")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 14
Self-Limited Primary Cutaneous Rosai-Dorfman Disease: A Case Report and Literature Review
Authors Zhang P , Liu F , Cha Y, Zhang X, Cao M
Received 16 October 2021
Accepted for publication 20 December 2021
Published 24 December 2021 Volume 2021:14 Pages 1879—1884
DOI https://doi.org/10.2147/CCID.S343815
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Jeffrey Weinberg
Pan Zhang,1 Fang Liu,1 Yin Cha,2 Xiuying Zhang,1 Mei Cao1
1Department of Dermatology, Beijing Chaoyang Hospital, Capital Medical University, Beijing, People’s Republic of China; 2Department of Dermatology, Dali Bai Autonomous Prefecture People’s Hospital, Dali, Yunnan Province, People’s Republic of China
Correspondence: Fang Liu
Department of Dermatology, Beijing Chaoyang Hospital, Capital Medical University, 8 Gongti South Road, Chaoyang District, Beijing, 100020, People’s Republic of China
Tel +86-10-85231688
Fax +86-10-85231217
Email [email protected]
Abstract: Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy (SHML), is a rare, benign, and self-limiting disease. Cutaneous Rosai-Dorfman disease (CRDD) is an exceedingly rare extranodal variant, which is strictly limited to the skin and is easily misdiagnosed. We present the case of a 47-year-old Chinese woman with primary CRDD and a review of relevant literature. The patient had red-yellow papules on her left cheek and chest for more than three months with occasional tenderness. Histopathology showed phagocytic histiocytes in the dermis with the typical presentation of emperipolesis. Immunohistochemistry (IHC) staining showed S100 (+), CD68 (+), and CD1a (-). Based on these findings and the negative systemic presentation, the patient was diagnosed with primary papulonodular-type CRDD, and no treatment regimen was initiated. After three months, the skin lesions partially disappeared, confirming the spontaneous regression of CRDD.
Keywords: primary cutaneous Rosai-Dorfman disease, sinus histiocytosis with massive lymphadenopathy, diagnosis, histopathology, immunohistochemistry, treatment, gene mutation
Introduction
Rosai-Dorfman disease (RDD) is an uncommon non-Langerhans histiocytosis of group R with a variable clinical presentation manifesting as peripheral lymphadenopathy with or without other organ involvement.1 RDD is also known as SHML and was first identified by Rosai and Dorfman as an independent disease in 1969.2 It is a benign and self-limiting disease with an incidence rate of 1:200,000,3 which mainly affects children and young people. The average age of RDD onset is 20.6 years. RDD is more common in Africa, and it has a higher prevalence among male patients than among females patients.3,4 The main clinical manifestations reported are painless enlargement of the bilateral cervical lymph nodes, mild fever, leukocytosis, and hyperglobulinemia.4 RDD histopathology is characterized by the proliferation and infiltration of sinusoidal histiocytes, which engulf intact lymphocytes. RDD pathology can be divided into three types: lymph nodal, extranodal, and mixed. Extranodal RDD accounts for approximately 40% of the reported cases and may involve the orbital tissue, skin, and central nervous system. When the condition presents with only skin lesions, without lymph node involvement, the disease is defined as cutaneous RDD (CRDD), which accounts for approximately 3% of all RDD cases.5,6 The 2016 revised histiocytosis classification has distinguished primary CRDD from the R (Rosai-Dorfman) group and classified it as a separate group (cutaneous group, C group) due to significant differences of primary CRDD from systemic RDD.3,4 The average age of onset is higher for CRDD than for RDD; Bruce-Brand4 and Ahmed7 reported the average age of patients at CRDD onset to be 43.5 years and 45.2 years, respectively. Additionally, the proportion of female patients with CRDD was higher than that of male patients, and a greater proportion of patients were of Asian and Caucasian descent compared with other groups.8 Molecular tests have been used widely in the investigation of RDD and CRDD.
