")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Self-assembled amphotericin B-loaded polyglutamic acid nanoparticles: preparation, characterization and in vitro potential against Candida albicans
Authors Zia Q, Khan AA, Swaleha Z, Owais M
Received 26 February 2014
Accepted for publication 8 May 2014
Published 5 March 2015 Volume 2015:10(1) Pages 1769—1790
DOI https://doi.org/10.2147/IJN.S63155
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Qamar Zia,1 Aijaz Ahmed Khan,2 Zubair Swaleha,3 Mohammad Owais1
1Interdisciplinary Biotechnology Unit, 2Department of Anatomy, 3Women’s College, Aligarh Muslim University, Aligarh, India
Abstract: In the present study, we developed a self-assembled biodegradable polyglutamic acid (PGA)-based formulation of amphotericin B (AmB) and evaluated its in vitro antifungal potential against Candida albicans. The AmB-loaded PGA nanoparticles were prepared in-house and had a mean size dimension of around 98±2 nm with a zeta potential of -35.2±7.3 mV. Spectroscopic studies revealed that the drug predominantly acquires an aggregated form inside the formulation with an aggregation ratio above 2. The PGA-based AmB formulation was shown to be highly stable in phosphate-buffered saline as well as in serum (only 10%–20% of the drug was released after 10 days). The AmB-PGA nanoparticles were less toxic to red blood cells (<15% lysis at an AmB concentration of 100 µg/mL after 24 hours) when compared with Fungizone®, a commercial antifungal product. An MTT assay showed that the viability of mammalian cells (KB and RAW 264.7) was negligibly affected at AmB concentrations as high as 200 µg/mL. Histopathological examination of mouse kidney revealed no signs of tissue necrosis. The AmB-PGA formulation showed potent antimicrobial activity similar to that of Fungizone against C. albicans. Interestingly, AmB-bearing PGA nanoparticles were found to inhibit biofilm formation to a considerable extent. In summary, AmB-PGA nanoparticles showed highly attenuated toxicity when compared with Fungizone, while retaining equivalent active antifungal properties. This study indicates that the AmB-PGA preparation could be a promising treatment for various fungal infections.
Keywords: polyglutamic acid, amphotericin B, toxicity, candidiasis
Introduction
Amphotericin B (AmB), a broad-spectrum antifungal agent, is a polyene-based compound that has a propensity to aggregate in an aqueous environment.1,2 Exploiting its exclusive physicochemical properties, a commercial antifungal product known as Fungizone® (AmB-D) was introduced to treat various life-threatening fungal infections.3,4 Fungizone is basically a micellar deoxycholate-based formulation of AmB.
Effective antifungal therapy often requires long treatment regimens lasting up to 30 days; this in turn leads to various toxic manifestations (eg, nausea, chills, fevers, vomiting).5,6 Moreover, clinical use of AmB-D is often marred with acute and chronic infusion-related toxicities, the most serious being dose-dependent nephrotoxicity.5,7,8 To circumvent AmB-D-related toxicity issues, various lipid formulations of AmB have been developed, including the commercially available AmBisome® (AmB-L), ie, liposomal AmB.2,9–11 AmB-L has a far superior therapeutic index with highly attenuated toxicity when compared with AmB-D. However, the complex process involved in the production of AmB-L,12,13 its high cost,14,15 and issues related to stability tend to offset its benefits and limit its utility, especially in third world countries. Since lipids are the principal constituent of the formulation, it is difficult to imagine AmB-L ever becoming affordable. Efforts to make generic forms that have efficacy comparable with existing formulations have met with limited success.16,17 Furthermore, the existing AmB formulations are not therapeutically bioequivalent and are reported to have clear differences in toxicity.16,18 Taking into consideration the various limitations associated with lipid formulations of AmB,19 it would be desirable to develop alternative drug delivery systems that can be used widely in the treatment of an array of fungal diseases.
