot be explained solely by these changes. Firstly, beneficial effects of SGLT2Is on HF are seen even among patients without T2DM.11 Secondly, some antidiabetic drugs with greater decreases in HbA1c show no cardioprotective effects.12 In addition, they cannot be ascribed to natriuretic effect, since diuretic therapy has been associated with increased risk of CVD and mortality in patients with HF.13–16 As a consequence, accumulating evidence has been gathered in an attempt to figure out the possible underlying mechanisms of cardioprotection. In view of the rapid decline in risk of HHF, it can be speculated that the benefits of SGLT2Is are due to the direct effects on the heart itself, namely improved DCM, instead of metabolic improvement. In this review, cardiac structural and functional improvements and related direct and potential molecular mechanisms of SGLT2Is for the treatment and prevention of DCM are summarized.
Diabetic Cardiomyopathy
An important complication in DM, DCM is a specific CM that causes high mortality and disability rates and seriously affects quality of life in patients with DM. The definition of DCM is based on changes in cardiac structure and systolic and diastolic function independently of coronary artery disease, hypertension, and severe valvular disease in patients with DM.2 It was first found in the autopsy results of four diabetic patients who showed obvious symptoms of HF in the absence of coronary artery disease and valvular disease. In its subclinical status, DCM is asymptomatic and only has structural abnormalities that cause left ventricle (LV) stiffness and hypertrophy. Then, it progresses to diastolic dysfunction. The main manifestation in the heart is an obvious rise in atrial filling and isovolumetric relaxation time and a decline in early diastolic filling.17 The development of systolic dysfunction occurs before LV dilation and symptomatic HF.17 Cardiomyocyte hypertrophy, apoptosis, fibrosis, and cardiac microvascular injury all lead to these cardiac functional and structural changes, which eventually contribute to clinical intractable HF and even malignant arrhythmia, cardiogenic shock.
Clinical trials have demonstrated that incidence of HF in patients with DM fluctuated from 19% to 26%.18–20 Another study showed the incidence of HF in diabetic patients (39%) was higher than that in nondiabetic patients (23%), and that after 43 months of observation the relative risk for developing HF was 1.3.21 A population-based observational study showed patients with DM had an increased diastolic and systolic dysfunction and also increased LV mass and LV-wall thickness.22–24 However, intensive glucose control cannot produce the expected benefits in CV complications in patients with DM. Other kinds of glucose-lowering drugs have no decline on the risk of HF events, even though they have greater glucose-lowering effects.14 Furthermore, a new trial (DAPA-HF) showed that dapagliflozin exerts a beneficial effect on the progression of HF in patients without DM.25 Therefore, there could be other potential mechanisms associated with improvement in CM.
The pathophysiology of DCM is quite complex, and potential mechanisms are still under investigation. Insulin resistance, hyperglycemia, and hyperlipidemia are key points in the pathophysiology of DCM. Lipid-metabolism disorders do great harm to the heart and often trigger cardiac dysfunction in DM patients. Excess fatty-acid uptake and lipotoxicity lead to cardiomyocyte steatosis and hypertrophy with the deposition of triacylglycerols.26 In addition, increased fatty-acid oxidation leads to more oxygen uptake and reactive oxygen species (ROS) and reactive nitrogen species overproduction.26 Calcium-handling dysfunction, mitochondrial dysfunction, microvascular dysfunction, increasing oxidative stress and inflammation, endoplasmic reticulum stress (ERS), activation of the renin–angiotensin–aldosterone system (RAAS), cardiac sympathetic nerve activity, and reduced nitric oxide bioavailability have all been involved in the development of DCM3 (Figure 1). At present, there are no effective preventive or therapeutic measures for DCM. The cardioprotective effects of glucose-lowering drugs are drawing attention. Interestingly, several glucose-lowering drugs in use, such as GLP1-receptor agonists and SGLT2Is, have been demonstrated to have beneficial effects on cardiac protection beyond control of blood glucose. Currently, more basic studies are focused on the molecular mechanisms of cardioprotection with these glucose-lowering drugs, in order to attenuate DCM.
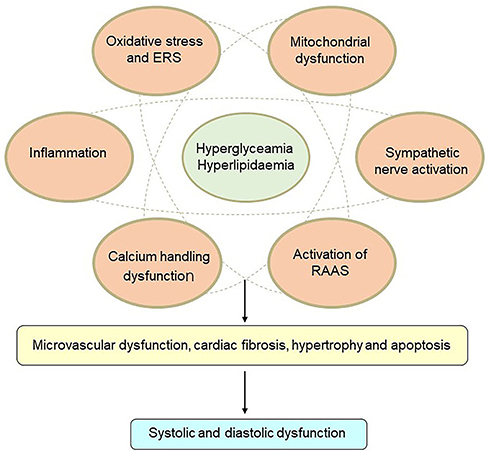 |
Figure 1 Mechanisms of diabetic cardiomyopathy. Hyperglycemia and hyperlipidemia induce metabolic changes in the heart that cause mitochondrial dysfunction, oxidative stress, inflammation, and endoplasmic reticulum (ER) stress in cardiomyocytes. Oxidative stress, ER stress, and inflammation can trigger the renin–angiotensin–aldosterone system (RAAS), enhance cardiac sympathetic nerve activity, and calcium-handling dysfunction. These changes mediate cardiac hypertrophy, apoptosis, fibrosis, and microvascular dysfunction, resulting in diastolic and systolic dysfunction. |
SGLT2 Inhibitors
There are two forms of SGLT expressed in the kidney: SGLT1 and SGLT2. In healthy conditions, all the filtered glucose (~180 g/day) is reabsorbed in the renal proximal tubule. SGLT2 is located in the early part of the proximal tubule (S1) and accounts for 80%–90% of filtered glucose reabsorption. SGLT1 is located in the later part of the proximal tubule (S2/S3) and accounts for the remaining 10%–20% of filtered glucose reabsorption.27–31 As a consequence, SGLT1 and SGLT2 are together in charge of glucose reabsorption in the renal proximal tubule under normal physiological conditions.27 SGLT2 is primarily detected in the kidney, while SGLT1 is expressed mainly in the kidney and gut, with a small amount expressed in lungs and the heart.31 In T1DM and T2DM, SGLT2 expression in the proximal tubule of the kidney is increased, which is consistent with the animal model of DM.32–36 Therefore, the regulation of SGLT2 has been confirmed as a new effective target for treatment of DM. Indeed, selective SGLT2Is prevent major reabsorption of glucose and increase urinary glucose and sodium excretion, thereby decreasing plasma-glucose levels in an insulin-independent manner and leading to a low risk of hypoglycemia.4,37 (Figure 2). Furthermore, the use of SGLT2Is can moderately reduce body weight, uric acid, and systolic and diastolic blood pressure in DM.38 Common adverse reactions of SGLT2Is include urinary tract infection, hypovolemia, and ketoacidosis. Among these, the risk of reproductive tract infection is completely controllable and the incidence of ketoacidosis very low. SGLT2Is available in China include canagliflozin, dapagliflozin, and empagliflozin. Other SGLT2Is include ipragliflozin, luseogliflozin, tofogliflozin, remogliflozin, sotagliflozin, bexagliflozin, and ertugliflozin. Notably and interestingly, empagliflozin has higher selectivity to bind to SGLT2 than SGLT1 (2,500-fold) than dapagliflozin (1,200-fold) and canagliflozin (413-fold).39
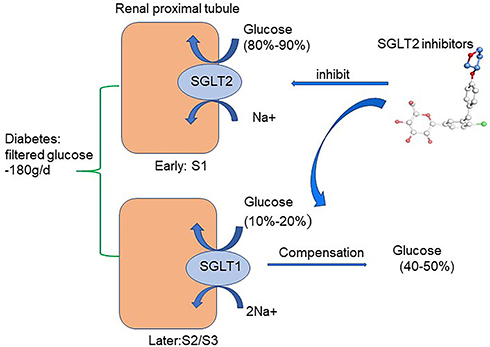 |
Figure 2 Glucose-lowering mechanisms of SGLT2 inhibitors. The renal proximal tubule accounts for the absorption of all the filtered glucose (~180 g/day) while SGLT2, which is located in the early part of the proximal tubule (S1), accounts for the 80%–90% of filtered glucose reabsorption. Therefore, SGLT2Is prevent major reabsorption (80–90%) of filtered glucose in the early proximal tubule and increase urinary glucose excretion. |
Large clinical trials have demonstrated that SGLT2Is have protective effects on the heart in patients with T2DM. EMPA-REG OUTCOME showed that empagliflozin may reduce the risk of CV mortality, all-cause mortality, and HHF by 38%, 32%, and 35%, respectively, during treatment of 3.1 years in 7,020 patients with T2DM who were at high CV risk.5 CANVAS showed canagliflozin lowered the 3P-MACE primary end point by 14% and incidence of HHF by 33% in T2DM patients.6 The mechanisms of cardioprotective effect of SGLT2Is are not fully clear. It is unlikely that HHF events are reduced by SGLT2Is due to mitigating coronary atherosclerosis, since these drugs do not decrease the risk of myocardial infarction and stoke.5 This benefit for HF could not be attributed to their promoting sodium excretion, since they produce decreased natriuretic peptides only modestly, and diuretics do not have favorable effects on the evolution of HF. Recently, DECLARE-TIMI 58 also demonstrated that another SGLT2I named dapagliflozin decreased the incidence of HHF and composite 3P-MACE primary end point by 17%.7 EMPRISE demonstrated that compared with sitagliptin, treatment with empagliflozin decreased risk of HHF among T2DM patients with or without CVD.8 It has been reported that SGLT2Is prevents deterioration of cardiac function in various models of HF.40–42 These studies suggest that SGLT2Is play an important role in prevention of DCM. It is very important to understand potential mechanisms of SGLT2Is in DCM to find new treatment targets in patients with DCM.
