")
Back to Journals » Journal of Blood Medicine » Volume 10
Serum homocysteine and disease severity in sickle cell anemia patients in Lagos
Authors Uche E, Adelekan O , Akinbami A, Osunkalu V , Ismail K, Ogbenna AA, Badiru M , Dosunmu A, Oluwole E , Kamson O
Received 15 December 2018
Accepted for publication 1 April 2019
Published 8 May 2019 Volume 2019:10 Pages 127—134
DOI https://doi.org/10.2147/JBM.S198316
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin Bluth
Ebele Uche,1 Oluwaseun Adelekan,2 Akinsegun Akinbami,1 Vincent Osunkalu,3 Kamal Ismail,1 Ann Abiola Ogbenna,3 Mulikat Badiru,1 Adedoyin Dosunmu,1 Esther Oluwole,4 Omolara Kamson5
1Department of Haematology and Blood Transfusion, Lagos State University College of Medicine, Lagos, Nigeria; 2Department of Haematology and Blood Transfusion, General Hospital Marina, Lagos, Nigeria; 3Department of Haematology and Blood Transfusion, College of Medicine, University of Lagos, Lagos, Nigeria; 4Department of Community Health and Primary Care, College of Medicine University of Lagos, Lagos, Nigeria; 5Department of Haematology and Blood Transfusion, Lagos State University Teaching Hospital, Lagos, Nigeria
Purpose: Hypercoagulability in sickle cell anemia (SCA) may be responsible for the increased development of vascular occlusion in certain organs as well as acute pain episodes. The causes of hypercoagulability in SCA are multifactorial and include raised homocysteine levels. This study, therefore, aimed to determine serum homocysteine levels in SCA patients in steady state and to correlate its levels with SCA disease severity.
Patients and Methods: This was a cross-sectional study done among SCA patients in steady state attending the Haematology Clinic of the Lagos State University Teaching Hospital (LASUTH). Matched age and sex HbAA controls were also recruited. Serum homocysteine of each participant was done with enzyme-linked immunosorbent assay and disease severity score assessed in every SCA patient using clinical and laboratory parameters.
Results: The mean value for homocysteine in the study group (SCA patients) was 19.80±19.75 μmol/L whilst that of the control group was 9.16±4.29 μmol/L. Thirty-nine out of 96 (46.6%) SCA patients had elevated homocysteine levels (>15 μmol/L) whilst all 96 participants in the control group had normal homocysteine levels. The difference in the means in the two groups was statistically significant with p=0.001. Majority (62.5%) of the SCA patients had a mild disease (severity score ≤3). There was a significant correlation between serum homocysteine levels and disease severity scores with p=0.04; χ2,=4.04.
Conclusion: Homocysteine levels were significantly higher in HbSS patients compared with matched HbAA controls and showed a positive correlation with disease severity scores in the SCA patients.
Keywords: sickle cell anemia, disease severity, homocysteine
Introduction
Sickle cell anemia (SCA) is the most common form of sickle cell disease1 and worldwide, it is one of the commonest inherited disorders.2–5
The prevalence of sickle cell disease is highest in sub-Saharan Africa.2,4,6 Current studies demonstrate that over 230,000 affected children are born in this region annually which is an estimated 80% of the global total.4,6
Globally, Nigeria has the highest prevalence of the disease with reported prevalence values of between 2% and 3%.1,4
Sickle cell disease is a multi-systemic disorder and2–5 occurs when an individual is homozygous for the sickle cell mutation (HbSS or SCA) or is compound heterozygote for the sickle hemoglobin and β-thalassemia, hemoglobin C, and some less common β-globin mutations.7 SCA is the most common form of sickle cell disease. Hemoglobin S is produced by a mutation in the HBB gene (Beta-globin gene) located on chromosome 11p15.5in which the 17th nucleotide is changed from adenine to thymine (GTG→GAG),7 resulting in the substitution of valine for glutamic acid normally at the sixth position of the amino terminus of the β chain of hemoglobin.
