")
Back to Archived Journals » Hypoxia » Volume 4
Sevoflurane mitigates shedding of hyaluronan from the coronary endothelium, also during ischemia/reperfusion: an ex vivo animal study
Authors Chen C, Chappell D, Annecke T, Conzen P, Jacob M, Welsch U, Zwissler B, Becker BF
Received 20 October 2015
Accepted for publication 21 January 2016
Published 15 April 2016 Volume 2016:4 Pages 81—90
DOI https://doi.org/10.2147/HP.S98660
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Congcong Chen,1,3 Daniel Chappell,2,3 Thorsten Annecke,2,3 Peter Conzen,2 Matthias Jacob,2,3 Ulrich Welsch,4 Bernhard Zwissler,2 Bernhard F Becker3
1Department of Anesthesiology, Second Affiliated Hospital of Zhejiang University, Hangzhou, People's Republic of China; 2Clinic of Anesthesiology, Ludwig-Maximilians-University, Munich, Germany; 3Walter-Brendel-Centre of Experimental Medicine, Ludwig-Maximilians-University, Munich, Germany; 4Institute of Anatomy, Ludwig-Maximilians-University, Munich, Germany
Abstract: Glycosaminoglycan hyaluronan (HA), a major constituent of the endothelial glycocalyx, helps to maintain vascular integrity. Preconditioning the heart with volatile anesthetic agents protects against ischemia/reperfusion injury. We investigated a possible protective effect of sevoflurane on the glycocalyx, especially on HA. The effect of pre-ischemic treatment with sevoflurane (15 minutes at 2% vol/vol gas) on shedding of HA was evaluated in 28 isolated, beating guinea pig hearts, subjected to warm ischemia (20 minutes at 37°C) followed by reperfusion (40 minutes), half with and half without preconditioning by sevoflurane. HA concentration was measured in the coronary effluent. Over the last 20 minutes of reperfusion hydroxyethyl starch (1 g%) was continuously infused and the epicardial transudate collected over the last 5 minutes for measuring the colloid extravasation. Additional hearts were fixed by perfusion after the end of reperfusion for immunohistology and electron microscopy. Sevoflurane did not significantly affect post-ischemic oxidative stress, but strongly inhibited shedding of HA during the whole period, surprisingly even prior to ischemia. Immunohistology demonstrated that heparan sulfates and SDC1 of the glycocalyx were also preserved by sevoflurane. Electron microscopy revealed shedding of glycocalyx caused by ischemia and a mostly intact glycocalyx in hearts exposed to sevoflurane. Coronary vascular permeability of the colloid hydroxyethyl starch was significantly decreased by sevoflurane vs the control. We conclude that application of sevoflurane preserves the coronary endothelial glycocalyx, especially HA, sustaining the vascular barrier against ischemic damage. This may explain beneficial effects associated with clinical use of volatile anesthetics against ischemia/reperfusion injury.
Keywords: endothelial glycocalyx, hyaluronan, ischemia-reperfusion injury, volatile anesthetics, preconditioning
Introduction
Recent studies have demonstrated that inhalational anesthetics, like sevoflurane, have cardioprotective effects, both experimentally and clinically.1–7 However, cellular and molecular mechanisms of action are far from understood.4–7 Patel et al proved that isoflurane modifies cardiac myocyte sarcolemmal membrane structure and composition, and that activation of phosphorylation of CAV1 contributes to cardiac protection.8 Marinovic et al showed that both sarcolemmal and mitochondrial K-ATP channels play essential and distinct roles in protection afforded by isoflurane.9 Feng et al concluded from their research that isoflurane had a protective effect against myocardial ischemic injury by preventing opening of the mitochondrial permeability transition pore.10 Also, leukocytes contribute essentially to reperfusion damage and there is some extent of neutrophil activation during open heart surgery with cardiopulmonary bypass.11 Kowalski et al found that sevoflurane, isoflurane, and halothane in concentrations of both 1 and 2 minimum alveolar concentration (MAC) reduced polymorphonuclear neutrophil adhesion in the reperfused heart.12 Furthermore, Heindl et al demonstrated that sevoflurane reduced polymorphonuclear neutrophil adhesion in the reperfused coronary system and thereby preserved cardiac function.13 Interestingly, only sevoflurane preconditioning of hearts, but not of platelets, reduced intracoronary platelet adhesion, indicating that the effect of sevoflurane was most likely mediated via an endothelial mechanism.14
A healthy vascular endothelium is coated by the glycocalyx, including a variety of transmembrane and membrane-attached molecules.15,16 Quantitatively important parts of the endothelial glycocalyx are transmembrane syndecans and membrane-anchored glypicans. Both have side chains carrying heparan sulfate (HS) and chondroitin sulfate.17 Along with attached chains of hyaluronan (HA) and plasma proteins, these constituents make up an endothelial surface layer approaching 1 μm in thickness.18 There are also many functionally important molecular components such as cell adhesion molecules located inside the endothelial glycocalyx.19,20 Altogether, the intact endothelial glycocalyx plays a key role, not only as barrier reducing the permeability of macromolecules, but also decreases leukocyte and platelet adhesion at the vascular wall.21,22 Treatment with sevoflurane has recently been shown to reduce shedding of syndecan and HS, thereby decreasing leukocyte and platelet adhesion.23,24
Our study aimed to show the extent of shedding of HA during the period of ischemia/reperfusion, and to examine the effects of sevoflurane on the glycocalyx under these conditions. The study was performed using a highly standardized and well characterized organ model: the isolated perfused, spontaneously beating heart (guinea pig Langendorff preparation).
Materials and methods
This investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institute of Health (NIH Publication No 85-23, revised 1996). The investigation was approved by the independent ethics committee of the State of Bavaria. Licensure and approval of the investigation were obtained from the Government of Upper Bavaria (file no 209.1/211-2531.3-3/99). The methods described below are well established and have been published before.23–26 The work was performed at: Clinic of Anesthesiology and Walter-Brendel-Centre of Experimental Medicine, Ludwig-Maximilians-University, Munich, Germany.