Here we report a Chinese female spontaneous resolution CRDD case that may assist other physicians in treating this kind of unusual disease.
Case Report
A 47-year-old woman presented to the dermatology department with rashes on her left cheek and chest. Initially, a rash developed on the left cheek three months prior, with no obvious inducing factor and the patient gradually developed a similar rash on her left chest, and the area gradually increased. The rash did not cause any symptoms such as itching, irritation, or pain. Little improvement was noted after one month of external treatment with tacrolimus and halometasone creams, but the patient developed occasional tenderness.
The patient was in good health and denied any history of chronic disease, drug allergies, or genetic diseases. Physical examination revealed no obvious abnormalities in the heart, lung, or abdomen, and no swelling of any superficial lymph nodes in the body.
Dermatological Examination
Clustered yellow papules (diameter 5 mm) with clear boundaries were found on the left cheek, covering an area of approximately 3 cm × 3 cm. The surface was smooth and had a waxy luster (Figure 1A). A similar rash was seen on the left chest, covering an area of approximately 6 cm × 3.5 cm with a clear boundary, smooth surface, waxy luster, mild infiltration and hypertrophy, normal skin temperature, and no tenderness (Figure 1B). Some of the papules were fused and varied in color (red, brown, deep red).
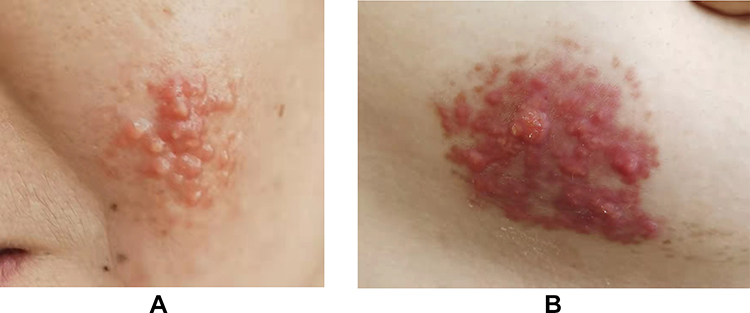 |
Figure 1 Skin lesions observed on the third visit before diagnosis. (A) Red to yellow papules on her left cheek, measuring approximately 3 cm × 3 cm. (B) Papules and erythema on her left chest, measuring approximately 6 cm × 3.5 cm. |
Blood test revealed that the total immunoglobulin E (IgE) level was 389.0 kU/L; the level of ferritin, a tumor biomarker, was below average at 9.70 µg/L. Blood analysis demonstrated negative results for hepatitis B, hepatitis C, syphilis and AIDS. Blood glucose levels were normal. High-resolution chest CT showed mild chronic inflammation in the left lung, multiple small lymph nodes in the mediastinum, and flocculent shadows in the anterior mediastinum in the region of the thymus. Abdominal ultrasonography revealed a hepatic cyst. Histopathological examination of the left chest skin lesion showed a roughly normal epidermis with cell masses in the dermis. The phagocytic histiocytes had deep irregular nuclei with pale and frothy cytoplasm. Lymphocytes, plasma cells, a small number of neutrophils and eosinophils were observed in the mass upon hematoxylin and eosin (HE) staining (Figure 2). Emperipolesis phenomenon was also observed (Figure 3). IHC staining showed S100 (+) (Figure 4A), CD68 (+) (Figure 4B), and CD1a (-) (Figure 4C).