The manifestations seen in SCA are as a result of two major pathophysiological processes: vaso-occlusion with ischemia-perfusion injury and hemolytic anemia.4
Hypercoagulability in SCA may be responsible for the increased development of vascular occlusion in certain organs7,8 and acute pain episodes.7,9 The causes of hypercoagulability in SCA are multifactorial. Factors such as increased plasma levels of homocysteine, reduced levels of natural anticoagulants like Protein C and S,10 increased levels of thrombin generation markers like thrombin-anti-thrombin complexes or protein fragment 1+2 (F1+2),11 increased D-dimer complexes, circulating antiphospholipid antibodies, increased tissue factor expression and adherence of sickled erythrocytes to antibodies, increased tissue factor expression and adherence of sickled erythrocytes to the vascular endothelium, Factor V Leiden and Prothrombin gene mutation have all been implicated.12,13
Homocysteine is a highly reactive sulfur-containing amino acid which is known to cause endothelial injury, endothelial dysfunction, and thrombin formation.14 Studies have revealed that elevated plasma total homocysteine may result from a deficiency of folate and vitamin B12 and could, therefore, be used as a surrogate marker of folate and cobalamin (vitamin B12) deficiency.15,16 Elevated plasma homocysteine levels are also associated with increased risk of cardiovascular diseases such as atherosclerosis, coronary artery disease, and ischaemic stroke.17,18 Homocysteine has also been proposed as a hemolytic toxin.19 Although the precise mechanism of the hemolytic effect of homocysteine is not clear, its pro-oxidant attributes have been suggested as a cause.
There are multiple mechanisms by which homocysteine may induce vascular injury. Oxidative stress by free radicals formed during the oxidation of reduced homocysteine may directly injure endothelial cells.20 The prothrombotic effects of homocysteine such as a reduction of endothelial cell tissue plasminogen activator binding sites, activation of factor VIIa and V, inhibition of protein C, increased fibrinopeptide, impaired thrombomodulin function, and increased blood viscosity have been demonstrated in acute coronary syndromes.21–23 Marked platelet accumulation may be secondary to direct pro-aggregatory effects of homocysteine or an impairment in endothelium-mediated platelet inhibition.24 Also, the thiolactone metabolite of homocysteine can combine with LDL-cholesterol to produce aggregates which are taken up by vascular macrophages in the arterial intima and are subsequently released lipid into atherosclerotic plaques.25
Homocysteine also increases smooth muscle cell proliferation and enhances collagen production. Furthermore, persistent exposure of endothelial cells to homocysteine reduces the activity of dimethylarginine dimethylaminohydrolase enzyme which degrades dimethylarginine, an endogenous inhibitor of nitric oxide synthase which impairs the production of nitric oxide which is a potent vasodilator.24 The effect of impaired nitric oxide release can then trigger and escalate atherothrombogenesis and oxidative stress.
In individuals with phenotype Hb SS, elevated homocysteine levels have been associated with an increased risk of hypercoagulability and subsequent thrombosis through the inhibition of protein C anticoagulant pathway which may contribute to the pathogenesis of several SCD-related complications such as stroke.26–30
Measurement of disease severity in individuals with SCA
There is currently a general lack of consensus on the definition of disease severity and the best way or “gold standard” to measure this concept.31,32 Some researchers have however suggested “severity scores”, though none adequately encompasses severity of disease.33–36 This is as a result of the complex relationships that exist between clinical and laboratory measures of disease expression and the need to identify genetic variants that impact the severity of the disease.37,38 Furthermore the effect of geographical, socio-economic, and environmental factors which are recognized modifiers of disease severity also have to be put into consideration.39
Furthermore, a systematic review done revealed that composite indices incorporating clinical events, laboratory tests, and treatment data have been used by several researchers to measure overall disease severity, though with variability observed in the methodological quality of these indices.32 The analysis revealed that about 55 indices were used but the five most frequently used variables were painful vaso-occlusive crises (87% of the indices), central nervous system abnormalities, ie, CVD, cerebral vasculopathy, seizures, encephalopathy (67%), aseptic/avascular necrosis of the bones (53%), acute chest syndrome (43%), and number of blood transfusions (40%). However, many of the indices used in this review were found to be confounded by factors beyond the severity of the disease especially health care interventions.32
A study conducted among children with SCA in south-western Nigeria over a one-year period, assessed a total of 15 parameters to reflect each patient’s present state their state during the previous one year and lifetime complications.39 These parameters were scored according to the frequency of occurrence and severity with scores ranging from 1 to 5. The total score was then calculated for each child (range from 0 to 34), and the disease was classified as mild when the total score was <8, moderate was 8–17, and severe when the total score was >17.39 Another study done among Nigerian subjects with SCA in steady states, estimated the disease severity scores using three indices; mean crisis per year, number of organ (system) complications, and degree of anemia after subjects were examined clinically and hospital records reviewed.40 The complications analyzed included: anemic heart failure, osteomyelitis, avascular necrosis of the femoral head, lobar pneumonia, pigment gall stones, growth retardation, liver failure, renal failure, and recurrent seizures after a cerebrovascular accident. The severity scores were categorized as mild, moderate, or severe. Subjects with a total severity score of ≤3 were considered to have a mild disease, 4–7 were classified as moderate and >7 deemed to have a severe disease.40
Therefore, it is imperative to have a universally accepted method of measuring disease severity in sickle cell patients which would allow a reliable prognosis and could guide therapeutic decision-making.