Heart preparation
Hearts of male guinea pigs (body weight 250–300 g) were isolated and perfused at 37°C using a modified Krebs-Henseleit buffer (116 mM NaCl, 23 mM NaHCO3, 3.6 mM KCl, 1.16 mM KH2PO4, 1.2 mM CaCl2, 0.58 mM MgSO4, 5.4 mM glucose, 0.3 mM pyruvate, and 2.8 U/L insulin, gassed with 94.6% oxygen and 5.4% carbon dioxide at 37°C, pH 7.40±0.05) in a modified Langendorff mode. Hearts were removed from the thorax and prepared as previously described. The oxygen is physically dissolved, guaranteeing adequate oxygen content in the coronary artery perfusate.25 Physiological, pulsatile coronary flow was achieved by maintaining the coronary perfusion pressure at 70 cm H2O throughout the whole procedure, except for the period of stopped-flow ischemia (see Experimental protocols). The veins entering the right atrium and pulmonary veins were ligated to ensure that all coronary effluent emerged via the right ventricle though the pulmonary artery.26,27 The latter was cannulated to collect the coronary venous effluent. Transudate, a mixture of interstitial and lymphatic fluids formed by net filtration and appearing on the epicardial surface, was collected from the apex of the heart.
Sevoflurane (Abbott Laboratories, Abbott Park, IL, USA) was added at 2% vol/vol, equivalent to 1 MAC, to the O2/CO2 gas mixture equilibrating the Krebs-Henseleit perfusate for one group of the hearts and sustained throughout the rest of the protocol. Addition was achieved by means of a calibrated vaporizer (Draeger, Luebeck, Germany) and monitored by a piezo electric gas detector (Datex, Helsinki, Finland), as described previously.14 To ensure equilibration of volatile anesthetic with the liquid phase, application of sevoflurane with the gas phase to the perfusate began 30 minutes before its use.
Experimental protocols
Animals were stunned by neck dislocation using a specially designed instrument. After explantation and preparation of the hearts, an equilibration interval of 15 minutes was allowed to establish steady state conditions, using the Krebs-Henseleit perfusate without sevoflurane (group A, n=14) or with 2% vol/vol sevoflurane in the aerating gas (group B, n=14). Baseline measurements of coronary effluent for HA content were performed in the last 3 minutes before inducing 20 minutes of warm (37°C), global stopped-flow ischemia. Warm ischemia models the situation pertaining in infarcted human myocardium. Reperfusion was conducted with Krebs-Henseleit buffer. After 20 minutes of reperfusion, hearts of both groups were perfused for another 20 minutes in the presence of 1% hydroxyethyl starch (HES). This was achieved by infusing 6% HES solution (molecular weight 130,000; degree of substitution 0.4; Fresenius AG, Bad Homburg, Germany) into the Krebs-Henseleit perfusate at a rate of one sixth of the total coronary flow. Infusion for 20 minutes has been shown to allow for steady state distribution of HES.15
Baseline measurements of coronary flow and transudate formation were performed 3 minutes before ischemia. Samples of effluent were collected between minutes 0–5, 5–10, 10–20, and 35–40 after onset of reperfusion. Transudate samples were collected over 5-minute intervals at 0–5, 5–10, 10–15, 15–20, 20–25, 25–30, 30–35, and 35–40 minutes after start of reperfusion.
At the end of each experiment, both atria and the large vessels were cut away, and the ventricles were weighed at once (wet weight). Weight of all hearts of each group was determined again after 24 hours at 60°C (dry weight) to establish a wet-to-dry weight ratio. This served as a quantitative measure for formation of edema.
Determination of HA, HES, lactate, uric acid, and purines
In both groups, samples of effluent were used for measuring shedding of HA, proceeding as described previously.26,28 Aliquots (4 mL) of samples were concentrated to 50 μL with 10 kD cutoff ultrafilters (EMD Millipore, Billerica, MA, USA). These concentrates were assayed using an enzyme-linked immunosorbent assay kit (Echelon Biosciences Incorporated, Salt Lake City, UT, USA) based on an enzyme-linked antibody against HA. On the basis of information given by the supplier, it is assumed that this enzyme-linked immunosorbent assay recognizes HA molecules independent of their molecular size.
Concentrations of HES were quantified in the samples of coronary transudate at 35–40 minutes after start of reperfusion, using a method described by Förster et al,29 as modified by Rehm et al.15 Basically, this involves hydrolysis of HES to glucose and subsequent enzymatic/photometric determination of the sugar.
Lactate, purines, and uric acid concentrations were determined by high performance liquid chromatography in samples of coronary effluent. Lactate is an indicator of the severity of ischemic challenge. Purine release, resulting from metabolism of adenine nucleotides and nucleoside, is directly related to the rate of energy consumption, and inversely related to the rate of energy production. Uric acid, the end product of enzymatic purine metabolism in the heart, serves as an indicator of oxidative stress, because it is subject to further oxidative chemical degradation. Therefore, hearts under oxidative stress typically release less urate relative to precursor purines than when oxidative stress is mitigated.30 For lactate, 10 μL of effluent were applied to a Nucleosil 100-5NH2 4×250 mm column (Macherey-Nagel, Dueren, Germany), 10 mM NH4H2PO4, pH 3.5, serving as eluent. Lactate was detected by its ultraviolet absorbance at 210 nm. Uric acid levels were determined in 10 μL of coronary effluent sample applied to a 5 μm C-18 Nucleosil 4×250 mm column (Macherey-Nagel). HClO4 (pH 2)/60% methanol at a ratio of 90:10 (vol%) served as eluent. Uric acid was detected by its ultraviolet absorbance at 280 nm. In the case of purines, all were transformed first to uric acid and then determined as such. This involved sequential enzymatic conversion of adenosine, inosine, hypoxanthine, and xanthine to urate.30
Rates of release were obtained by multiplying the concentration values with the respective rates of coronary flow or, in the case of HES, transudate formation.