Much preclinical research on polymeric nanoparticles has suggested some potential for delivery of active drug substances.20 Polymer nanoparticulate technologies can be used to improve water solubility and bioavailability of the active drug component.21,22 Nanoparticles shield the drug from interacting directly with normal healthy cells, thus minimizing its detrimental effects to a considerable extent. Biodegradable polymers have also been shown to optimize pharmacokinetics (including controlled and prolonged release of the drug with tissue retention), improve efficacy, and address drug safety issues.23
Our earlier efforts to develop a biodegradable polymer drug complex based on PLGA (poly[lactic-co-glycolic acid]) have been quite encouraging,24,25 and led us to hypothesize that a delivery vehicle based on an analogous polymer, ie, polyglutamic acid (PGA), would have potential to circumvent the toxicity of AmB to a greater extent. PGA was selected for its biodegradable, nontoxic, and biocompatible properties. Pilot studies established PGA to be nonimmunogenic and well tolerated, even at high doses. Moreover, nanoparticles composed of PGA have been shown to immobilize proteins, peptides, and other related chemicals onto their surfaces and/or interior bulk.26,27 PGA has also been extensively evaluated in cancer chemotherapy and been implied at various stages of clinical trials.28–30 Moreover, the development of PGA based nanoparticles involves simple and easy preparation protocol, making it a superb strategy for the encapsulation of AmB. AmB-bearing PGA nanoparticles prepared in-house were investigated for their physical characteristics, stability, safety (both in vitro and in vivo), and in vitro antifungal efficacy in comparison with two commonly used formulations of AmB, ie, AmBisome and Fungizone.
Materials and methods
Chemicals
Poly-l-glutamic acid sodium salt (20–40 kDa and 50–70 kDa; catalog numbers G0546 and G0421, respectively), AmB (European Pharmacopoeia reference standard), polyvinyl alcohol (PVA), Roswell Park Memorial Institute (RPMI) 1640 medium, and dimethyl sulfoxide were purchased from Sigma Aldrich (St Louis, MO, USA). The antifungal agents AmBisome (Gilead Sciences Inc, San Dimas, CA, USA) and Fungizone (Apothecon, Princeton, NJ, USA) were reconstituted according to the manufacturers’ instructions. KB cells (HeLa contaminant, cervical adenocarcinoma-derived epithelial cells) and mouse leukemia RAW 264.7 cells (macrophage-like, Abelson leukemia virus transformed cell line derived from BALB/c mice) were sourced from the American Type Culture Collection (ATCC, Manassas, VA, USA). Detection kits for liver and kidney function test parameters were purchased from Span Diagnostics Ltd (Mumbai, India). The rest of the chemicals were of analytical grade of purity and sourced locally.
Fungal isolates
The test strain of Candida albicans (ATCC 18804) was obtained from the ATCC while Candida glabrata (MTCC 3019) was sourced from the Microbial Type Culture Collection (MTCC, Chandigarh, India). Sabouraud dextrose agar/broth was used for growing both the fungal strains. The C. albicans and C. glabrata strains were characterized using Hi-Chrome agar as well as germ tube formation with a standard protocol prior to its use in various studies. C. albicans (ATCC 10231) was used as a quality control strain.
Animals
Inbred female Balb/c mice (aged 6–8 weeks, mean weight 20±2 g) were obtained from the animal facility at the Indian Institute of Toxicological Research, Lucknow, India. The animals were then acclimatized for a few days under standard husbandry conditions, ie, room temperature (22°C±3°C), relative humidity (65%±10%), and a 12-hour light/dark cycle. They had free access to a standard dry pellet diet and water ad libitum under strict hygienic conditions. The animals were anesthetized with ketamine 5 mg/kg and xylazine 4 mg/kg prior to being euthanized. Humane endpoints were applied for mice that survived at the conclusion of the experiment. In all experimental procedures, efforts were made to minimize pain and suffering. All animal experiments were reviewed and approved by the Institutional Animal Ethics Committee of the Interdisciplinary Biotechnology Unit, Aligarh Muslim University, India and were performed according to the national regulatory guidelines issued by Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India (approval identifier 332/CPCSEA).
Preparation of AmB–PGA nanoparticles
Preparation of PGA nanoparticles was achieved following a published method that was modified in our laboratory.31 Briefly, 10 mg of PGA and 5 mg of AmB were dissolved in 10 mL of dimethyl sulfoxide in a 30 mL sample vial. The above solution was then added dropwise to the PVA solution (10 mg/0.5 mL) under constant stirring. The AmB/PGA mixtures were stirred at room temperature for 1 hour. To prepare the nanoparticles, the solution was dialyzed against deionized water (500 mL) for 24 hours (Spectra/Por® membrane tubing, molecular weight cutoff 2 kDa, Spectrum Labs, Rancho Dominguez, CA, USA). After dialysis, the particles were centrifuged at 20,000 g for 1 hour and the nanoparticles were washed with deionized water several times to remove free PVA. The precipitated mass was probe-sonicated briefly, followed by dialysis to remove free AmB. The solution was then filtered using a Millipore Express Plus Membrane microsyringe filter (hydrophilic, polyethersulfone, 0.22 μm) (Millipore, Bangalore, India). Finally, the solution was freeze-dried to yield a yellow fluffy product.