This study aimed to determine serum homocysteine levels in HbSS patients in steady state as well as in age and sex-matched controls. It also determined the disease severity scores of the patients and assessed the relationship between serum homocysteine levels in HbSS patients and disease severity.
Material and methods
Study area and population
The study was done among SCA patients attending the adult Haematology Clinic of the Lagos State University Teaching Hospital (LASUTH) Lagos, Nigeria. Apparently healthy individuals were used as controls.
Study design
This study was cross sectional and hospital based involving SCA patients in steady state and matched HbAA controls.
Study period
The study was carried out over a period of eight months from May 2016 to December 2016
Sampling technique
Purposive non-probability sampling technique was used to recruit presumably healthy staff and students of LASUTH Ikeja with hemoglobin phenotype AA. Only individuals with hemoglobin phenotype AA were used as the control population. A sampling frame containing the list of individuals with SCA attending the adult Haematology Clinic in LASUTH was drawn and subjects were selected randomly. However, only individuals with a hemoglobin electrophoresis result showing the S band only were enrolled in the study.
Inclusion criteria
- Age between 18 and above.
- Known SCA patients (screened by the hemoglobin S solubility test and diagnosed by cellulose acetate electrophoresis at pH 8.6) who were in steady state. Steady state was defined as the period free of crisis extending from at least three weeks since the last clinical event and three months or more since the last blood transfusion, to at least one week before the start of a new clinical event.41
- Hemoglobin AA persons (confirmed by cellulose acetate electrophoresis at pH 8.6).
- Those who met the above criteria and agreed to participate in the study by signing the informed consent form.
Exclusion criteria
- Confirmed Hb phenotype AC, AS.
- Individuals with a history of acute or chronic illness eg, febrile illness, hypertension, epilepsy, asthma, diabetes mellitus.
- Intravenous drug abusers.
- Participants who refused to give consent.
- SCA patients in crisis
Ethical considerations
Ethical approval was obtained from the Health Research and Ethics Committee of the Lagos State University Teaching Hospital prior to the commencement of the study and conducted in accordance with the Declaration of Helsinki.
Participants’ informed consent