Potential Mechanisms of SGLT2 Inhibitors in DCM
As known, SGLT2 is located mainly in the renal proximal tubule, responsible for absorption of glucose. SGLT2 mRNA has also been detected in testes, cerebral arteries, and the cerebellum.43,44 However, SGLT2 is not expressed in normal or failing myocardium,45 indicating direct benefits of SGLT2Is for hearts is not through inhibition of SGLT2.
SGLT2 Inhibitors and Ion Homeostasis in DCM
Myocardial Ca2+ and Na+ homeostasis are essential for cardiomyocyte energy production and respiration, proper cardiac signal transduction, and heart-rhythm regulation.46,47 Ca2+ homeostasis is mainly regulated by ion channels and exchangers, such as the Na+–Ca2+ exchanger (NCX), L-type Ca2+ channels, ryanodine receptors, and sarcoplasmic reticulum calcium ATPase 2a (SERCA2a), which is responsible for contraction and relaxation of cardiomyocytes.46–48 Meanwhile, Na+ homeostasis is usually in charge of the Na+–H+ exchanger (NHE), NCX, and the Na+–K+ pump, which directly affects cardiomyocyte Ca2+ concentrations.49,50 Both calcium and sodium concentrations are changed in DCM.49,51–53 Changes in Na+ transport in the diabetic heart are attributed to declined NCX activities but increased NHE activity, leading to excess cytoplasmic Na+.53–58 Increased intracellular Na+ and Ca2+ lead to mitochondrial dysregulation, energy imbalance, excess ROS production, and cardiac hypertrophy and fibrosis.59
Recent studies have suggested that SGLT2Is have an important effect on ion homeostasis in the diabetic heart (Figure 3). A study demonstrated that 1–10µM dapagliflozin increased the amplitude of shortening and Ca2+ transience in cardiomyocytes of streptozotocin (Stz)-induced diabetic rats.60 Dapagliflozin decreased only cardiomyocyte systolic Ca2+, with no change in intracellular diastolic Ca2+, while there was no such effect in the diabetic group. These changes happened after only 5 minutes’ intervention with dapagliflozin, indicating dapagliflozin exerted acute negative inotropic effects on diabetic cardiomyocytes through the mechanisms of Ca2+ transport.60 Baartscheer et al demonstrated that empagliflozin (0.25–1 µM) reduced cardiomyocyte sodium ([Na+]c) and calcium ([Ca2+]c) and increase mitochondrial calcium ([Ca2+]m) independently of SGLT2 inhibition itself through inhibition of NHE activity.61 With the exposure of 5 mM and 11 mM glucose, empagliflozin both inhibited NHE and reduced [Na+]c, which supported the view that changes in extracellular glucose concentration had no relationship with these direct effects in cardiomyocytes by empagliflozin. In addition, dapagliflozin (1 µM) and canagliflozin (3 µM) also have a direct cardiac effect by decreasing NHE1 activity and reducing [Na+]c, indicating a direct class effect of SGLT2Is on cardiomyocytes.62 The interaction between SGLT2Is and NHE1 was further proved by molecular binding study, which showed that these SGLT2Is had strong binding capacity with the extracellular Na+-binding point of NHE.62 SGLT2Is may have an extra off-target effect on NHE1, which could explain the CV benefits. A subsequent study showed that dapagliflozin (0.4 μM) attenuated the upregulation of cardiac NHE1 also in lipopolysaccharide- induced mouse cardiac fibroblasts through AMPK activation.63 Therefore, the impact of SGLT2Is on NHE may be different in different cardiac cells. Two studies recently showed SGLT2Is improve diastolic function in a T2DM mouse model. The first study showed that empagliflozin improved Ca2+ homeostasis by increasing phospholamban phosphorylation and enhancing SERCA2α function, thus further contributing to improvement in cardiac relaxation and diastolic function in diabetic mice.64 Since calcium plays an important role in activating cardiac hypertrophic signaling pathways, including MAPKs, empagliflozin also activated the three MAPK-signaling pathways in diabetic hearts by modulation of intracellular Ca2+, leading to cardiac hypertrophy.64,65 In the second study, empagliflozin ameliorated LV diastolic function by decreasing CaMKII Thr286 activation with subsequent lower phosphorylation of ryanodine receptors in a model of DCM, indicating that improvement in diastolic function was likely mediated by reduced spontaneous diastolic sarcoplasmic reticulum calcium leakage.66
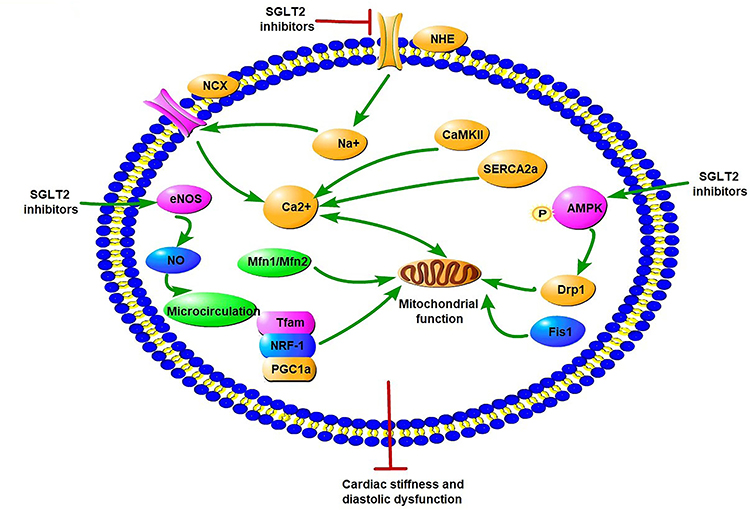 |
Figure 3 Potential molecular mechanisms of SGLT2 inhibitors on iron homeostasis, mitochondrial function, and cardiac microvasculature in diabetic cardiomyopathy. SGLT2Is can reduce cardiomyocyte sodium (Na+) and calcium (Ca2+) by inhibition of NHE activity in cardiomyocytes. In addition, SGLT2Is also regulate Ca2+ through enhancing SERCA2α function and CaMKII activation. SGLT2Is can enhance mitochondrial function by improvement of mitochondrial fusion–fission proteins, such as Mfn1:Mfn2 ratio and Fis1, as well as Drp1, which is dependent of AMPK activation. Activation of the PGC1α–NRF1–Tfam signaling pathway by SGLT2Is play a crucial role in regulation of mitochondrial biogenesis in the diabetic heart. Microcirculation can be improved by SGLT2Is by eNOS phosphorylation and NO-dependent improvement of endothelial function. All these changes have beneficial effects on attenuation of cardiac stiffness and diastolic dysfunction |
SGLT2 Inhibitors and Mitochondrial Function in DCM
Mitochondria are important for the maintenance of cardiac physiological function by their actions in ROS production, Ca2+ handling, cell apoptosis, and energy production.67 The mechanisms of cardiac mitochondrial dysfunction in DCM involve altered energy metabolism, oxidative stress, impaired mitochondrial calcium handling, cell apoptosis, and altered mitochondrial dynamics and biogenesis.68 Mitochondria are dynamic structures that maintain balance through constant fusion and division.69 Mitochondrial fusion contributes to the composition of normal mitochondria and maintenance of mitochondrial function. Mitochondrial fission is a mechanism for the elimination of dysfunctional mitochondria through autophagy in cells.70 Mitochondrial fusion is mediated by the interaction between Mfn1 and Mfn2 in the outer membrane of mitochondria and Opa1 in the inner membrane of mitochondria.71 On the other hand, mitochondrial division is recruited to specific sites of the outer membrane of mitochondria by the role of Drp1, Fis1, and MFF.72 Mitochondrial dysfunction, impaired dynamics, and cardiac contractile dysfunction are observed in diabetic patients.69