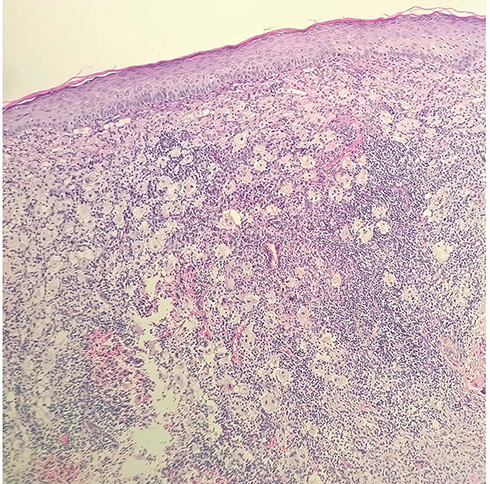 |
Figure 2 Epidermis is generally normal. Cell masses can be seen infiltrating the dermis. Phagocytosis of histiocytes can be seen; the cytoplasm appears pale and frothy, and some nuclei are deeply stained and irregular in shape. (40X). |
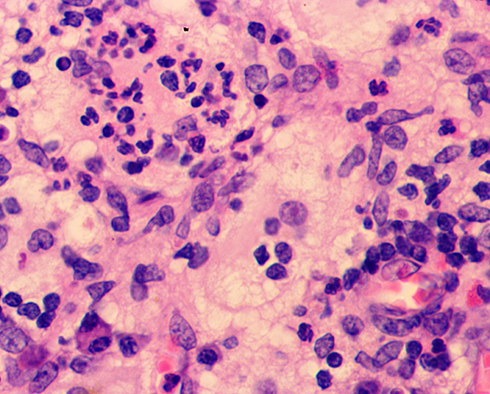 |
Figure 3 Highly magnified (400×) microscopic image shows emperipolesis. |
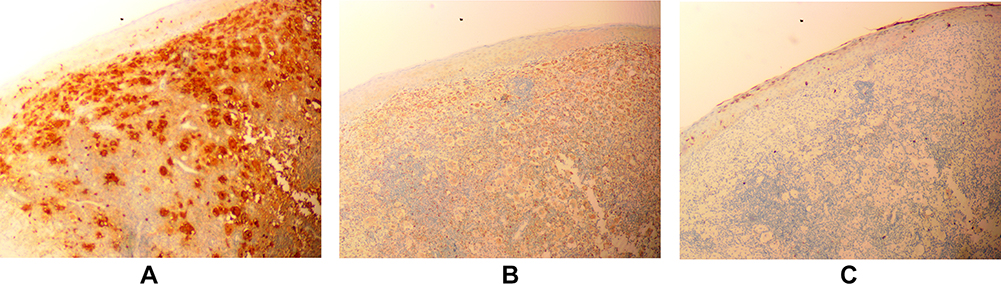 |
Figure 4 Immunohistochemical (staining) findings. (A) S100 (+) (40×). (B) CD68 (+) (40×). (C) CD1a (-) (40×). |
Diagnosis
After relevant auxiliary examinations and whole-body physical and hematological examinations, it was confirmed that the patient had no lymph node enlargement or visceral involvement. Thus, the diagnosis of primary papulonodular-type CRDD was made.
Treatment
No treatment was initiated and the skin lesions partially disappeared three months after diagnosis (Figure 5A and B). The patient is still being monitored via follow-up examinations (Figure 6).