A written informed consent was obtained from all the participants before being recruited into the study.
Questionnaire administration and history taking
Each participant was interviewed to obtain relevant demographic and clinical data with the use of a questionnaire. The questionnaire was administered to each participant by the researcher. Information on the subjects and their medical history including disease complications were retrieved from their case notes.
Assessment of disease severity score
The disease severity score was assessed using a modified scoring system by Hedo et al.42,43 (Table 1).
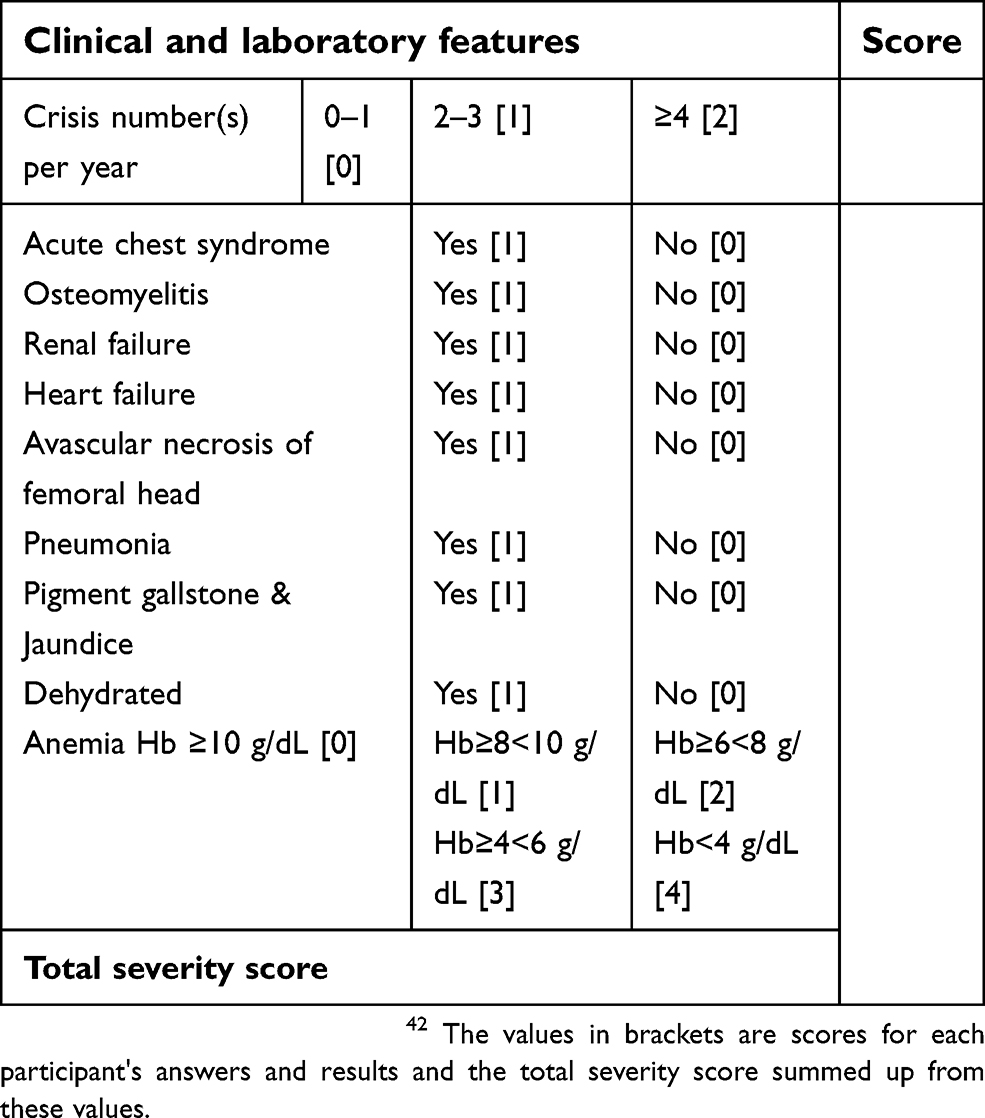 | Table 1 Modified disease severity scoring system for sickle cell anemia |
The total severity score was calculated as Mild SCA [≤3], Moderate SCA [>3 but ≤7], and Severe SCA [>7].42,43
Specimen collection and storage
Eight milliliters of venous blood was drawn from each subject after informed consent. This was done from an intravenous access (the antecubital vein was mostly used) and under aseptic conditions with the use of a disposable vacutainer needle. Five milliliters of this was dispensed into sodium ethylene diamine tetra-acetate (EDTA) specimen bottles. This sample was used for a full blood count using the automated Sysmex autoanalyzer machine. This was analyzed within 2 hrs of collection. Some of the blood collected in the EDTA specimen bottle was used for cellulose acetate electrophoresis test to confirm the Hb phenotypes of all the subjects.
Three milliliters of blood was transferred into plain tubes and put on ice for homocysteine estimation using enzyme-linked immunosorbent assay kit from Elabscience with manufacturer’s instructions strictly followed.
Results
Age and gender
This was a cross-sectional study of 192 participants consisting of 96 individuals with SCA (cases) and 96 with HbAA genotype (controls). A total of 202 participants were initially recruited into the study. Ten participants were excluded from the study altogether, seven on the basis of their hemoglobin genotype while the others had incompletely filled questionnaires.
Overall, the minimum age was 18 years and the maximum 59 years with a mean of 30.48±11.16 years. A total of 102 were females (53.1%) while 90 (42.9%) were males. Amongst the cases, 51 (53.1%) were females and 45 were males (46.9%) and their mean age was 29.33±10.37 years. The control group was exactly the same in number and gender distribution with the cases (Table 2).