Accumulating evidence suggests that SGLT2Is may regulate mitochondrial function and dynamics in DCM (Figure 3). DM exhibits a marked expansion of myocardial mitochondrial ultrastructure, including intermyofibrillar mitochondria, decreasing matrix electron density, loss and fusion of cristae, and mitochondrial fragmentation in cardiac tissue, while these ultrastructural anomalies are improved by empagliflozin treatment.54 In a rat model of metabolic syndrome, dapagliflozin preserved depolarized mitochondrial membrane potential and suppressed prolonged ventricular repolarization and oxidative stress by improvement of mitochondrial fusion–fission proteins, eg, through normalization of the Mfn1:Mfn2 ratio and Fis1, beyond a glucose-lowering effect.73 Empagliflozin inhibits mitochondrial fission by increasing Drp1S637 phosphorylation and decreasing Drp1S616 phosphorylation through the activation of AMPK, ultimately leading to suppressed ROS and reserved cardiac microvascular function.74 Another study showed that empagliflozin inhibited mitochondrial fission by suppressing Fis1 upregulation after myocardial infarction in diabetic hearts and contributed to normalization of the size and number of mitochondria and promotion of autophagy in the diabetic rat heart. As a consequence, empagliflozin reduced diabetic myocardial infarction size.75 As such, SGLT2Is have an important role in regulation of mitochondrial dynamics, including mitochondria fusion and fission. However, the exact role of mitochondrial fission and fusion in the occurrence and progression of DCM remains unclear. In addition, the detailed molecular mechanism of SGLT2Is regulating mitochondrial fusion and fission is uncertain. It is still uncertain whether AMPK activation, one of the important regulators in mitochondrial fission, is a class effect of SGLT2Is or a drug-specific effect of empagliflozin. Future studies are needed.
As is known, DCM is related to high incidence of atrial fibrillation (AF). A recent study showed that empagliflozin can ameliorate atrial structural and electrical remodeling through improving mitochondrial respiratory function and mitochondrial biogenesis in a high-fat diet in Stz-induced diabetic rats.76 ThePGC1α–NRF1–Tfam signaling pathway plays a crucial role in regulation of mitochondrial biogenesis in diabetic rats.76 Empagliflozin upregulates PGC1α, NRF1, and Tfam in the diabetic heart. PGC1α is regulated by AMPK and conducted by NRF1 and Tfam.77,78 As such, empagliflozin prevents the atrial fibrillation inducibility in diabetic rats. Inhibition of atrial remodeling in DM may be a potential benefit in DCM.
SGLT2 Inhibitors and Cardiac Microvasculature in DCM
Coronary microvascular dysfunction plays an important role in development and progression of DCM. It influences coronary blood flow, cardiac blood perfusion, and diastolic and systolic function.79 Structural abnormalities of coronary artery microcirculation include inflammatory infiltration, lumen obstruction, perivascular fibrosis, and vascular remodeling.80 Regarding functional aspects, coronary microcirculation abnormalities include dysfunction of endothelial cells and smooth-muscle cells and ischemic reperfusion and dysfunction of vascular relaxation and constriction.80 Cardiac microvascular endothelial cells (CMECs), the main cells in cardiac microcirculation, suffer hyperglycemic injury much more easily than cardiomyocytes.81 The progression of DM contributes to depletion of CMEC viability and metabolic defects in vascular smooth-muscle cells. Thickening of the basement membrane in small coronary vessels and microvessels leads to the occurrence and development of coronary microvascular disease and myocardial fibrosis, as well as a decrease in myocardial perfusion, which affects diastolic and contractile functioning in the heart.82
In a recent study, empagliflozin had a direct protective effect on diabetic coronary microcirculation through two mechanisms: enhancement of endothelial function and decreased vascular remodeling. Empagliflozin improved eNOS phosphorylation and endothelium-dependent contraction and relaxation in cardiac microvasculature, thereby reducing endothelial inflammation and vascular permeability74,83 (Figure 3). In addition, empagliflozin improved cardiac microcirculation perfusion through increased microvessel density and lessened vascular remodeling.74 Empagliflozin also increased CMEC survival through inhibition of mitochondrial ROS production and oxidative stress.74 Through these changes, empagliflozin enhanced myocardial blood circulation and perfusion. Consistently with these results, Adingupu et al demonstrated that empagliflozin improved coronary microvascular function in terms of coronary flow–velocity reserve via NO-dependent improvement in endothelial function in prediabetic ob/ob−/− mice, since empagliflozin raised the L-arginine:ADMA ratio, a marker of endothelial function. Contractile function measured by fractional area change was also improved by empagliflozin.84 Another study reported CMEC and cardiomyocyte function in a novel coculture system. CMECs promoted cardiomyocyte contraction and relaxation. Inhibition of NO production in CMECs lessened the beneficial effect of CMECs on contraction and relaxation function of cardiomyocytes. Empagliflozin decreased excess mitochondrial and cytoplasmic ROS production in CMECs, thus enhancing NO bioavailability and leading to enhancement of cardiomyocyte relaxation and contraction.85 Then, a question was raised as to whether the molecular mechanisms of SGLT2Is on CMECs are specific to the inhibition of SGLT2. SGLT1 has been found in the heart, while SGLT2 has been found in the kidneys.86,87 Uthman et al showed that empagliflozin (1 µM) and dapagliflozin (1 µM) restored NO bioavailability by inhibiting ROS generation in TNFα-induced human coronary arterial endothelial cells (HCAECs) independently of SGLT2 inhibition, as SGLT2 mRNA was in fact not detectable in HCAECs.88 In order to clarify the molecular mechanism of SGLT2I effects on CMECs, further studies on gene knockdown by SGLT2 siRNA in CMECs or isolated endothelial cells are needed urgently.