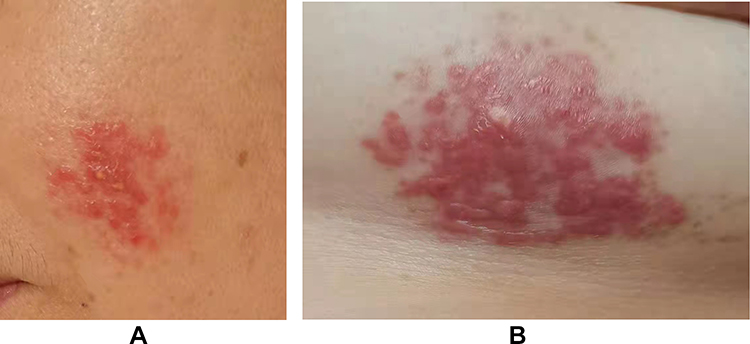 |
Figure 5 Three months after diagnosis, skin lesions partially disappeared. (A) Papules on the left cheek disappeared, but erythema (approximately 3 cm × 3 cm) is noted. (B) Papules on left chest were smaller in size (approximately 5 cm × 3 cm). |
 |
Figure 6 Timeline. |
Discussion
Histiocytoses are a group of proliferative disorders of macrophage–monocyte origin. Histiocytoses are currently classified into five groups (L, C, R, M, H) with each group having a different mutation in the mitogen-activated protein kinase pathway (MAPK).1,3 In RDD, the BRAFV600E mutation is generally absent (as it is characteristic of Langerhans cell histiocytosis and Erdheim-Chester syndrome).1 Other kinase mutations, such as KRAS, NRAS, ARAF, and MAP2K1, have been reported in nodal and extranodal RDD, and a recent study involving a small group of patients with CRDD confirmed the prevalence of the NRAS mutation in four out of seven patients (51.7%).9 The presence of clonal mutations in some cases of RDD suggests that the disease can be considered to be more neoplastic than inflammatory.1
The pathogenesis of CRDD was previously poorly understood; it may be related to infections, immune deficiencies, genetic factors, or tumors,8 however, recent evidence demonstrating clonality in a subset of cases raises the possibility of a neoplastic process. A possible association with IgG4-related disease remains controversial.4,6
Head and face are the most commonly affected regions in patients of all ages, followed by the trunk and limbs.8 After analyzing 25 Chinese patients with CRDD, Kong et al divided the disease into three types: papulonodular type (79.5%), indurated plaque type (12.8%), and tumor-like type (7.7%).10 Kong described the clinical characteristics of each type in detail: while most patients have no symptom, a few experience mild discomfort or pain as a result of improper treatment. The shape of the rash correlates with the disease duration, and the lesion gradually may develop from the papulonodular type to the plaque type and, finally, to the tumor-like type.10 Additionally, some patients can relapse multiple times or can develop skin lesions at multiple sites.11 Due to the pleomorphism of skin lesions and the complexity of the disease course, CRDD is easily misdiagnosed or missed.
Routine laboratory and imaging examinations of CRDD have not revealed specific manifestations. Therefore, the diagnosis of CRDD predominantly depends on histopathology and IHC findings. This disease has four important histopathological features:10
- Microscopic examination (high magnification) reveals a distribution of deeply stained areas and lightly stained areas throughout the dermis. The lightly stained areas were situated close to the basal layer of the epidermis and composed of large, polygonal, round, or oval histiocytes distributed in patches. The membranes of these histiocytes are thin, pale, and rich in cytoplasm. They can also be foamy and have unclear boundaries, large nuclei, and prominent nucleoli.10 These cells are called RDD cells. The deeply stained area is primarily composed of lymphocytes, plasma cells, and neutrophils.7,10
- The presence of complete lymphocytes and plasma cells in the cytoplasm of histiocytes is called “emperipolesis” or lymphocytophagocytosis which has great diagnostic significance for CRDD.1–14
- IHC results typically indicate S100 and CD68 positivity, and CD1a negativity.1–14 Birbeck particles are not visible on electron microscopic examination.10
- Lymphocytes and plasma cells are polyclonal, and this rules out malignant diseases such as lymphomas.10
CRDD can be differentiated from other diseases based on these characteristics. As there is no obvious specific site of onset and clinical manifestation, CRDD should be differentiated from cutaneous fibroma, xanthoma, Tangier disease, Langerhans cell histiocytosis, xanthogranuloma, multicenter reticulohistiocytosis, leprosy, carinal fibrosarcoma, and malignant melanoma. Differential diagnosis should be based on histopathology and IHC.1–14
Although most of the mutations found in RDD are absent in CRDD, there are reports of NRAS mutations that need to be further investigated—especially the possibility of progression to extracutaneous disease. We aware that the lack of mutation screening in our case is a limitation; however histological and IHC examinations supported the diagnosis.