Electron microscopy and immunohistochemistry
Electron microscopy of hearts to visualize the glycocalyx was performed using perfusion-fixation with a lanthanum nitrate/glutaraldehyde solution in modification of a method described by Vogel et al31 and Rehm et al.15 Immunohistochemical characterization of hearts was conducted to gain insight into the composition of the glycocalyx. To compare staining of two key components of glycocalyx between groups, hearts were perfused with 4% formalin. Whole hearts were paraffin-embedded, sliced (5 μm thickness), and slices stained with monoclonal antibody against HS (Seikagaku Corporation, Tokyo, Japan) or SDC1 (Abcam, Cambridge, UK). The primary antibodies, applied for generating an avidin-biotin horseradish peroxidase complex with the Vectostain kit (Vector, Burlingame, CA, USA), were diluted and handled as follows: anti-HS 1:100, tissue pre-incubation with 0.2% trypsin at 37°C; anti-SDC1 1:50, and tissue pretreatment by microwave irradiation. Controls, in which the primary antibody was replaced by buffer, were treated identically. Diaminobenzidine or aminoethyl carbazole was used as chromogen. Since controls without primary antibody did not develop color, they are not shown in results.
Statistical analysis
Data dealing with rates of flow or of release are expressed per gram of heart wet weight. All data in figures are presented as mean ± standard error of the mean, those in Table 1 as mean ± standard deviation. The number of samples (n) =14 for both, controls without sevoflurane (group A) and those with sevoflurane (group B). Comparisons between the two groups were made using Student’s t-test. Student’s t-test was used to make intragroup comparisons, with Bonferroni correction applied when multiple comparisons were involved. P<0.05 was considered to be significant. The statistical software used to conduct the analyses was SigmaStat 3.5 (Systat Software Inc., San Jose, CA, USA).
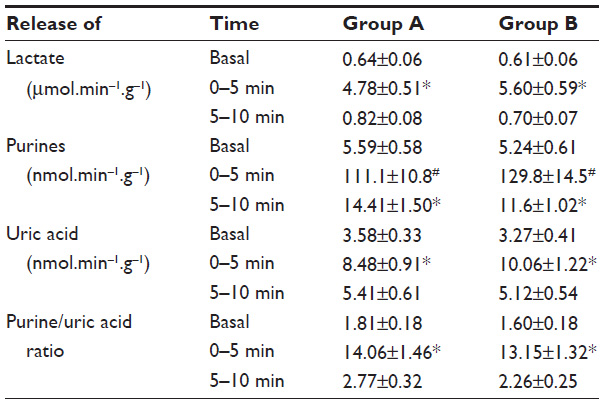 | Table 1 Comparison of metabolite release during reperfusion |
Results
Coronary flow
The perfusion pressure of all hearts was maintained constant at 70 cm H2O. Thus, increases in flow correspond to dilatation, whilst decreased flow reflects coronary constriction. As shown in Figure 1, coronary flow rate in group A remained unchanged within the first 5 minutes of reperfusion (7.23±0.88 mL.min–1.g–1 basal vs 7.57±0.91 mL.min–1.g–1 first 5 minutes of reperfusion, P>0.05, Student’s t-test). This indicates an inability to develop reactive hyperemia after 20 minutes of ischemia. In contrast, there was an initial reactive hyperemia in group B treated with sevoflurane (7.59±0.91 mL.min–1.g–1 basal vs 8.62±1.03 mL.min–1.g–1 first 5 minutes of reperfusion, P<0.05, Student’s t-test). In fact, coronary flow was significantly higher in group B than in group A during the first 5 minutes of reperfusion (P<0.05; Figure 1). With ongoing reperfusion, coronary flow decreased significantly by ~20% vs basal in both groups. Treatment with sevoflurane was unable to prevent this.
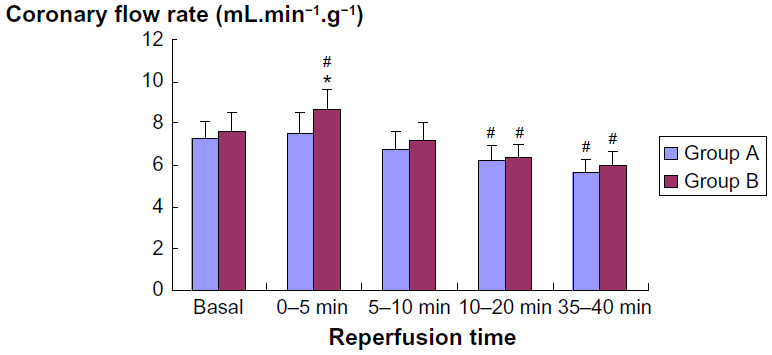 | Figure 1 Coronary flow in relation to time during reperfusion. |
Transudate and tissue edema formation
Transudate formation, the direct result of net fluid filtration in the intact coronary bed of the isolated hearts, amounted to ~5% of coronary flow under basal conditions. Figure 2 presents transudate formation standardized to the individual heart weight. The baseline values measured before ischemia showed no difference between groups.