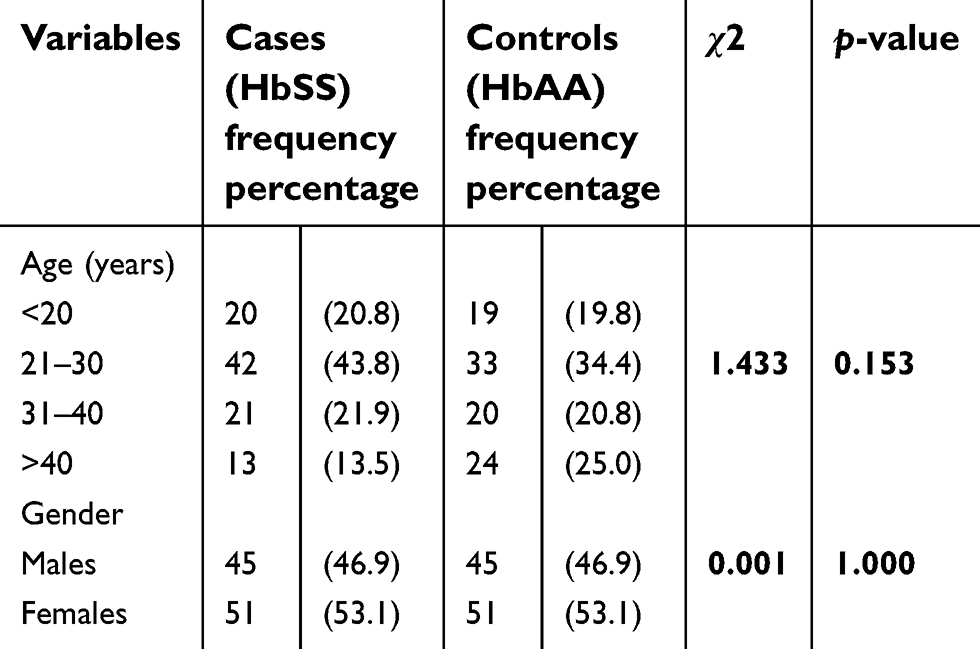 | Table 2 Age and gender distribution of study participants |
Serum homocysteine levels
The mean homocysteine value amongst the SCA patients was 19.80±19.75 µmol/L and 39 out of 96 of them (46.6%) had elevated homocysteine levels (>15 µmol/L). All subjects in the control group (100%) had normal homocysteine levels and the mean value in this group was 9.16±4.29 µmol/L. The mean values of both cases and controls were statistically significant with a p-value of 0.001 (Table 3)
 | Table 3 Comparison of proportions and mean homocysteine levels of cases and controls |
Disease severity scores
Majority of the subjects 60 (62.5%) had a disease severity score ≤3 (mild), while the remaining 36 (37.5%) had a moderate disease severity score of 4–7 (Table 4).
 | Table 4 Distribution of disease severity scores among individuals with sickle cell anemia |
Homocysteine and disease severity scores in individuals with SCA
A statistically significant relationship was established between serum homocysteine levels and disease severity scores in subjects with SCA with a p-value of 0.04; χ2value of 4.04 (Table 5).
 | Table 5 The relationship between serum homocysteine levels and disease severity scores in HbSS subjects |
Discussion
SCA is a genetic disease characterized by a hypercoagulable state, and the pathogenesis is considered to be multifactorial. Elevated homocysteine levels have been identified as an independent risk factor for thrombo-embolic complications observed in this subset of individuals. Abnormal homocysteine levels have been reported in some published data in patients with SCA.29 This study reported statistically significantly higher serum homocysteine levels among the HbSS subjects (19.80±19.75 µmol/L) in comparison to that of HbAA controls (9.16±4.29 µmol/L) with a p-value of 0.001. This is similar to a study conducted among adults with SCA receiving care at a New York Hospital which reported significantly higher homocysteine levels in patients with SCA compared to control subjects.44 Another study45 by Lowenthal et al, also observed elevated concentration of homocysteine in patients with SCA in spite of their elevated plasma folate and vitamin B12 levels which was similar to that found in controls. These studies, however, are at variance with a survey in Ibadan by Olaniyi et al46 in which 60 HbSS subjects were studied, and the mean plasma homocysteine level was found to be significantly lower in HbSS subjects when compared with the HbAA control group (p<0.001). Similarly, a study47 which evaluated children with SCA reported homocysteine levels similar to that of control subjects, and there was no correlation between their levels of homocysteine and red cell folate concentration and clinical or laboratory measures. Therefore, in this present study, it is plausible that the concentration of folate required to normalize homocysteine levels in individuals with SCA may be higher than in normal subjects since they have a higher nutritional requirement for folic acid than the general population.
In addition, the kidneys play an important role in the metabolism of homocysteine,48, therefore, an impaired renal function which is not an uncommon finding in sickle cell anemia patients could be a cause of increased homocysteine levels reported in this study.
Individuals with SCA are known to demonstrate great clinical diversity. Indeed, variability can also occur between patients and even within the same individual. The clinical spectrum of the disease could range from mild asymptomatic disease to persistent severe life- threatening conditions associated with multiple organ dysfunction. Our study calculated the disease severity state based on 10 parameters derived from physical examination, laboratory investigations and findings retrieved from hospital records. The parameters analyzed were a number of crisis per year, acute chest syndrome, osteomyelitis, renal failure, heart failure, avascular necrosis of the femoral head, pneumonia, pigment gallstones and jaundice, dehydration and hemoglobin levels. The total severity score was calculated as Mild SCA [≤3], Moderate SCA [>3 but ≤7], and Severe SCA [>7].42,43 Most (62.5%) of the subjects in our study had a mild disease severity score. This finding is contrary to observations from a study40 done among 73 steady-state HbSS individuals attending the Sickle cell disease clinic at the University College Hospital, Ibadan, Nigeria which reported a mild disease severity score in only about 25% of the patients. In evaluating the clinical severity of SCA in Nigerian children, Adegoke and Kuti39 assessed a total of 15 parameters to reflect each patient’s present clinical state, their clinical state during the previous one year and lifetime complications. They reported a moderate disease severity score in most of the children.