SGLT2 Inhibitors and Oxidative and Endoplasmic Reticulum Stress in DCM
In DM, hyperglycemia and insulin resistance promote ROS production and activation of ERS, including dysfunction of Ca2+ handling and excess unfolded proteins.89 These metabolic abnormalities all lead to the occurrence and progression of DCM.90–92 The interaction of oxidative stress and ERS promotes cardiomyocyte apoptosis, necrosis, and autophagy, and thus affects the systolic and diastolic functions of the myocardium.17 Studies have reported the potential role of SGLT2Is as cardiac antioxidant modulators through inhibition of oxidative stress beyond their glucose-lowering effects.93–95 Suppression of oxidative stress by SGLT2Is leads to attenuation of cardiac fibrosis and hypertrophy in DM.93,96 Several studies have explored potential molecular mechanisms of antioxidative stress in SGLT2Is (Figure 4). First, changes in Zn2+ of cardiomyocytes were associated with antioxidative effects and attenuation of LV-developed pressure in insulin-resistant metabolic syndrome rats.97 Acute and chronic hyperglycemia both induced increased oxidative stress and rising levels of free Zn2+ in cardiomyocytes, which contributed to cardiac dysfunction.98,99 Dapagliflozin increased the protein levels of the Zn2+ transporters ZIP7 and ZIP14, which account for Zn2+ influx into cardiomyocytes. On the contrary, dapagliflozin decreased ZnT7, which is in charge of Zn2+ efflux in cardiomyocytes, thus resulting in Zn2+ upregulation in cardiomyocyte and cardiac antioxidative effects.97 NADPH oxidase is one of the main mechanisms of ROS production in the diabetic heart.100 Li et al showed that empagliflozin enhanced cardiac structure and function and alleviated excessive oxidative stress by reducing NOX4 and increasing Nrf2 and HO1 expression in cardiac tissue of diabetic mice. It seems that empagliflozin causes translocation of Nrf2 to the cell nucleus and activates the Nrf2–ARE signal to suppress oxidative stress in the diabetic heart.101
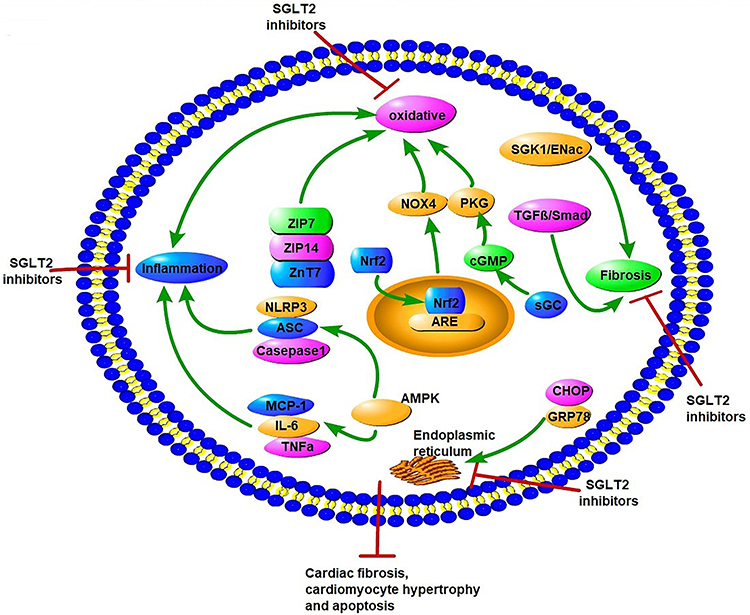 |
Figure 4 Potential molecular mechanisms of SGLT2 inhibitors on oxidative stress, endoplasmic reticulum stress, inflammation factors, and fibrosis in diabetic cardiomyopathy. SGLT2Is can inhibit oxidative stress by rising levels of free Zn2+ in cardiomyocytes, translocation of Nrf2 to the cell nucleus, activation of the Nrf2–ARE signal, and activation of the sGC–cGMP–PKG pathway. SGLT2Is also inhibit ERS through inhibition of CHOP and GRP78. Inflammation factors, such as NLRP3, ASC, caspase 1, IL6, TNFα, and MCP1, are all attenuated by SGLT2Is dependent of AMPK activation. SGLT2Is prevent cardiac fibrosis through the SGK1–ENac and TGFβ–Smad pathways. All these changes by SGLT2Is lead to attenuation of cardiomyocyte apoptosis, hypertrophy, and fibrosis. |
Nrf2 is a target of SGLT2Is in suppression of oxidative stress in DCM. Empagliflozin reverses these changes by activation of the sGC–cGMP–PKG pathway. Furthermore, with inhibition of the sGC–cGMP–PKG pathway by sGC siRNA in hearts, the cardiac benefits in DCM of empagliflozin are reversed.102 Consistently, a study proved that in cardiomyocytes from patients with HF with preserved ejection fraction and obese diabetic rats, empagliflozin reduced inflammatory and oxidative stress in both cardiomyocyte cytosol and mitochondria in HF with preserved ejection fraction and improved the NO–sGC–cGMP cascade and PKGIα activity via reduced PKGIα oxidation and polymerization, leading to reduced cardiomyocyte passive stiffness.103 This may lead to enhancement of cardiac diastolic function. These studies indicate that sGC–cGMP–PKG may be a promising therapeutic option for DCM. In addition to antioxidative stress, Zhou et al demonstrated empagliflozin protected against DCM by inhibition of the ERS pathway. Empagliflozin downregulated the expression of CHOP and GRP78, a marker reflecting activation of ERS, and decreased mRNA levels of ERS-associated factors, including ATF4, TRAF2, and XBP1, in the myocardia of diabetic rats. As a consequence, empagliflozin decreased caspase 3 expression and cardiomyocyte apoptosis, and eventually enhanced cardiac function in Stz-induced diabetic rats.104
SGLT2 Inhibitors and Inflammation Factors and Cardiac Fibrosis in DCM
Inflammatory response has been implicated in the development of DCM. Activation and expression of proinflammatory cytokines, such as IL6 and IL8, TNFα, MCP1, VCAM1, and ICAM1, all lead to cardiac oxidative stress, fibrosis, apoptosis, and cardiac dysfunction.17 These inflammatory responses exist in various kinds of cardiac cells, including cardiomyocytes, coronary endothelial cells, and cardiac fibroblasts.
Anti-inflammation effects of SGLT2Is play a role in protection against DCM. Several studies have demonstrated that SGLT2Is lessened cardiac macrophage infiltration and decreased inflammatory cytokines (TNFα and TGFβ) in diabetic mice.93,96,105 Insulin resistance and hyperglycemia can activate a molecular marker, NLRP3, in DCM, leading to activation of procaspase 1, IL1β, and IL18.106 A study proved that dapagliflozin attenuated NLRP3, ASC, IL1β, IL6, and caspase 1, as well as TNFα mRNA levels, markedly through the activation of AMPK in hearts of diabetic mice.107 In cardiac fibroblasts in vitro, dapagliflozin (0.3–0.5 μM) attenuated lipopolysaccharide-induced upregulation of the NLRP3 inflammasome, while phlorizin (an SGLT1 and SGLT2 inhibitor, 100 μM)) alone had no effect on the NLRP3 inflammasome, indicating that the anti-inflammation effects of dapagliflozin were independent of SGLT1 and SGLT2 inhibition.107 In a T2DM rat model, empagliflozin activated AMPK and promoted autophagy in the heart, consequently decreasing levels of cardiac proinflammatory cytokines, including IL6, TNFα, and MCP1, supporting the theory of a direct effect of empagliflozin in attenuation of cardiac inflammation.108 As such, AMPK activation may be a key point in inhibition of the anti-inflammatory effects of dapagliflozin. Moreover, dapagliflozin also activates another key molecule — mTOR — a mediator of survival and metabolism, leading to activation of Akt and FOXO3 and attenuating the progression of DCM.109 These effects suggestd that SGLT2Is can be beneficial for cardiac inflammation in DCM.
Beneficial effects of SGLT2Is on diastolic function can be mediated by improved antifibrotic effects. Empagliflozin attenuates cardiac fibrosis associated with the amelioration of cardiac oxidative stress and inflammation.93,96 Empagliflozin can inhibit the SGK1–epithelial sodium channel profibrosis signaling pathway, leading to decreased myocardial interstitial fibrosis in a female rodent model of DM.110 It has been reported that SGK1 is directly associated with cardiac profibrotic/hypertrophic effects and is highly expressed in the heart under hyperglycemia.111,112 Another study showed that empagliflozin suppressed cardiac fibrosis through inhibition of the TGFβ–Smad pathway in T2DM mice.101 Empagliflozin significantly attenuates TGFβ1-induced fibroblast activation and cell-mediated ECM remodeling, together with suppression of critical profibrotic markers, including COL1A1, ACTA2 (αSMA), CTGF, FN1, and MMP2 in vitro, which suggests antifibrotic effects with empagliflozin.113
SGLT2 Inhibitors and Renin–Angiotensin–Aldosterone System in DCM
The RAAS plays an important role in the pathogenesis of DCM. Inappropriate activation of the RAAS exacerbates cardiac remodeling, interstitial fibrosis, and diastolic dysfunction in DCM.114 As SGLT2Is contribute to cardiorenal protection in DM, the RAAS may be a potential target. SGLT2Is can result in natriuresis, which can trigger elevated plasma angiotensin II (AngII) as compensation for the sodium and water loss.115,116 The AngII levels by raised SGLT2Is could act through AngII type II receptors, thus leading to RAAS blockade and subsequent vasodilation, anti-inflammatory effects, and positive systolic effects.117,118 As such, these drugs may act via nonclassic RAAS pathways. However, the action of SGLT2Is on the renal RAAS is uncertain. Angiotensinogen (AGT) is an important biomarker that reflects renal RAAS activity. Intervention with elevated glucose increases AGT mRNA levels in cultured human proximal tubular cells.119 Therefore, SGLT2Is may lower blood-glucose levels and cause a decrease in AGT levels in human early proximal tubular cells. On the contrary, SGLT2Is can increase glucose excretion in the distal proximal tubule and lead to an increase in AGT production. The different effects of SGLT2Is on early proximal tubular and later proximal tubular cells determines the different impacts on renal RAAS. A study showed treatment with dapagliflozin expressively decreased urinary AngII and AGT levels in T2DM rats.120 Canagliflozin prevented intrarenal AGT upregulation in T2DM mice and mitigated renal inflammation and fibrosis.121 The evidence that SGLT2Is activate the intrarenal RAAS in DM is poor. Future study is needed to evolve better biomarkers for the intrarenal RAAS. RAAS inhibition of SGLT2Is presumably plays an important role in cardiorenal protection from DM that is the same as RAAS inhibitors.