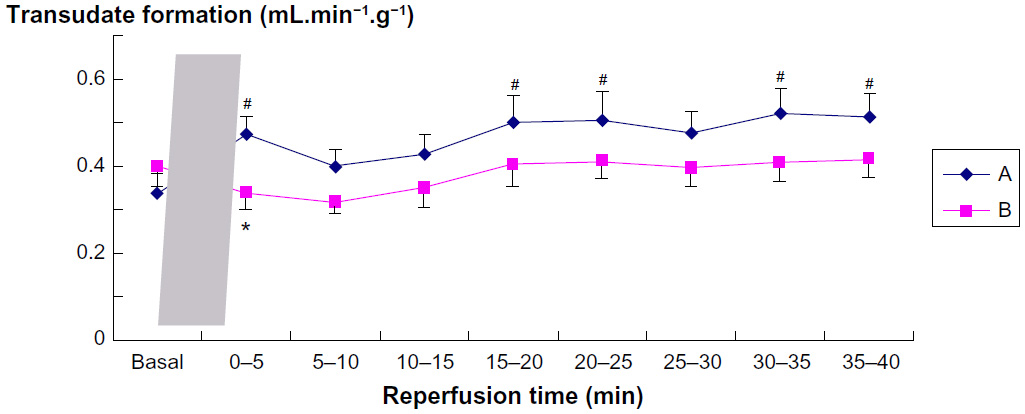 | Figure 2 Transudate formation in relation to sampling period. |
In the control group (group A), the formation of transudate increased within the first 5 minutes and then remained at higher level than basal during the course of reperfusion (Student’s t-test with Bonferroni correction, P<0.05). Pretreatment with sevoflurane (group B) completely prevented this increase in post-ischemic coronary leak (Figure 2). In fact, the formation of transudate significantly decreased in the first minutes of reperfusion in group B vs group A (P<0.05).
The mean wet-to-dry weight ratio of isolated hearts at the end of experimentation in group A was 8.4±0.5. Administration of sevoflurane to the hearts before ischemia yielded a lower ratio of 7.9±0.3 in group B. However, this decrease did not attain statistical significance (P>0.05 vs group A, unpaired Student’s t-test).
Rates of release of lactate, purines, and uric acid
Metabolic parameters of hearts subjected to ischemic stress are listed in Table 1. Lactate release was identical in groups A and B during equilibration. There was a large increase in both groups after the start of reperfusion (Student’s t-test, both P<0.05), independent of the treatment. The rate of lactate release decreased rapidly during further reperfusion, returning to near basal values in both groups (Table 1).
Basal purine release was identical in groups A and B. During the first minutes of reperfusion, purine release increased 20- to 30-fold (Student’s t-test, both P<0.01), independent of the treatment (Student’s t-test for unpaired samples, P>0.05). Purine release decreased again during reperfusion, but there was no significant difference between groups (Table 1).
Uric acid was released in comparable amounts in groups A and B during equilibration. There was a highly significant increase in uric acid release in the first minutes of reperfusion after ischemia (Student’s t-test, both P<0.05), with rates again comparable between the groups. After the peak, rates of release declined significantly to similar values at 5–10 minutes after the beginning of reperfusion (Table 1).
The ratio of release of precursor purines to urate release gives an indication of the degree of oxidative stress. For groups A and B, the ratio of basal release of purines to that of urate was approximately equal (Table 1). Purine release increased over-proportionally upon onset of reperfusion, giving rise to an almost 10-fold higher ratio in the first 5 minutes. The ratio was consistently ~10%–20% lower in the sevoflurane treated group, but there was no statistical difference between groups. This indicates the presence of, but no global reduction in oxidative stress in hearts reperfused after treatment with sevoflurane (Table 1).
Extravasation of HES
Net extravasation of the colloid HES was calculated as the product of transudate formation per gram of wet heart weight and HES concentration in the transudate. The values were determined after equilibration of hearts with HES, 35–40 minutes after beginning of reperfusion. Hearts subjected to ischemia without sevoflurane (group A) showed significantly higher leak of HES from the coronary bed than those in the sevoflurane-treated group B (Student’s t-test for unpaired samples, P<0.05, Figure 3).
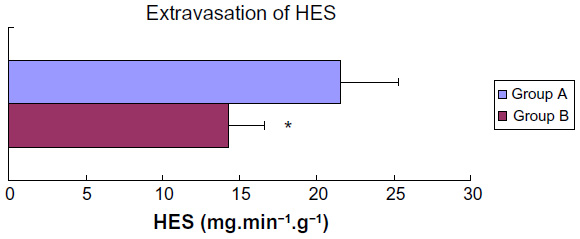 | Figure 3 Extravasation of HES into the transudate, period 35–40 minutes after the beginning of reperfusion. |
Measurement of HA
Although HA is not exclusively localized to endothelial cells in the myocardium, its appearance in coronary effluent may be taken as a sign of shedding mainly from the endothelial glycocalyx. HA was detected in the effluent of all hearts, already during the basal period of perfusion. However, significantly less was shed by hearts being pretreated with sevoflurane (Figure 4). Surprisingly, ischemia/reperfusion did not appreciably influence shedding of HA from the glycocalyx in either group. Although washout of HA declined in the course of reperfusion, the amount released from control hearts always exceeded that of hearts treated with sevoflurane (Figure 4).
 | Figure 4 Hyaluronan release in coronary venous effluent is shown at baseline and at two sampling periods during reperfusion. |
Electron microscopy and immunohistochemistry
Electron microscopic photographs showing the state of the endothelial glycocalyx of coronary vessels after ischemia and 40 minutes of reperfusion are depicted in Figure 5. Practically no glycocalyx could be visualized in control group A (Figure 5A and B). On the other hand, a distinct glycocalyx was seen in the sevoflurane-treated group B (Figure 5C and D). Perivascular tissue edema was observed in both cases (Figure 5).