Similar to our study, research done among children with SCA in Yemen,49 Senegal,50 and Saudi Arabia51 reported that most had a mild disease severity score.
Possible reasons for this difference in disease severity scores could be that some of the study participants may have taken vaccinations against pneumococcal infections which reduces the incidence of pneumonia, or some could be on hydroxyurea which decreases painful crisis. It could also possibly be related to an inheritance of β-globin haplotypes associated with a mild disease or a co-inheritance with α-thalassemia also associated with mild disease.
We established a statistically significant relationship between serum homocysteine levels and disease severity scores in subjects with SCA. This is similar to a study by Houston et al, who documented a statistically significant relationship between plasma homocysteine levels and disease severity in individuals with SCA.26
Similarly, a direct correlation between plasma homocysteine levels and frequency of vaso-occlusive crisis and disease severity was seen in an evaluation of 30 patients with SCA recruited from Ibn-Al-Baldy Hospital in Iraq.14
However, in contrast, Rugani MA in his study did not report a significant correlation between serum homocysteine and severity of the vaso-occlusive crisis in sickle cell disease.52
It could be inferred that elevated homocysteine levels observed in this present study could be a contributory factor to the development of hypercoagulability and vaso-occlusion resulting complications seen in the HbSS subjects. However, it is known that patients with nutrient deficiencies eg, vitamin B12 and B6, as well as folate deficiency, have elevated levels of homocysteine. Patients with elevated homocysteine levels may need to be evaluated regularly and treatment with combination B-vitamin therapy instituted in order to prevent the myriad of complications associated with hyperhomocysteinuria.
However, presently no clinically salient pro-atherogenic homocysteine concentration threshold has been identified for improved targeting of individuals who may benefit from this combination therapy.53
Study limitations
- This study measured serum homocysteine levels among the subjects as an indirect index of folate lack; the effects of other nutrients deficiency such as vitamin B12 and riboflavin were outside the scope of this work and could have impacted on the results obtained in this study.
- Furthermore, in the folate metabolism pathway, there are other metabolic enzymes and co-factors, which are coded by genes and whose mutations could result in elevated serum homocysteine levels which may hypothetically contribute to the severity or differential phenotypic expression in adults with phenotype Hb SS but were also outside the scope of this study.
Conclusion
Subjects with SCA had significantly higher mean serum homocysteine when compared with controls. A significant relationship was established between serum homocysteine levels and disease severity scores in subjects with SCA.
Acknowledgments
The authors are grateful to Mr. Sola Ojewunmi who coordinated the ELISA analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Akinyanju OO. Profile of sickle cell disease in Nigeria. Ann NY Acad Sci. 1989;565:126–136.
2. Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86:480–487.
3. Rees D, Williams T, Gladwin M. Sickle-cell disease. Lancet. 2010;376:2018–2031. doi:10.1016/S0140-6736(10)61029-X
4. Serjeant GR. Sickle-cell disease. Lancet. 1997;350:725–730.
5. Taylor TD, Noguchi H, Totoki Y, et al. Human chromosome 11 DNA sequence and analysis including novel gene identification. Nature. 2006;440:491–500. doi:10.1038/nature04614
6. Rahimi Z, Parsian A. Sickle cell disease and venous thromboembolism. Mediterr J Hematol Infect Dis. 2011;3:1–7. doi:10.4084/mjhid.2011.024
7. Steinberg MH. Sickle cell anaemia, the first molecular disease: overview of molecular etiology, pathophysiology, and therapeutic approaches. ScientificWorld J. 2008;8:1295–1324. doi:10.1100/tsw.2008.157
8. Hebbel R, Robert P. Ischemia-reperfusion injury in sickle cell anaemia: relationship to acute chest syndrome, endothelial dysfunction, arterial vasculopathy, and inflammatory pain. Hematol Oncol Clin North Am. 2014;28:181–198. doi:10.1016/j.hoc.2013.11.005
9. Colombatti R, De Bon E, Bertomoro A, et al. Coagulation activation in children with sickle cell disease is associated with cerebral small vessel vasculopathy. PLoS One. 2013;8:311–318.