As an inert and benign disease, CRDD has a good prognosis and does not require active treatment.5–11,14 Treatment options include steroids, methotrexate, dapsone, thalidomide, isotretinoin, and surgery, and the reported efficacies of these treatments vary.11–13 Surgical resection is reportedly the most effective intervention with a cure rate of 80%.11,12
Conclusion
As the patient had asymptomatic primary CRDD and her condition was stable and showed spontaneous resolution, no intervention is currently required. The patient will continue to be monitored periodically, molecular testing and surgical resection will be considered if the patient’s condition deteriorates rapidly.
Abbreviations
RDD, Rosai-Dorfman disease; SHML, sinus histiocytosis with massive lymphadenopathy; CRDD, Cutaneous Rosai-Dorfman disease; IHC, immunohistochemistry; IgE, immunoglobulin E; HE staining, hematoxylin and eosin staining.
Ethical Statement
Signed consent was obtained from the patient for the publication of case details and the accompanying images. The publication of the case details was approved by Beijing Chaoyang Hospital, Capital Medical University.
Funding
The authors declare that no funding was received for this study or manuscript preparation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gawdzik A, Ziarkiewicz-Wróblewska B, Chlebicka I, Jankowska-Konsur A, Szepietowski JC, Maj J. Cutaneous Rosai–Dorfman disease: a treatment challenge. Dermatol Ther. 2021;11(4):1443–1448. doi:10.1007/s13555-021-00557-1
2. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969;87(1):63–70.
3. Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672–2681. PMID: 26966089; PMCID: PMC5161007. doi:10.1182/blood-2016-01-690636
4. Bruce-Brand C, Schneider JW, Schubert P. Rosai–Dorfman disease: an overview. J Clin Pathol. 2020;73(11):697–705. doi:10.1136/jclinpath-2020-206733
5. Fayne R, Rengifo SS, Gonzalez I, et al. Primary cutaneous Rosai–Dorfman disease; a case-based review of a diagnostically and therapeutically challenging rare variant. Ann Diagn Pathol. 2020;45:151446. doi:10.1016/j.anndiagpath.2019.151446
6. Zhang Y, Chen H, Jiang YQ, et al. Clinicopathological features of cutaneous Rosai–Dorfman disease and its relationship to IgG4-related disease: a retrospective study. Br J Dermatol. 2019;181(4):844–845. doi:10.1111/bjd.17939
7. Ahmed A, Crowson N, Magro CM. A comprehensive assessment of cutaneous Rosai–Dorfman disease. Ann Diagn Pathol. 2019;40:166–173. doi:10.1016/j.anndiagpath.2019.02.004
8. Al-Khateeb TH. Cutaneous Rosai–Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofac Surg. 2016;74(3):528–540. doi:10.1016/j.joms.2015.09.017
9. Wu KJ, Li SH, Liao JB, Chiou CC, Wu CS, Chen CC. NRAS mutations may be involved in the pathogenesis of cutaneous Rosai Dorfman disease: a pilot study. Biology. 2021;10(5):396. PubMed: 34063325. PubMed Central: PMC8147632. doi:10.3390/biology10050396
10. Kong YY, Kong JC, Shi DR, et al. Cutaneous Rosai–Dorfman disease: a clinical and histopathologic study of 25 cases in China. Am J Surg Pathol. 2007;31(3):341–350. doi:10.1097/01.pas.0000213387.70783.b6
11. Michaeli O, Elassa M, Williams R, Baltazar G. Recurrent cutaneous Rosai–Dorfman disease. Cureus. 2019;11(12):e6289. doi:10.7759/cureus.6289
12. Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-destombes disease. Blood. 2018;131(26):2877–2890. doi:10.1182/blood-2018-03-839753
13. Gupta GK, Pittaluga S, Cowen EW. Image gallery: primary cutaneous Rosai–Dorfman disease. Br J Dermatol. 2019;181(2):e34. doi:10.1111/bjd.17999
14. Gaul M, Chang T. Cutaneous Rosai–Dorfman disease. Cutis. 2019;103(3):171–173.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.