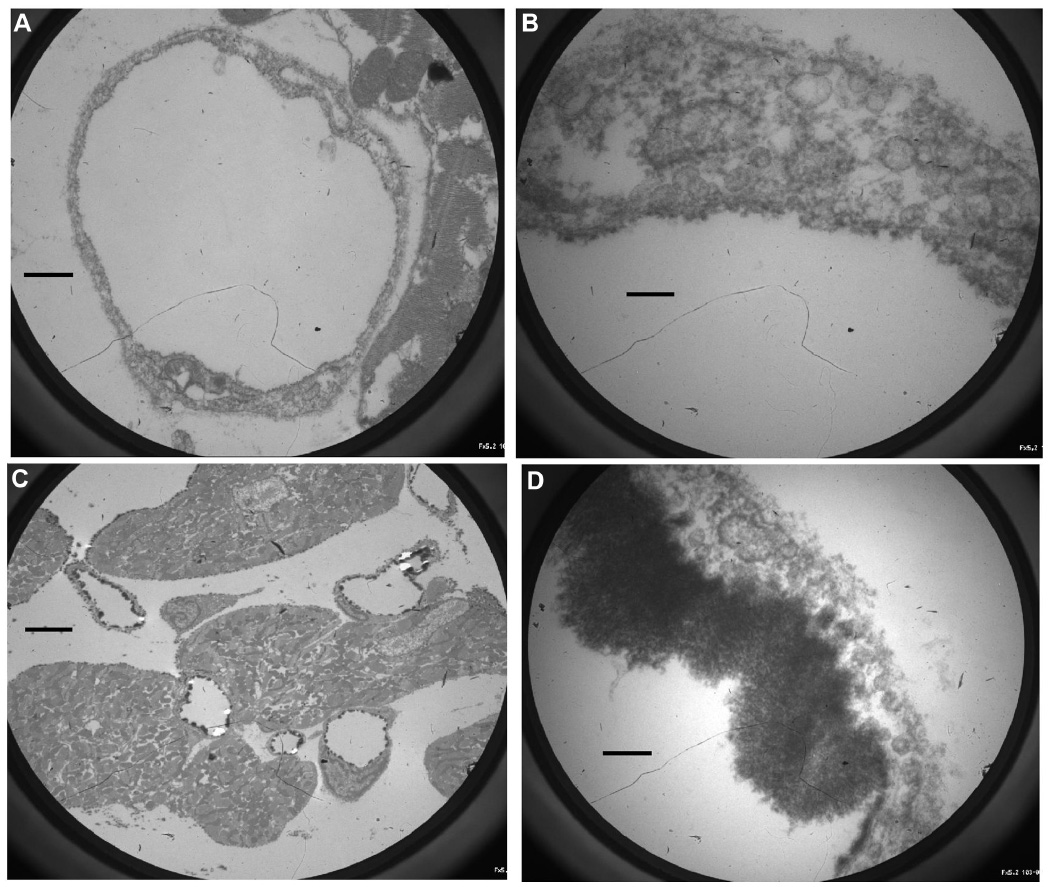 | Figure 5 Electron microscope images of hearts stained to reveal the glycocalyx. |
Immunohistochemical staining of these hearts after fixation with formaldehyde was carried out to reveal the presence of HS (Figure 6) and SDC1 (Figure 7) at the vascular surface. Despite prolonged staining, there was clearly not more HS-positive material in the walls of vessels than in the parenchyma of hearts of control group A (Figure 6A). In contrast, sevoflurane preserved significantly more HS (brown stain) in vessels of various calibers in hearts of group B with sevoflurane (Figure 6B). Likewise, SDC1-positive material did not stain appreciably more in vessels than in parenchyma of hearts of group A (Figure 7A), while there was strong vascular evidence (brown stain) in hearts of group B (Figure 7B).
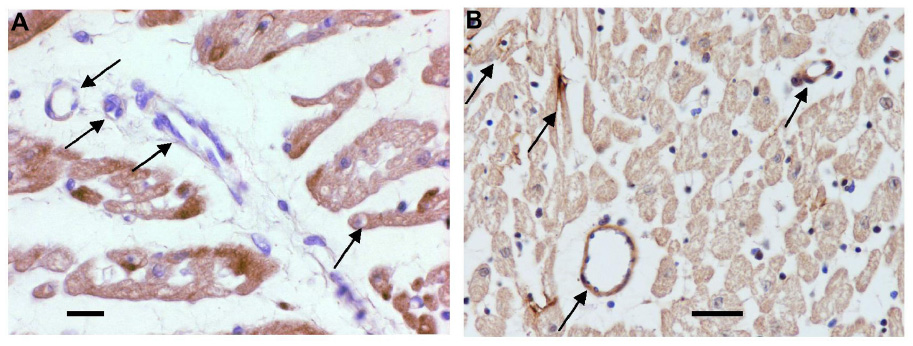 | Figure 6 Immunohistochemical staining of heparan sulfate (brown coloration). |
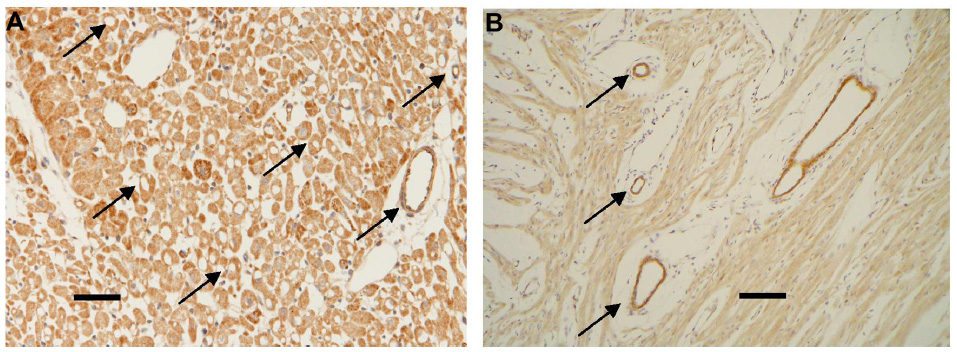 | Figure 7 Immunohistochemical staining of SDC1 (brown coloration). |
Discussion
One main result of the present study was substantiating the ability of sevoflurane to preserve the glycocalyx and the vascular filtration barrier in the situation of ischemia/reperfusion of isolated guinea pig hearts. The more novel finding, however, relates to the mitigation of loss of HA from the endothelial glycocalyx, already under conditions of normoxic perfusion with crystalloid solution (Krebs-Henseleit buffer).