10. El-Hazmi MA, Warsy AS, Bahakim H. Blood proteins C and S in sickle cell disease. Acta Haematol. 1993;90:114–119. doi:10.1159/000204390
11. Liesner R, Mackie I, Cookson J, et al. Prothrombotic changes in children with sickle cell disease: relationships to cerebrovascular disease and transfusion. Br J Haematol. 1998;103:1037–1044. doi:10.1046/j.1365-2141.1998.01121.x
12. Ataga KI. Hypercoagulability and thrombotic complications in hemolytic anaemias. Haematologica. 2009;94:1481–1484. doi:10.3324/haematol.2009.013672
13. Ataga KI, Cappellini MD, Rachmilewitz EA. β-thalassemia and sickle-cell anaemia as paradigms of hypercoagulability. Br J Haematol. 2007;139:3–13. doi:10.1111/j.1365-2141.2007.06740.x
14. Sati‘Abbas S, Abul–Razak N, Mustafa N, Abd Ali R. Homocysteine, folic acid, vitamin B12 and pyridoxine: effects on vaso-occlusive crisis in sickle cell anemia and sickle –thalassemia. Ipmj. 2011;10:473–479.
15. Chao-Hung HO The influence of age, sex, vitamin B12, folate levels and methylenetetrahydrofolate reductase C677T genetic mutations on plasma homocysteine in the Chinese population. Haematologica. 2000;85:1051–1054.
16. Klee GG. Cobalamin and folate evaluation; measurement of methylmalonic acid and homocysteine vs vitamin B12 and folate. Clin Chem. 2000;46:1277–1283.
17. Toole JF, Malinow MR, Chambless LE, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA. 2004;291:565–575. doi:10.1001/jama.291.5.565
18.
19. Ventura P, Panini R, Tremosini S, Salvioli G. A role for homocysteine increase in haemolysis of megaloblastic anaemias due to vitamin B(12) and folate deficiency: results from an in vitro experience. Biochim Biophys Acta. 2004;1739:33–42. doi:10.1016/j.bbadis.2004.08.005
20. Mansoor MA, Bergmark C, Svardal AM, Lonning PE, Ueland PM. Redox status and protein binding of plasma homocysteine and other aminothiols in patients with early-onset peripheral vascular disease. Homocysteine and peripheral vascular disease. Arterioscler Thromb Vasc Biol. 1995;15:232.
21. Al-Obaidi MK, Philippou H, Stubbs PJ, et al. Relationships between homocysteine, factor VIIa, and thrombin generation in acute coronary syndromes. Circulation. 2000;101:372.
22. Nappo F, De Rossa N, Marfella R, et al. Impairment of endothelial functions by acute hyperhomocysteinemia and reversal by antioxidant vitamins. JAMA. 1999;281:2113. doi:10.1001/jama.281.22.2113
23. Hajjar KA. Homocysteine-induced modulation of tissue plasminogen activator binding to its endothelial cell membrane receptor. J Clin Invest. 1993;91:2873. doi:10.1172/JCI116532
24. Stamler JS, Osborne JA, Jaraki O, et al. Adverse vascular effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. J Clin Invest. 1993;91:308.
25. McCully KS. Homocysteine and vascular disease. Nat Med. 1996;2:386. doi:10.1038/nm0496-386
26. Houston PE, Rana S, Sekhsaria S, Perlin E, Kim KS, Castro OL. Homocysteine in sickle cell: relationship to stroke. Am J Med. 1997;103:192–196.
27. Van der Dijis FLP, Schong J, Brouwer DAJ, et al. Elevated homocysteine levels indicate suboptimal folate status in paediatric sickle cell patients. Am J Hematol. 1998;59:192–198.
28. Prengler M, Pavlakis SG, Prohovik I, Adams RJ. Sickle cell disease: the neurological complications. Ann Neurol. 2002;51:543–552. doi:10.1002/ana.10192
29. Al-Maktari L, Al-Nuzaily M, Bamashmoos S, Taresh S, Ali F. Thrombotic events in pateints with sickle cell anaemia: relationship to protein C, S and total homocysteine levels. Int J Curr Res Aca Rev. 2014;2:17–24.
30. Pandey S, Pandey HR, Mishra RM, Pandey S, Saxena R. Increased homocysteine level in Indian sickle cell patients. Ind J Clin Biochem. 2012;27:103–104.
31. Driss A, Asare KO, Hibbert JM, Gee BE, Adamkiewicz TV, Stiles JK. Sickle cell disease in the post genomic era: a monogenic disease with a polygenic phenotype. Genomic Insights. 2009;2:23–48. doi:10.4137/GEI.S2626
32. Van Den Tweel XW. Measurement of disease severity in patients with sickle cell disease: a systematic review [PhD Thesis] 2009; 42–52.