Others
Decreased ketogenesis is often seen in T2DM related to insulin resistance and hyperinsulinemia.122 Ketones may play a crucial role in balancing energy metabolism in prevention of DCM, instead of glucose utilization.123 SGLT2Is can increase ketone concentration in both DM and non-DM.124,125 Increasing ketone levels promotes ketone oxidation and improves cardiac energy utilization in mice with DCM.126 It has been demonstrated that ketones, such as 3-hydroxybutyrate, can promote cardiac contractility and exert beneficial hemodynamic effects in HF.127 As such, SGLT2Is enhance ketone-body metabolism and have a beneficial effect in myocardium energy metabolism. SGLT2Is have shown favorable effects on lipid profile. An increase in LDL-C and HDL-C levels and a decrease in triglycerides and small, dense LDL levels have been seen by application of SGLT2Is.128 Furthermore, a study showed empagliflozin induced significant decrease in cardiac lipotoxicity, such as sphingolipids and glycerophospholipids, as well as the fatty-acid CD36 in T2DM rats.129 Lipid metabolism in the heart, which has a crucial role in the development of DCM and HF,130 is improved by empagliflozin.
Studies have shown that the density and number of sympathetic nerve fibers are significantly increased in diabetic hearts, suggesting overactivation of sympathetic nerves playing an important role in DCM.131,132 Overactivation of the sympathetic nervous system promotes cardiac hypertrophy, fibrosis, cardiomyocyte apoptosis, and cardiac dysfunction.133 Dapagliflozin decreases tyrosine-hydroxylase density and myocardial norepinephrine content, contributing to attenuation of myocardial damage and fibrosis, indicating that improved sympathetic nerve activity is a potential mechanism for the cardiac protection of SGLT2Is in DCM.105 Chronic activation of the hexosamine biosynthetic pathway and subsequent excessive O-GlcNAcylation is associated with DM and affects heart function in hyperglycemia. O-GlcNAcylation impairs cardiac Ca2+ homeostasis, mitochondrial function, and ERS by modulating key protein targets, such as phospholamban, calmodulin kinase II, and FOXO1, in the diabetic heart.134–137 Dapagliflozin prevents DCM by decreasing O-GlcNAcylated protein levels in diabetic lipodystrophic mice.138 The study establishing this specifically highlighted that O-GlcNAcylated levels of FOXO1 decreased by SGLT2Is contributed to improvement in cardiac function and attenuation of DCM.138 Targeting the O-GlcNAcylation of specific proteins by SGLT2Is seems to be a possible method in the prevention and treatment of DCM.
Conclusion
At present, there is no effective method to prevent the development of DCM. Simply glucose-lowering cannot prevent progression of DCM and subsequent HF. CV-outcome trials have revealed unexpectedly strong cardioprotective effects of SGLT2Is in patients with T2DM independently of glucose lowering. More and more studies are focusing on the direct potential mechanism of SGLT2Is on the heart itself in a hyperglycemic state. In this review, related studies on SGLT2Is in protection from and treatment of DCM were summarized. Restorative cardiac iron homeostasis, improved mitochondrial function, and microcirculation, anti-inflammation, antifibrosis, and antioxidative stress, enhanced ketone-body metabolism and beneficial metabolic effects, declined sympathetic nerve activity, O-GlcNAcylation, and RAAS inhibition are implicated in the cardioprotective effects of SGLT2Is. Among all the molecules, the AMPK pathway is the most important and closely related to SGLT2Is in DCM, which is activated by SGLT2Is in various kinds of cardiac cells, such as cardiomyocytes, cardiac fibroblasts, immune cells, and vascular endothelial cells.139 The mitochondrion is the determinant organelle in the heart for SGLT2Is actions. SGLT2Is improve impaired mitochondrial respiratory function, mitochondrial biogenesis, and mitochondrial ultrastructure and dynamics in DCM. It is possible that multiple mechanisms are involved in the action of SGLT2Is on DCM. Further studies are essential to improve our understanding of molecular mechanisms of SGLT2Is in DCM, which provides a new treatment strategy for DCM.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data, took part in drafting the article or revising it critically for important intellectual content, agreed to submit to the current journal, gave final approval to the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflict of interest.
References
1. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34(1):29–34. doi:10.1016/0002-9149(74)90089-7
2. Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30(6):595–602. doi:10.1016/0002-9149(72)90595-4
3. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018;122(4):624–638. doi:10.1161/CIRCRESAHA.117.311586
4. Abdul-Ghani MA, Norton L, Defronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev. 2011;32(4):515–531. doi:10.1210/er.2010-0029
5. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type2 diabetes. N Engl J Med. 2015;373:2117–2128.
6. Neal B, Perkovic V, Mahaffey KW, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644–657.
7. Wiviott SD, Raz I, Bonaca MP, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380(4):347–357.
8. Patorno E, Pawar A, Franklin JM, et al. Empagliflozin and the risk of heart failure hospitalization in routine clinical care: a first analysis from the empagliflozin comparative effectiveness and safety (EMPRISE) study. Circulation. 2019;139(25):2822–2830.
9. Gallo LA, Wright EM, Vallon V. Probing SGLT2 as a therapeutic target for diabetes: basic physiology and consequences. Diab Vasc Dis Res. 2015;12:78–79.
10. Scheen AJ. Cardiovascular effects of new oral glucose-Lowering agents DPP-4 and SGLT-2 inhibitors. Circ Res. 2018;122(10):1439–1459. doi:10.1161/CIRCRESAHA.117.311588
11. McMurray JJV, Solomon SD, Inzucchi SE, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381(21):1995–2008.
12. Packer M. Heart failure: the most important, preventable, and treatable cardiovascular complication of type 2 diabetes. Diabetes Care. 2018;41(1):11–13. doi:10.2337/dci17-0052
13. Packer M. Autophagy stimulation and intracellular sodium reduction as mediators of the cardioprotective effect of sodium–glucose cotransporter 2 inhibitors. Eur J Heart Fail. 2020;22(4):618–628. doi:10.1002/ejhf.1732
14. Packer M. Autophagy-dependent and -independent modulation of oxidative and organellar stress in the diabetic heart by glucose-lowering drugs. Cardiovasc Diabetol. 2020;19(1):62. doi:10.1186/s12933-020-01041-4
15. Packer M. Critical examination of mechanisms underlying the reduction in heart failure events with SGLT2 inhibitors: identification of a molecular link between their actions to stimulate erythrocytosis and to alleviate cellular stress. Cardiovasc Res. 2020;cvaa064. doi:10.1093/cvr/cvaa064.
16. Packer M. Role of deranged energy deprivation signaling in the pathogenesis of cardiac and renal disease in states of perceived nutrient overabundance. Circulation. 2020;141(25):2095–2105. doi:10.1161/Circulation.119.045561.
17. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. 2016;12(3):144–153. doi:10.1038/nrendo.2015.216
18. Rydén L, Armstrong PW, Cleland JG, et al. Efficacy and safety of high-dose lisinopril in chronic heart failure patients at high cardiovascular risk, including those with diabetes mellitus. Results from the ATLAS trial. Eur Heart J. 2000;21(23):1967–1978. doi:10.1053/euhj.2000.2311
19. Shindler DM, Kostis JB, Yusuf S, et al. Diabetes mellitus, a predictor of morbidity and mortality in the studies of left ventricular dysfunction (SOLVD) trials and registry. Am J Cardiol. 1996;77(11):1017–1020. doi:10.1016/S0002-9149(97)89163-1
20. Thrainsdottir IS, Aspelund T, Thorgeirsson G, et al. The association between glucose abnormalities and heart failure in the population-based Reykjavik study. Diabetes Care. 2005;28(3):612–616. doi:10.2337/diacare.28.3.612
21. Aronow WS, Ahn C. Incidence of heart failure in 2737 older persons with and without diabetes mellitus. Chest. 1999;115(3):867–868. doi:10.1378/chest.115.3.867
22. Lee M, Gardin JM, Lynch JC, et al. Diabetes mellitus and echocardiographic left ventricular function in free-living elderly men and women: the cardiovascular health study. Am Heart J. 1997;133(1):36–43. doi:10.1016/S0002-8703(97)70245-X
23. Devereux RB, Roman MJ, Paranicas M, et al. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation. 2000;101(19):2271–2276. doi:10.1161/01.CIR.101.19.2271
24. Bertoni AG, Goff DC
25. McMurray JJ, Solomon SD, Inzucchi SE, et al. DAPA-HF trial committees and investigators. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381(21):1995–2008. doi:10.1056/NEJMoa1911303
26. Fuentes-Antrás J, Picatoste B, Ramírez E, et al. Targeting metabolic disturbance in the diabetic heart. Cardiovasc Diabetol. 2015;14(1):17. doi:10.1186/s12933-015-0173-8
27. Wright EM, Turk E. The sodium/glucose cotransport family SLC5. Pflugers Arch. 2004;447(5):510–518. doi:10.1007/s00424-003-1202-0
28. Norton L, Shannon CE, Fourcaudot M, et al. Sodium-glucose cotransporter (SGLT) and glucose transporter (GLUT) expression in the kidney of type 2 diabetic subjects. Diabetes Obes Metab. 2017;19(9):1322–1326. doi:10.1111/dom.13003
29. Vallon V, Platt KA, Cunard R, et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol. 2011;22(1):104–112.
30. Lee YJ, Lee YJ, Han HJ. Regulatory mechanisms of Na (+)/glucose cotransporters in renal proximal tubule cells. Kidney Int Suppl. 2007;72:27–35. doi:10.1038/sj.ki.5002383
31. Abdul-Ghani MA, Norton L, DeFronzo RA. Renal sodium-glucose cotransporter inhibition in the management of type 2 diabetes mellitus. Am J Physiol Ren Physiol. 2015;309(11):889–900. doi:10.1152/ajprenal.00267.2015
32. Vestri S, Okamoto MM, de Freitas HS, et al. Changes in sodium or glucose filtration rate modulate expression of glucose transporters in renal proximal tubular cells of rat. J Membr Biol. 2001;182(2):105–112. doi:10.1007/s00232-001-0036-y
33. Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes. 2005;54(12):3427–3434. doi:10.2337/diabetes.54.12.3427
34. Tabatai NM, Sharma M, Blumenthal SS, Petering DH. Enhanced expressions of expressions of sodium-glucose cotransporters in the kidneys of diabetic Zucker rats. Diabetes Res Clin Pract. 2009;83(1):e27–30. doi:10.1016/j.diabres.2008.11.003
35. Vallon V. The mechanisms and therapeutic potential of SGLT2 inhibitors in diabetes mellitus. Annu Rev Med. 2015;66(1):255–270. doi:10.1146/annurev-med-051013-110046
36. Vallon V, Gerasimova M, Rose MA, et al. SGLT2 inhibition empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol. 2014;306(2):194–204. doi:10.1152/ajprenal.00520.2013
37. Chao EC, Henry RR. SGLT2 inhibition–a novel strategy for diabetes treatment. Nat Rev Drug Discov. 2010;9(7):551–559. doi:10.1038/nrd3180
38. Kaplan A, Abidi E, El-Yazbi A, Eid A, Booz GW, Zouein FA. Direct cardiovascular impact of SGLT2 inhibitors: mechanisms and effects. Heart Fail Rev. 2018;23(3):419–437. doi:10.1007/s10741-017-9665-9
39. Grempler R, Thomas L, Eckhardt M, et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012;14(1):83–90. doi:10.1111/j.1463-1326.2011.01517.x
40. Byrne NJ, Parajuli N, Levasseur JL, et al. Empagliflozin prevents worsening of cardiac function in an experimental model of pressure overload-induced heart failure. JACC Basic Trans Sci. 2017;2(4):347–354. doi:10.1016/j.jacbts.2017.07.003
41. Borghetti G, von Lewinski D, Eaton DM, Sourij H, Houser SR, Wallner M. Diabetic cardiomyopathy: current and future therapies beyond glycemic control. Front Physiol. 2018;9:1514.
42. Shi X, Verma S, Yun J. Effect of empagliflozin on cardiac biomarkers in a zebrafish model of heart failure: clues to the EMPA-REG OUTCOME trial? Mol Cell Biochem. 2017;433(1):97–102. doi:10.1007/s11010-017-3018-9
43. Chen J, Williams S, Ho S, et al. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010;1(2):57–92. doi:10.1007/s13300-010-0006-4
44. Vrhovac I, Balen Eror D, Klessen D, et al. Localizations of Na(+)-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch. 2015;467(9):1881–1898.
45. Di Franco A, Cantini G, Tani A, et al. Sodium-dependent glucose transporters (SGLT) in human ischemic heart: a new potential pharmacological target. Int J Cardiol. 2017;15(243):86–90. doi:10.1016/j.ijcard.2017.05.032
46. Shattock MJ, Ottolia M, Bers DM, et al. Na+/Ca2+ exchange and Na+/K+ ATPase in the heart. J Physiol. 2015;593(6):1361–1382. doi:10.1113/jphysiol.2014.282319
47. Kho C, Lee A, Hajjar RJ. Altered sarcoplasmic reticulum calcium cycling—targets for heart failure therapy. Nat Rev Cardiol. 2012;9(12):717–733.
48. Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415(6868):198–205. doi:10.1038/415198a
49. Despa S, Bers DM. Na (+) transport in the normal and failing heart—remember the balance. J Mol Cell Cardiol. 2013;61:2–10.
50. Cingolani HE, Ennis IL. Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation. 2007;115(9):1090–1100.
51. Lambert R, Srodulski S, Peng X, Margulies KB, Despa F, Despa S. Intracellular Na+ concentration ([Na+]i) is elevated in diabetic hearts due to enhanced Na+-glucose cotransport. J Am Heart Assoc. 2015;4(9):e002183.
52. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. 2014;57(4):660–671.
53. Hattori Y, Matsuda N, Kimura J, et al. Diminished function and expression of the cardiac Na+-Ca2+ exchanger in diabetic rats: implication in Ca2+ overload. J Physiother. 2000;527(Pt 1):85–94.
54. Anzawa R, Bernard M, Tamareille S, et al. Intracellular sodium increase and susceptibility to ischaemia in hearts from type 2 diabetic db/db mice. Diabetologia. 2006;49(3):598–606.
55. Chattou S, Diacono J, Feuvray D. Decrease in sodium calcium exchange and calcium currents in diabetic rat ventricular myocytes. Acta Physiol Scand. 1999;166(2):137–144.
56. Darmellah A, Baetz D, Prunier F, Tamareille S, Rucker-Martin C, Feuvray D. Enhanced activity of the myocardial Na+/H+ exchanger contributes to left ventricular hypertrophy in the GotoKakizaki rat model of type 2 diabetes: critical role of Akt. Diabetologia. 2007;50(6):1335–1344.
57. Hansen PS, Clarke RJ, Buhagiar KA, et al. Alloxan-induced diabetes reduces sarcolemmal Na+-K+ pump function in rabbit ventricular myocytes. Am J Physiol Cell Physiol. 2007;292(3):1070–1077.
58. Kjeldsen K, Braendgaard H, Sidenius P, Larsen JS, Norgaard A. Diabetes decreases Na+-K+ pump concentration in skeletal muscles, heart ventricular muscle, and peripheral nerves of rat. Diabetes. 1987;36(7):842–848.
59. Bay J, Kohlhaas M, Maack C. Intracellular Na+ and cardiac metabolism. J Mol Cell Cardiol. 2013;61:20–27.
60. Hamouda NN, Sydorenko V, Qureshi MA, Alkaabi JM, Oz M, Howarth FC. Dapagliflozin reduces the amplitude of shortening and Ca(2+) transient in ventricular myocytes from streptozotocin-induced diabetic rats. Mol Cell Biochem. 2015;400(12):57–68.
61. Baartscheer A, Schumacher CA, Wüst RC, et al. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia. 2017;60:568–573.
62. Uthman L, Baartscheer A, Bleijlevens B, et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na (+)/H (+) exchanger, lowering of cytosolic Na (+) and vasodilation. Diabetologia. 2018;61(3):722–726.