Sevoflurane has previously been found to help sustain the endothelial glycocalyx in the face of ischemia/reperfusion.23,24 An earlier study demonstrated shedding of the endothelial glycocalyx due to ischemia in an animal model.32 Furthermore, Rehm et al provided evidence for shedding of the endothelial glycocalyx during ischemia/reperfusion procedures in humans.33 Preservation of the endothelial glycocalyx would not only maintain the endothelial permeability barrier,15 but also prevent leukocyte34,35 and platelet adhesion22 and, thus, should play important roles in inflammation processes and ischemia/reperfusion injury.16,20
In the present ex vivo study, 20-minute warm no-flow ischemia and reperfusion, a situation akin to that of human myocardial infarction with prompt clinical revascularization, severely damaged the coronary endothelial glycocalyx. This caused a significant increase in formation of transudate, a direct parameter of coronary fluid leak. Sevoflurane pretreatment significantly lowered formation of transudate and, in addition, led to less colloid extravasation at the end of reperfusion, ie, significantly improved post-ischemic coronary vascular integrity. Above that, sevoflurane-treated hearts showed significant reactive hyperemia in the early stage of reperfusion compared to hearts without sevoflurane. This indicates that application of sevoflurane accelerates recovery of heart function during the reperfusion period and maintains endothelial function.16
To shed more light on the mechanism behind cardioprotection by sevoflurane under these conditions, the present study focused on the shedding of HA, a constituent of the glycocalyx. Measurements of HA in the coronary effluent showed consistent release, both before and after ischemic injury in control hearts (Figure 4). Previous studies on isolated hearts have demonstrated basal release also of HS and SDC1.23,24,26 Presumably, some shedding of glycocalyx is already initiated by heart preparation and may be perpetuated by perfusion with an albumin-free crystalloid buffer, as in our experiments.28 HA, receptor bound, is known to be among the first markers for glycocalyx shedding.36 Surprisingly, ischemia/reperfusion did not enhance washout of HA, although this has been observed for SDC1 and HS.23,26 This distinction may result from the type of membrane anchoring. The transmembrane syndecans are the major core protein families on the endothelial cell membrane and are presumably cleaved by metalloproteinases.7 Shedding of HS requires the action of heparinases or heparanases, the latter found in perivascular mast cells.7,16 HA, which is a much longer disaccharide polymer than the HS, is synthesized on the cell surface and is not covalently attached to a core protein. HA weaves through the glycocalyx and is anchored to the plasma membrane by interacting with its receptor, the transmembrane CD44 that localizes in caveolae, and by interacting with the chains of syndecans.37 Accordingly, shedding of HA occurring already before ischemic challenge may reflect ongoing detachment of HA from its membrane receptor CD44 in the course of perfusion with Krebs-Henseleit buffer.
Besides this, the electron microscopic photos of hearts of this study revealed extensive destruction of the glycocalyx as an aspect of damage caused by ischemia/reperfusion. Similarly, immunohistochemistry of control hearts detected almost no SDC1 or HS staining on the surface of vessels after the ischemia-reperfusion procedure. Yet, sevoflurane treatment significantly decreased the shedding of glycocalyx, as evidenced both by electron microscopy and immunohistochemistry (Figures 5–7). As shown by quantitative measurement in the coronary effluent, sevoflurane treated hearts also shed far less HA after ischemia (Figure 4).
Uric acid, a powerful physiologic antioxidant, is subject to oxidative chemical degradation.30 Therefore, a high purine/uric acid ratio in coronary effluent indicates high oxidative stress. During the first few minutes of reperfusion, there are significantly higher purine/uric acid ratios vs basal in both groups, with values ~15% lower in the sevoflurane group. However, this difference did not attain statistical significance. Thus, global oxidative stress was not alleviated by sevoflurane, although local effects at the vessel wall cannot be discounted. The high lactate washout detected in early reperfusion indicates high ischemic stress, lactate being the product of anaerobic metabolism. In the current ex vivo model, the ischemic impact on the hearts was similar in both groups, irrespective of application of sevoflurane. These data indicate that our ischemia/reperfusion protocol causes strong oxidative and metabolic stress, but, since sevoflurane treatment does not globally influence either, some other mechanism or a regional effect must be responsible for the protective action on the glycocalyx.
Previous research demonstrated a protective effect of NO on the glycocalyx, presumably via an antioxidative action.38 In turn, endogenous generation of NO in hearts requires an intact glycocalyx for mediating shear stress.37,39 By preventing washout of HA, as demonstrated for the first time in this work, sevoflurane will clearly augment this mechanism.
In the case of sevoflurane, it has been demonstrated that reactive oxygen species (ROS) and NO, or reaction products including peroxynitrite, mediate an organ-protective effect. Cardioprotection by sevoflurane preconditioning can be abolished by NO synthase inhibitors or by scavengers of ROS.40,41 However, the data on total myocardial urate and purine release presented in Table 1 suggest that, should any augmentation of ROS or NO by sevoflurane take place, this can only occur in a strictly localized, regional manner. It has also been observed that sevoflurane can bind to specific sites on proteins,42 especially albumin, which is known to be closely associated with and to stabilize the glycocalyx.7,20,24 In another study performed on isolated perfused guinea pig hearts, evidence was found of a protective effect of sevoflurane against ischemia/reperfusion-induced glycocalyx shedding, the mechanism seeming to involve attenuation of lysosomal CTSB release.23 Thus, binding of sevoflurane to the glycocalyx or to some protease(s) may prevent enzymatic shedding during post-ischemic reperfusion of the heart, besides possible mediation of protection by NO or ROS.