Determination of entrapment efficiency of PGA-intercalated AmB
The entrapment efficiency (EE) of AmB in the PGA formulation was determined by dissolving an aliquot of the AmB-PGA formulation in methanol. To rule out the possibility that the observed results were due to the AmB-PGA nanoparticles only and not because of the PGA molecule itself, we included plain PGA nanoparticles that were not loaded with AmB as a control throughout the study; these are referred to herein as “sham PGA” nanoparticles.
The AmB content was determined using a high performance liquid chromatography method following the published procedure.32 A standard calibration curve of the drug was plotted at 405 nm by determining the area under the curve corresponding to the increasing amount of drug (3.125–200 ng of AmB equivalent in a 20 μL sample injection volume). The highest AmB concentration was 10 μg/mL (equivalent to 200 ng of AmB in 20 μL). Dilutions of AmB were performed in methanol to ensure that AmB remained in the monomeric form. Details of the high-performance liquid chromatography procedure are provided in the Supplementary materials section.
The percent EE was calculated as the percentage of the ratio between the entrapped AmB and the total amount of AmB added at the beginning during preparation of the formulation:
|
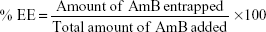
Determination of particle size and zeta potential of AmB–PGA nanoparticles
The zeta potential gives an indication of the charge acquired by a particulate system on its suspension in aqueous medium. The zeta potential of the nanoparticles was determined using DTS software (Malvern Instruments Limited, Malvern, UK) based on M3-PALS technology. The concentration of the AmB-PGA formulation used was 10−4 M. Particle size was determined by photon correlation spectroscopy (90Plus/BI-MAS, Brookhaven Instruments, Holtville, NY, USA). Samples were filtered using a 0.22 μm syringe filter (polyethersulfone, Millipore) to remove contamination with dust particles. Dynamic light scattering measurements were carried out using DynaPro-TC-04 equipment (Protein Solutions, Wyatt Technology, Santa Barbara, CA, USA) equipped with a temperature-controlled microsampler. The size and surface morphology of the AmB-loaded nanoparticles were characterized using an electron microscope (CM-10, Philips, Hamburg, Germany). For transmission electron microscopy studies, the AmB-PGA formulation was used at a concentration of 10 μg/mL AmB equivalents in double-distilled water. The details of these procedures are provided in the Supplementary materials section.
PGA nanoparticle-mediated transfer of entrapped calcein to macrophages
Successful elimination of the intracellular pathogen C. albicans requires effective delivery of a drug pay load to the infected macrophages. We determined the uptake of PGA-encapsulated fluorescent dye (calcein) by macrophages to assess the ability of the newly developed PGA formulation to home the intercalated drug to C. albicans-harboring macrophages. The details of the uptake study are provided in the Supplementary materials section.
Spectrophotometric analysis of AmB–PGA formulation
Ultraviolet spectroscopic analysis of various formulations of AmB was carried out in the range 300–450 nm on a double-beam spectrophotometer (model V-750, Jasco Inc, Easton, MD, USA) operated at a resolution of 1 nm. AmB is known to give four absorption peaks. The aggregation ratio was calculated as the ratio of peak I (315–340 nm) to peak IV (410 nm).33 Fungizone and the AmB-PGA formulation were both scanned at the same concentration (10−5 M).
In vitrorelease kinetic studies
In vitro release of AmB from the AmB-PGA complex in phosphate-buffered saline (PBS) and serum was evaluated by dialysis,34 as standardized in our laboratory. The quantification was carried out using high-performance liquid chromatography32 as described in the Supplementary materials section. In order to determine the release kinetics, a stock solution of AmB-PGA nanoparticles was prepared in water at a concentration of 1,000 mg/L AmB equivalents. The aqueous solution of nanoparticles (1 mL) was added to either 20 mM sterile PBS (pH 7.4) or serum (1 mL, containing 5% dimethyl sulfoxide). The final concentration of AmB-PGA was 500 μg/mL AmB equivalents (total amount of AmB, 1 mg in 2 mL). The PBS and serum also contained a small amount of sodium azide to avoid microbial growth. The AmB-PGA solution (2 mL) was placed in a dialysis bag (molecular weight cutoff 12–14 kDa) and dialyzed against 50 mL of PBS (pH 7.4) containing 5% dimethyl sulfoxide at 37°C. The dialysis medium was completely removed at regular time intervals for analysis of drug concentration and replaced with fresh medium to prevent drug saturation (maintaining strict sink conditions throughout the experiment). Release runs were continued for 240 hours. The absorbance of the collected samples was measured at 405 nm by high-performance liquid chromatography in aqueous methanol (50% v/v). The AmB concentration was extrapolated from the calibration curve of AmB in aqueous methanol (50% v/v). The calculated amount of the released drug was plotted against time. The results recorded are the mean of three runs carried out independently.