63. Ye Y, Jia X, Bajaj M, Birnbaum Y. Dapagliflozin attenuates Na (+)/H (+) exchanger-1 in cardiofibroblasts via AMPK activation. Cardiovasc Drugs Ther. 2018;32(6):553–558.
64. Hammoudi N, Jeong D, Singh R, et al. Empagliflozin improves left ventricular diastolic dysfunction in a genetic model of type 2 diabetes. Cardiovasc Drugs Ther. 2017;31:233–246.
65. Liang L, Jiang J, Frank SJ. Insulin receptor substrate-1-mediated enhancement of growth hormone-induced mitogen-activated protein kinase activation. Endocrinology. 2000;141:3328–3336.
66. Moellmann J, Klinkhammer BM, Droste P, et al. Empagliflozin improves left ventricular diastolic function of db/db mice. Biochim Biophys Acta Mol Basis Dis. 2020;1866(8):165807.
67. Hernandez-Resendiz S, Buelna-Chontal M, Correa F, Zazueta C. Targeting mitochondria for cardiac protection. Curr Drug Targets. 2013;14(5):586–600.
68. Duncan JG. Mitochondrial dysfunction in diabetic cardiomyopathy. Biochim Biophys Acta. 2011;1813(7):1351–1359.
69. Montaigne D, Marechal X, Coisne A, et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. 2014;130(7):554–564.
70. Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The role of autophagy in the heart. Annu Rev Physiol. 2018;80:1–26.
71. Maejima Y, Chen Y, Isobe M, Gustafsson AB, Kitsis RN, Sadoshima J. Recent progress in research on molecular mechanisms of autophagy in the heart. Am J Physiol Heart Circ Physiol. 2015;308:H259–268.
72. Shirakabe A, Zhai P, Ikeda Y, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload induced mitochondrial dysfunction and heart failure. Circulation. 2016;133:1249–1263.
73. Durak A, Olgar Y, Degirmenci S, Akkus E, Tuncay E, Turan B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc Diabetol. 2018;17(1):144.
74. Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018;15:335–346.
75. Mizuno M, Kuno A, Yano T, et al. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol Rep. 2018;6:e13741.
76. Shao Q, Meng L, Lee S, et al. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc Diabetol. 2019;18:165.
77. Wang Y, Zhao X, Lotz M, Terkeltaub R, Liu-Bryan R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor gamma coactivator 1alpha. Arthritis Rheumatol. 2015;67(8):2141–2153.
78. Yan W, Zhang H, Liu P, et al. Impaired mitochondrial biogenesis due to dysfunctional adiponectin-AMPK-PGC-1alpha signaling contributing to increased vulnerability in diabetic heart. Basic Res Cardiol. 2013;108(3):329.
79. Sandesara PB, O’Neal WT, Kelli HM, et al. The prognostic significance of diabetes and microvascular complications in patients with heart failure with preserved ejection fraction. Diabetes Care. 2018;41:150–155.
80. Shome JS, Perera D, Plein S, Chiribiri A. Current perspectives in coronary microvascular dysfunction. Microcirculation. 2017;24:e12340.
81. Zhou H, Hu S, Jin Q, et al. Mff dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J Am Heart Assoc. 2017;6(3):e005328.
82. Sawada N, Jiang A, Takizawa F, et al. Endothelial PGC-1alpha mediates vascular dysfunction in diabetes. Cell Metab. 2014;19(2):246–258.
83. Katakam PV, Wappler EA, Katz PS, et al. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase, arterioscler. Thromb Vasc Biol. 2013;33(4):752–759.
84. Adingupu DD, Göpel SO, Grönros J, et al. SGLT2 inhibition with empagliflozin improves coronary microvascular function and cardiac contractility in prediabetic ob/ob-/- mice. Cardiovasc Diabetol. 2019;18(1):16.
85. Juni RP, Kuster DWD, Goebel M, et al. Cardiac microvascular endothelial enhancement of cardiomyocyte function is impaired by inflammation and restored by empagliflozin. J Am Coll Cardiol Basic Trans Sci. 2019;4:575–591.
86. Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91:733–794.
87. Zhou L, Cryan EV, D’Andrea MR, Belkowski S, Conway BR, Demarest KT. Human cardiomyocytes express high level of Naþ/glucose cotransporter 1 (SGLT1). J Cell Biochem. 2003;90:339–346.
88. Uthman L, Homayr A, Juni RP, et al. Empagliflozin and dapagliflozin reduce ROS generation and restore NO bioavailability in tumor necrosis factor α-stimulated human coronary arterial endothelial cells. Cell Physiol Biochem. 2019;53(5):865–886.
89. Aroor AR, Mandavia CH, Sowers JR. Insulin resistance and heart failure: molecular mechanisms. Heart Fail Clin. 2012;8:609–617.
90. Mandavia CH, Pulakat L, DeMarco V, Sowers JR. Over-nutrition and metabolic cardiomyopathy. Metabolism. 2012;61:1205–1210.
91. Dhalla NS, Liu X, Panagia V, Takeda N. Subcellular remodeling and heart dysfunction in chronic diabetes. Cardiovasc Res. 1998;40:239–247.
92. Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86(4):1133–1149.
93. Kusaka H, Koibuchi N, Hasegawa Y, Ogawa H, Kim-Mitsuyama S. Empagliflozin lessened cardiac injury and reduced visceral adipocyte hypertrophy in prediabetic rats with metabolic syndrome. Cardiovasc Diabetol. 2016;15(1):157.
94. Lee TM, Chang NC, Lin SZ. Dapagliflozin, a selective SGLT2 inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic Biol Med. 2017;104:298–310.
95. Tanajak P, Sa-Nguanmoo P, Sivasinprasasn S, et al. Cardioprotection of dapagliflozin and vildagliptin in rats with cardiac ischemia-reperfusion injury. J Endocrinol. 2018;236(2):69–84.
96. Lin B, Koibuchi N, Hasegawa Y, et al. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc Diabetol. 2014;13(1):148.
97. Olgar Y, Turan B. A sodium-glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin comparison with insulin shows important effects on Zn2+-transporters in cardiomyocytes from insulin-resistant metabolic syndrome rats through inhibition of oxidative stress. Can J Physiol Pharmacol. 2019;97(6):528–535.
98. Ayaz M, Turan B. Selenium prevents diabetes-induced alterations in [Zn2+]i and metallothionein level of rat heart via restoration of cell redox cycle. Am J Physiol Heart Circ Physiol. 2006;290(3):H1071–80.
99. Tuncay E, Turan B. Intracellular Zn (2+) increase in cardiomyocytes induces both electrical and mechanical dysfunction in heart via endogenous generation of reactive nitrogen species. Biol Trace Elem Res. 2016;169(2):294–302.
100. Teshima Y, Takahashi N, Nishio S, et al. Production of reactive oxygen species in the diabetic heart. Roles of mitochondria and NADPH oxidase. Circ J. 2014;78(2):300–306. doi:10.1253/circj.CJ-13-1187
101. Li C, Zhang J, Xue M, et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol. 2019;18(1):15. doi:10.1186/s12933-019-0816-2
102. Xue M, Li T, Wang Y, et al. Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice. Clin Sci. 2019;133(15):1705–1720. doi:10.1042/CS20190585
103. Kolijn D, Pabel S, Tian Y, et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc Res. 2020:cvaa123. doi:10.1093/cvr/cvaa123.
104. Zhou Y, Wu W. The sodium–glucose co-transporter 2 inhibitor, empagliflozin, protects against diabetic cardiomyopathy by inhibition of the endoplasmic reticulum stress pathway. Cell Physiol Biochem. 2017;41(6):2503–2512. doi:10.1159/000475942
105. Hussein AM, Eid EA, Taha M, et al. Comparative study of the effects of GLP1 analog and SGLT2 inhibitor against diabetic cardiomyopathy in type 2 diabetic rats: possible underlying mechanisms. Biomedicines. 2020;8(3):43. doi:10.3390/biomedicines8030043
106. Pal PB, Sonowal H, Shukla K, Srivastava SK, Ramana KV. Aldose reductase mediates NLRP3 inflammasome-initiated innate immune response in hyperglycemia-induced Thp1 monocytes and male mice. Endocrinology. 2017;158:3661–3675.