Limitations of the ex vivo model are apparent. For one, the crystalloid perfusate lacks formed and dissolved blood constituents. In particular, leukocytes, blood platelets, complement factors and enzymes will further influence reperfusion damage.7,16,20 Also, the low oncotic pressure and the absence of albumin lead to gradual deterioration of the glycocalyx.27,28,39 To minimize this, the perfusion protocol was kept to a duration of under 2 hours. Obviously, this precludes drawing any conclusions about long-term effects of treatment with sevoflurane on post-infarction myocardial recovery. Interestingly, the volatile anesthetic isoflurane has been found to reduce infarct size and to improve long-term regeneration of the heart in an in vivo animal model.43
It may well be that additional blood constituents will modify the action of sevoflurane in vivo as opposed to the findings presented here. Because sevoflurane attenuates intravascular activation of leukocytes and platelets,12–14,24 it is possible that augmentation of action occurs, enabling sevoflurane to retain the ability to substantially mitigate shedding of the glycocalyx. Thus, further studies, ultimately on patients undergoing major operations associated with vital organ ischemia/reperfusion, are necessary to substantiate our findings and, furthermore, to clarify the mechanisms underlying sevoflurane’s beneficial activity. However, already at this stage, use of sevoflurane as anesthetic in the situation of ischemia-reperfusion seems to be promising.
Conclusion
This study showed that application of sevoflurane at a concentration of 1 MAC prior to ischemia and throughout early reperfusion has a protective effect against ischemia/reperfusion injury to the endothelium in an ex vivo, isolated guinea pig heart model. Moreover, it reduces shedding of HA during the whole period of perfusion, irrespective of warm ischemia/reperfusion. Since vascular permeability depends on a double barrier, the endothelial glycocalyx acting as a competent component besides the barrier formed by endothelial cells themselves,14 preservation of the glycocalyx, especially of HA, and of endothelial cell integrity may be assumed.
Acknowledgments
Financial support and sponsorship: this work was supported by the Department of Anesthesiology and the Walter-Brendel-Centre of Experimental Medicine, Ludwig-Maximilians-University, Munich, Germany; and a scholarship from the Deutscher Akademischer Austauschdienst (DAAD) through the Sandwich Program (to Congcong Chen).
Disclosure
Peter Conzen has held lectures supported by Abbott, Germany. Thorsten Annecke has received a research grant from Abbott, Germany. The authors report no other conflicts of interest in this work.
References
Krolikowski JG, Bienengraeber M, Weihrauch D, et al. Inhibition of mitochondrial permeability transition enhances isoflurane-induced cardioprotection during early reperfusion: the role of mitochondrial KATP channels. Anesth Analg. 2005;101(6):1590–1596. | |
Obal D, Dettwiler S, Favoccia C, et al. The influence of mitochondrial KATP-channels in the cardioprotection of preconditioning and postconditioning by sevoflurane in the rat in vivo. Anesth Analg. 2005;101(5):1252–1260. | |
De Hert SG, Van der Linden PJ, Cromheecke S, et al. Cardioprotective properties of sevoflurane in patients undergoing coronary surgery with cardiopulmonary bypass are related to the modalities of its administration. Anesthesiology. 2004;101(2):299–310. | |
Tanaka K, Ludwing LM, Kersten JR, Pagel PS, Warltier DC. Mechanisms of cardioprotection by volatile anesthetics. Anesthesiology. 2004;100(3):707–721. | |
Liu H, Wang L, Eaton M, Schaefer S. Sevoflurane preconditioning limits intracellular/mitochondrial Ca2+ in ischemic newborn myocardium. Anesth Analg. 2005;101(2):349–355. | |
Landoni G, Biondi-Zoccai GG, Zangrillo A, et al. Desflurane and sevoflurane in cardiac surgery: a meta-analysis of randomized clinical trials. J Cardiothorac Vasc Anesth. 2007;21(4):502–511. | |
Becker BF, Jacob M, Leipert S, Salmon AH, Chappell D. Degradation of the endothelial glycocalyx in clinical settings: searching for the sheddases. Br J Clin Pharmacol. 2015;80(3):389–402. | |
Patel HH, Tsutsumi YM, Head BP, et al. Mechanisms of cardiac protection from ischemia/reperfusion injury: a role for caveolae and caveolin-1. FASEB J. 2007;21(7):1565–1574. | |
Marinovic J, Bosnjak ZJ, Stadnicka A. Distinct roles for sarcolemmal and mitochondrial adenosine triphosphate-sensitive potassium channels in isoflurane-induced protection against oxidative stress. Anesthesiology. 2006;105(1):98–104. | |
Feng J, Lucchinetti E, Ahuja P, et al. Isoflurane postconditioning prevents opening of the mitochondrial permeability transition pore through inhibition of glycogen synthase kinase 3beta. Anesthesiology. 2005;103(5):987–995. | |
Dreyer WJ, Michael LH, Millman EE, Berens KL. Neutrophil activation and adhesion molecule expression in a canine model of open heart surgery with cardiopulmonary bypass. Cardiovasc Res. 1995;29(6):775–781. | |
Kowalski C, Zahler S, Becker BF, et al. Halothane, isoflurane, and sevoflurane reduce postischemic adhesion of neutrophils in the coronary system. Anesthesiology. 1997;86(1):188–195. | |
Heindl B, Reichle FM, Zahler S, Conzen PF, Becker BF. Sevoflurane and isoflurane protect the reperfused guinea pig heart by reducing postischemic adhesion of polymorphonuclear neutrophils. Anesthesiology. 1999;91(2):521–530. | |