33. Steinberg MH, Dreiling BJ, Morrison FS, Necheles TF. Mild sickle cell disease. Clinical and Laboratory studies. JAMA. 1973;224:317–321.
34. Odenheimer DJ, Sarnaik SA, Whitten CF, Rucknagel DL, Sing CF. The relationship between fetal hemoglobin and disease severity in children with sickle cell anaemia. Am J Med Genet. 1987;27:525–535. doi:10.1002/ajmg.1320270305
35. Bray GL, Muenz L, Makris N, Lessin LS. Assessing clinical severity in children with sickle cell disease. Preliminary results from a cooperative study. Am J Pediatr Hematol Oncol. 1994;16:50–54.
36. Miller ST, Sleeper LA, Pegelow CH, et al. Prediction of adverse outcomes in children with sickle cell disease. N Engl J Med. 2000;342:83–89. doi:10.1056/NEJM200001133420203
37. Steinberg MH. Predicting clinical severity in sickle cell anaemia. Br J Haematol. 2005;129:465–481. doi:10.1111/j.1365-2141.2005.05411.x
38. Sebastiani P, Nolan VG, Baldwin CT, et al. A network model to predict the risk of death in sickle cell disease. Blood. 2007;110:2727–2735. doi:10.1182/blood-2007-04-084921
39. Adegoke SA, Kuti BP. Evaluation of clinical severity of sickle cell anaemia in Nigerian children. J Appl Haematol. 2013;4:58–64.
40. Hedo CC, Okpala IE, Aken‘Ova YA. Foetal haemoglobin levels in Nigerians with sickle cell anaemia. A revisitation. Trop Geogr Med. 1993;45:162–164.
41. Akinola NO, Stevens SM, Franklin IM, Nash GB, Stuart J. Subclinical ischaemic episodes during the steady state of sickle cell anaemia. J Clin Pathol. 1992;45:902–906.
42. Hedo CC, Aken'ova YA, Okpala IE, Durojaiye AO, Salimonu LS. Acute phase reactants and severity of homozygous sickle cell disease. J InternMed. 1993;233:467–470.
43. Akinlade KS, Atere AD, Rahamon SK, Olaniyi JA. Serum levels of copeptin, C-reactive protein and cortisol in different severity groups of sickle cell anaemia. Niger J Physiol Sci. 2013;28:159–164.
44. Dhar M, Bellevue R, Brar S, Carmel R. Mild hyperhomocysteinemia in adult patients with sickle cell disease: a common finding unrelated to folate and cobalamin status. Am J Hemat. 2004;76:114–120. doi:10.1002/ajh.20073
45. Lowenthal EA, Mayo MS, Cornwell PE, Thornley-Brown D. Homocysteine elevation in sickle cell disease. J Am Coll Nutri. 2000;24:374–379.
46. Olaniyi J, Akinlade K, Atere A, Arinola O. Plasma homocysteine, methyl-malonic acid, vitamin B12 and folate levels in adult Nigerian sickle cell anaemia patients. Br J Med Med Res. 2014;4:1327–1334. doi:10.9734/BJMMR/2014/3989
47. Rodriguez-Cortes HM, Griener JC, Hyland K, et al. Plasma Homocysteine levels. J Pediatri Hemat/Onco. 1999;21(3):173–174.
48. Friedman AN, Boston AG, Selhub J, Levey AS, Rosenberg IH, Kidney T. Homocysteine metabolism. J Am Soc Neph. 2001;12(1):2181–2189.
49. Al-Saqladi AM, Delpisheh A, Bin-Gadeem HA, Brabin BJ. Severity of sickle cell disease in Yemeni children. J Trop Pediat. 2009;55:208–209. doi:10.1093/tropej/fmn109
50. Diagne I, Ndiaye O, Moreira C, et al. Sickle cell disease in children in Dakar, Senegal. Arch Pediatr. 2000;7(1):16–24.
51. El-Hazmi MA. Clinical and haematological diversity of sickle cell disease in Saudi children. J Trop Pediatr. 1992;38:106–112. doi:10.1093/tropej/38.3.106
52. Rugani MA. Association of Homocysteine with Vaso-Occlusive Crisis in Sickle Cell Disease. Brazil:Fluminense Federal university;2008. Available from:
53. Bradley AM, Joseph L. The treatment of hyperhomocysteinemia. Annu Rev Med. 2009;60:39–54. doi:10.1146/annurev.med.60.041807.123308
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.