107. Ye Y, Bajaj M, Yang HC, Perez-Polo JR, Birnbaum Y. SGLT-2 inhibition with dapagliflozin reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. Further augmentation of the effects with saxagliptin, a DPP4 inhibitor. Cardiovasc Drugs Ther. 2017;31(2):119–132. doi:10.1007/s10557-017-6725-2
108. Aragón-Herrera A, Feijóo-Bandín S, Otero Santiago M, et al. Empagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of zucker diabetic fatty rats. Biochem Pharmacol. 2019;170:113677.
109. Chen H, Tran D, Yang HC, Nylander S, Birnbaum Y, Ye Y. Dapagliflozin and ticagrelor have additive effects on the attenuation of the activation of the NLRP3 inflammasome and the progression of diabetic cardiomyopathy: an AMPK–mTOR interplay. Cardiovasc Drugs Ther. 2020;34(4):443–461. doi:10.1007/s10557-020-06978-y.
110. Habibi J, Aroor AR, Sowers JR, et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc Diabetol. 2017;16(1):9. doi:10.1186/s12933-016-0489-z
111. Das S, Aiba T, Rosenberg M, et al. Pathological role of serum- and glucocorticoid-regulated kinase 1 in adverse ventricular remodeling. Circulation. 2012;126(18):2208–2219. doi:10.1161/CIRCULATIONAHA.112.115592
112. Aoyama T, Matsui T, Novikov M, Park J, Hemmings B, Rosenzweig A. Serum and glucocorticoid-responsive kinase-1 regulates cardiomyocyte survival and hypertrophic response. Circulation. 2005;111(13):1652–1659. doi:10.1161/01.CIR.0000160352.58142.06
113. Kang S, Verma S, Hassanabad AF, et al. Direct effects of empagliflozin on extracellular matrix remodelling in human cardiac myofibroblasts: novel translational clues to explain EMPA-REG OUTCOME results. Can J Cardiol. 2020;36(4):543–553. doi:10.1016/j.cjca.2019.08.033
114. Jia G, Habibi J, DeMarco VG, et al. Endothelial mineralocorticoid receptor deletion prevents diet-induced cardiac diastolic dysfunction in females. Hypertension. 2015;66(6):1159–1167. doi:10.1161/HYPERTENSIONAHA.115.06015
115. Cherney DZ, Perkins BA, Soleymanlou N, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation. 2014;129(5):587–597. doi:10.1161/CIRCULATIONAHA.113.005081
116. Tanaka H, Takano K, Iijima H, et al. Factors affecting canagliflozin-induced transient urine volume increase in patients with type 2 diabetes mellitus. Adv Ther. 2017;34(2):436–451. doi:10.1007/s12325-016-0457-8
117. Muskiet MH, van Raalte DH, van Bommel EJ, et al. Understanding EMPA-REG OUTCOME. Lancet Diabetes Endocrinol. 2015;3:928–929.
118. Filippatos TD, Liontos A, Papakitsou I, Elisaf MS. SGLT2 inhibitors and cardioprotection: a matter of debate and multiple hypotheses. Postgrad Med. 2019;131(2):82–88. doi:10.1080/00325481.2019.1581971
119. Wang J, Shibayama Y, Kobori H, et al. High glucose augments angiotensinogen in human renal proximal tubular cells through hepatocyte nuclear factor-5. PLoS One. 2017;12(10):e0185600. doi:10.1371/journal.pone.0185600
120. Shin SJ, Chung S, Kim SJ, et al. Effect of sodium-glucose co-transporter 2 inhibitor, dapagliflozin, on renal renin-angiotensin system in an animal model of type 2 diabetes. PLoS One. 2016;11(11):e0165703. doi:10.1371/journal.pone.0165703
121. Woods TC, Satou R, Miyata K, et al. Canagliflozin prevents intrarenal angiotensinogen augmentation and mitigates kidney injury and hypertension in mouse model of type 2 diabetes mellitus. Am J Nephrol. 2019;49(4):331–342. doi:10.1159/000499597
122. Kruljac I, Ćaćić M, Ćaćić P, et al. Diabetic ketosis during hyperglycemic crisis is associated with decreased all-cause mortality in patients with type 2 diabetes mellitus. Endocrine. 2017;55(1):139–143. doi:10.1007/s12020-016-1082-7
123. Pawlak M, Baugé E, Lalloyer F, Lefebvre P, Staels B. Ketone body therapy protects from lipotoxicity and acute liver failure upon PPARα deficiency. Mol Endocrinol. 2015;29(8):1134–1143. doi:10.1210/me.2014-1383
124. Al Jobori H, Daniele G, Adams J, et al. Determinants of the increase in ketone concentration during SGLT2 inhibition in NGT, IFG and T2DM patients. Diabetes Obes Metab. 2017;19(6):809–813. doi:10.1111/dom.12881
125. Ferrannini E, Baldi S, Frascerra S, et al. Shift to fatty substrates utilization in response to sodium glucose co-transporter-2 inhibition in nondiabetic subjects and type 2 diabetic patients. Diabetes. 2016;65(5):1190–1195. doi:10.2337/db15-1356
126. Verma S, Rawat S, Ho KL, et al. Empagliflozin increases cardiac energy production in diabetes: novel translational insights into the heart failure benefits of SGLT2 inhibitors. J Am Coll Cardiol Basic Trans Sci. 2018;3:575–587.
127. Nielsen R, Møller N, Gormsen LC, et al. Cardiovascular effects of treatment with the ketone body 3-hydroxybutyrate in chronic heart failure patients. Circulation. 2019;139(18):2129–2141. doi:10.1161/CIRCULATIONAHA.118.036459
128. Tentolouris A, Vlachakis P, Tzeravini E, Eleftheriadou I, Tentolouris N. SGLT2 inhibitors: a review of their antidiabetic and cardioprotective effects. Int J Environ Res Public Health. 2019;16(16):2965. doi:10.3390/ijerph16162965
129. Aragón-Herrera A, Feijóo-Bandín S, Otero Santiago M, et al. Empagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of Zucker diabetic fatty rats. Biochem Pharmacol. 2019;12(170):113677. doi:10.1016/j.bcp.2019.113677
130. Schulze PC, Drosatos K, Goldberg IJ. Lipid use and misuse by the heart. Circ Res. 2016;118(11):1736–1751. doi:10.1161/CIRCRESAHA.116.306842
131. Bakovic M, Filipovic N, Hamzic LF, Kunac N, Zdrilic E, Uljevic MV. Changes in neurofilament 200 and tyrosine hydroxylase expression in the cardiac innervation of diabetic rats during aging. Cardiovasc Pathol. 2018;32:38–43. doi:10.1016/j.carpath.2017.11.003
132. Yehya YM, Hussein AM, Ezam K, et al. Blockade of renin angiotensin system ameliorates the cardiac arrhythmias and sympathetic neural remodeling in hearts of type 2 DM rat model. Endocr Metab Immune Disord Drug Targets. 2020;20(3):464–478. doi:10.2174/1871530319666190809150921
133. Bisognano JD, Weinberger HD, Bohlmeyer TJ, et al. Myocardial-directed overexpression of the human beta(1)-adrenergic receptor in transgenic mice. J Mol Cell Cardiol. 2000;32(5):817–830. doi:10.1006/jmcc.2000.1123
134. Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022.
135. Yokoe S, Asahi M, Takeda T, et al. Inhibition of phospholamban phosphorylation by O-GlcNAcylation: implications for diabetic cardiomyopathy. Glycobiology. 2010;20(10):1217–1226. doi:10.1093/glycob/cwq071
136. Erickson JR, Pereira L, Wang L, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502(7471):372–376. doi:10.1038/nature12537
137. Ducheix S, Magré J, Cariou B, Prieur X. Chronic O-GlcNAcylation and diabetic cardiomyopathy: the bitterness of glucose. Front Endocrinol (Lausanne). 2018;9:642. doi:10.3389/fendo.2018.00642
138. Joubert M, Jagu B, Montaigne D, et al. The SGLT2 inhibitor dapagliflozin prevents cardiomyopathy in a diabetic lipodystrophic mouse model. Diabetes. 2017;66(4):1030–1040. doi:10.2337/db16-0733
139. Uthman L, Baartscheer A, Schumacher CA, et al. Direct cardiac actions of sodium glucose cotransporter 2 inhibitors target pathogenic mechanisms underlying heart failure in diabetic patients. Front Physiol. 2018;9:1575. doi:10.3389/fphys.2018.01575
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.