Heindl B, Conzen PF, Becker BF. The volatile anesthetic sevoflurane mitigates cardiodepressive effects of platelets in reperfused hearts. Basic Res Cardiol. 1999;94(2):102–111. | |
Rehm M, Zahler S, Lötsch M, et al. Endothelial glycocalyx as an additional barrier determining extravasation of 6% hydroxyethyl starch or 5% albumin solutions in the coronary vascular bed. Anesthesiology. 2004;100(5):1211–1223. | |
Becker BF, Chappell D, Jacob M. Endothelial glycocalyx and coronary vascular permeability: the fringe benefit. Basic Res Cardiol. 2010;105(6):687–701. | |
Mulivor AW, Lipowsky HH. Inflammation- and ischemia-induced shedding of venular glycocalyx. Am J Physiol Heart Circ Physiol. 2004;286(5):H1672–H1680. | |
Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflugers Archi. 2000;440(5):653–666. | |
Platts SH, Duling BR. Adenosine A3 receptor activation modulates the capillary endothelial glycocalyx. Circ Res. 2004;94(1):77–82. | |
Becker BF, Chappell D, Bruegger D, Annecke T, Jacob M. Therapeutic strategies targeting the endothelial glycocalyx: Acute deficits, but great potential. Cardiovasc Res. 2010;87(2):300–310. | |
Mulivor AW, Lipowsky HH. Role of glycocalyx in leukocyte-endothelial cell adhesion. Am J Physiol Heart Circ Physiol. 2002;283(4):H1282–H1291. | |
Vink H, Constantinescu AA, Spaan JA. Oxidized lipoproteins degrade the endothelial surface layer: implications for platelet-endothelial cell adhesion. Circulation. 2000;101(13):1500–1502. | |
Annecke T, Chappell D, Chen C, et al. Sevoflurane preserves the endothelial glycocalyx against ischaemia-reperfusion injury. Br J Anaesth. 2010;104(4):414–421. | |
Chappell D, Heindl B, Jacob M, et al. Sevoflurane reduces leukocyte and platelet adhesion after ischemia-reperfusion by protecting the endothelial glycocalyx. Anesthesiology. 2011;115(3):483–491. | |
Mobert J, Becker BF. Cyclooxygenase inhibition aggravates ischemia-reperfusion injury in the perfused guinea pig heart: Involvement of isoprostanes. J Am Coll Cardiol. 1998;31(7):1687–1694. | |
Chappell D, Jacob M, Hofmann-Kiefer K, et al. Hydrocortisone preserves the vascular barrier by protecting the endothelial glycocalyx. Anesthesiology. 2007;107(5):776–784. | |
Jacob M, Bruegger D, Rehm M, et al. Contrasting effects of colloid and crystalloid resuscitation fluids on cardiac vascular permeability. Anesthesiology. 2006;104(6):1223–1231. | |
Jacob M, Bruegger D, Rehm M, et al. The endothelial glycocalyx affords compatibility of Starling’s principle and high cardiac interstitial albumin levels. Cardiovasc Res. 2007;73(3):575–586. | |
Förster H, Wicarkzyk C, Dudziak R. Bestimmung der Plasmaelimination von Hydroxyethylstärke und von Dextran mittels verbesserter analytischer Methode. [Determination of the plasma elimination of hydroxyethyl starch and dextran using improved analytical methods]. Infusionsther Klin Ernahr. 1981;8(2):88–94. German. | |
Becker BF. Towards the physiological function of uric acid. Free Radic Biol Med. 1993;14(6):615–631. | |
Vogel J, Sperandio M, Pries AR, et al. Influence of the endothelial glycocalyx on cerebral blood flow in mice. J Cereb Blood Flow Metab. 2000;20(11):1571–1578. | |
Kurzelewski M, Czarnowska E, Beresewicz A. Superoxide- and nitric oxide-derived species mediate endothelial dysfunction, endothelial glycocalyx disruption, and enhanced neutrophil adhesion in the post-ischemic guinea-pig heart. J Physiol Pharmacol. 2005;56(2):163–178. | |
Rehm M, Bruegger D, Christ F, et al. Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation. 2007;116(17):1896–1906. | |
Constantinescu AA, Vink H, Spaan JA. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler Thromb Vasc Biol. 2003;23(9):1541–1547. | |
Chappell D, Brettner F, Doerfler N, et al. Protection of the glycocalyx decreases platelet adhesion after ischaemia/reperfusion: an animal study. Eur J Anaesthesiol. 2014;31(9):474–481. | |
Nieuwdorp M, van Haeften TW, Gouverneur MC, et al. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes. 2006;55(2):480–486. | |
Tarbell JM, Ebong EE. The endothelial glycocalyx: a mechano-sensor and -transducer. Sci Signal. 2008;1(40):pt8. | |
Bruegger D, Rehm M, Jacob M, et al. Exogenous nitric oxide requires an endothelial glycocalyx to prevent postischemic coronary vascular leak in guinea pig hearts. Crit Care. 2008;12(3):R73. | |
Jacob M, Rehm M, Loetsch M, et al. The endothelial glycocalyx prefers albumin for evoking shear stress-induced, nitric oxide-mediated coronary dilatation. J Vasc Res. 2007;44(6):435–443. | |
Riess ML, Kevin LG, McCormick J, et al. Anesthetic preconditioning: the role of free radicals in sevoflurane-induced attenuation of mitochondrial electron transport in Guinea pig isolated hearts. Anesth Analg. 2005;100(1):46–53. | |
Novalija E, Kevin LG, Eells JT, Henry MM, Stowe DF. Anesthetic preconditioning improves adenosine triphosphate synthesis and reduces reactive oxygen species formation in mitochondria after ischemia by a redox dependent mechanism. Anesthesiology. 2003;98(5):1155–1163. | |
Dubois BW, Cherian SF, Evers AS. Volatile anesthetics compete for common binding sites on bovine serum albumin: a 19F-NMR study. Proc Natl Acad Sci U S A. 1993;90(14):6478–6482. | |
Agnic I, Filipovic N, Vukojevic K, et al. Effects of isoflurane postconditioning on chronic phase of ischemia-reperfusion heart injury in rats. Cariovasc Pathol. 2015;24(2):94–101. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.