Drug toxicity studies
In vitro red blood cell lysis test
Fresh blood (15–20 mL) was kindly provided by the blood bank at Jawaharlal Nehru Medical College, Aligarh Muslim University, Aligarh, and collected in anticoagulant (ethylenediamine tetraacetic acid 1 mg/mL, at 10% w/v) syringes, followed by centrifugation at 1,000 g for 15 minutes at 4°C. Plasma was carefully aspirated and the exposed buffy coat was removed and discarded. The red blood cells were washed three times by centrifugation (1,000 g for 15 minutes at 4°C) and suspended in five volumes of normal saline (NaCl 0.9% w/v). The cells were then resuspended in 4 mL of saline, counted in a Neubauer™ chamber (Splabor, São Paulo, Brazil), and resuspended again until the desired concentration (5×107 cells/mL) was achieved. AmB-D, AmB-L, and empty PGA vesicles were included as controls and prepared according to the manufacturers’ protocol in double-distilled sterile water (Sigma-Aldrich).
To study the extent of hemolysis, 1 mL of red blood cells (5×107 cells/mL) was incubated with 1 mL of the various AmB formulations (containing 1, 5, 10, 50, and 100 μg/mL AmB equivalents) at 37°C for 1 hour and for 24 hours. The free form of AmB was dissolved in 50 μL of dimethyl sulfoxide and made up to a final volume of 1 mL with PBS (final 5% dimethyl sulfoxide). After the stipulated periods of time, the reaction mixture was centrifuged at 1,200 g. The supernatant was then collected and analyzed by ultraviolet-visible spectroscopy (λmax =576 nm) for released hemoglobin. Triton X-100 (nonionic surfactant) at a concentration of 0.1% was used as a positive control for 100% cell lysis. The result was expressed graphically as a percentage of 100% cell lysis and was determined by the following equation:
|
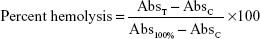
where AbsT is the absorbance of the supernatant from samples incubated with the drugs, AbsC is the absorbance of the supernatant from the control (PBS), and Abs100% is absorbance in the presence of 0.1% Triton X-100. The results are the mean of three independent experiments.
In vitro cytotoxicity test
KB cells and RAW 264.7 cells were plated in 96-well flat bottom tissue culture plates (NalgeNunc International, Rochester, NY, USA) at a density of 4×104 cells/mL and 1×105 cells/mL, respectively, in RPMI 1640 medium and 10% fetal calf serum and allowed to adhere overnight at 37°C in 5% CO2. The cells were treated by adding 100 μL of the AmB-PGA formulation at various concentrations (1, 5, 10, 50, 100, and 200 μg/mL AmB equivalents) into the respective well. AmB-D, AmB-L, and pure AmB used in preparation of the complex were taken as the controls. The plates were then incubated for 24 hours at 37°C in 5% CO2. Cell viability was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay, as follows. After washing the cells, 200 μL of a 0.5 mg/mL MTT solution in Dulbecco’s Modified Eagle’s Medium was added to each well and incubated at 37°C for 2 hours. The supernatants were removed, and 200 μL of extraction solution was added in order to solubilize the formazan salt. After 30 minutes at room temperature, the absorbance of each well was measured at 550 nm and the percentage of living cells was calculated using the following formula:
|
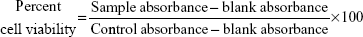
where blank absorbance is the medium alone and control absorbance is cells with medium alone. All assays were carried out in quadruplicate and the results are expressed as the mean.
Liver and kidney function test
A single injection of AmB-PGA (containing 5, 10, and 15 mg/kg body weight AmB equivalent in 200 μL), was administered via the lateral tail vein to healthy female Balb/c mice. AmB-D was administered at a concentration of 5 mg/kg body weight. Twenty-four hours later, blood was collected by retro-orbital puncture from the mice in the various experimental groups into lithium heparin Microtainers® (BD Biosciences, Franklin Lakes, NJ, USA) and spun at 2,000 g for 10 minutes at 4°C to isolate the plasma. The samples were frozen at −80°C until use. Serum chemistry analysis was performed for investigation of creatinine, alanine aminotransferase, aspartate aminotransferase, and blood urea nitrogen levels as per the respective guidelines provided by the manufacturer of each detection kit. The data recorded are the mean of three experiments carried out independently.
Histopathological studies
The toxic effects of AmB-PGA nanoparticles on the kidneys of the experimental animals were also assessed by histopathological studies. The kidneys of experimental mice from the control (PBS), AmB-D, AmB-L, and AmB-PGA nanoparticle-treated groups were processed as reported previously.35 The details of the experimental procedure used are provided in the Supplementary materials section.
In vitro susceptibility testing
The broth microdilution method, as described by the Clinical and Laboratory Standards Institute (M27-A2), was used to determine the anti-Candida activity of the various AmB formulations.36 The antifungal activity of AmB was evaluated over a final concentration range of 0.015–8 μg/mL. The wells containing fungal inoculum with various concentrations of drugs and appropriate controls (drug-free as well as inoculum-free) were incubated at 35°C for 48 hours. For AmB, the endpoint is the lowest concentration that inhibits visual growth. An endpoint score of 0 (100% inhibition) was taken as the minimum inhibitory concentration (MIC). Minimum fungicidal concentrations (MFCs) were determined by seeding the entire volumes from all clear MIC wells with the highest inoculum onto Sabouraud dextrose agar plates. The MFC is the lowest drug concentration that kills 99.9% (with less than three colonies remaining) of the final inoculum. All assays were performed in duplicate and repeated at least twice. Details of the procedure are provided in the Supplementary materials section.
Time-kill analysis
C. albicans was grown overnight at 35°C on Sabouraud dextrose agar. Isolated colonies were selected and suspended in 0.9% NaCl to a turbidity equivalent to that of a 0.5 McFarland standard. Wells containing RPMI 1640 medium (buffered with morpholinepropanesulfonic acid 0.165 M to pH 7.0) plus AmB-PGA nanoparticles or AmB at 4×the MIC or no antibiotic (growth control) were inoculated with the yeast suspension to a final concentration of ~105 colony forming units (CFU) per mL. The cultures were incubated at 35°C for up to 24hours. At the indicated times, aliquots were removed and the numbers of CFU/mL were determined on Sabouraud dextrose agar after serial dilutions in PBS. The assay was performed in quadruplicate. Each result is representative of at least three separate experiments.
In vitro antifungal activity of AmB–PGA against intracellular C. albicans
RAW 264.7 cells were cultured in 24-well plates at a density of 5×105 cells/well. After 2 hours, an inoculum containing 1×106 CFU of C. albicans was cocultured with the adhered monolayer. After a 30-minute incubation period,37 the noningested yeast was removed by washing the monolayer three times with Hank’s Balanced Salt Solution (Sigma-Aldrich). The macrophages were reincubated in the presence of the AmB-PGA formulation at various concentrations (1, 5, 10, and 50 μg/mL AmB equivalent). AmB-D and AmB-L were included as controls. Percentage of infection was calculated by lysing the macrophages (after 24 hours of treatment) with Triton X-100 (0.2%) and subculturing in Sabouraud dextrose agar plates, and compared with an untreated control. Experiments were carried out in triplicate and the results are expressed as mean of the data obtained.
Biofilm formation
Biofilms were formed in the wells of microtiter plates as previously described.38 Fresh C. albicans cells grown overnight in an orbital shaker at 30°C were harvested and washed in sterile PBS (10 mM phosphate buffer, 2.7 mM potassium chloride, 137 mM sodium chloride, pH 7.4). The cells were resuspended in RPMI 1640 medium supplemented with l-glutamine and buffered with morpholinepropanesulfonic acid to a cellular density equivalent to 1×106 cells/mL. All experiments were performed on commercially available, presterilized, polystyrene, flat-bottomed, 96-well microtiter plates (Corning Incorporated, Corning, NY, USA). Biofilms were formed by pipetting 100 μL of the standardized cell suspensions into selected wells of the microtiter plate and incubating the plate for 48 hours at 37°C. After biofilm formation, the medium was discarded and nonadherent cells were removed by thoroughly washing the biofilms three times in sterile PBS.
Visualization of inactivation of C. albicans cells in biofilm
Preliminary microscopic visualization of C. albicans biofilms after treatment with the AmB-PGA formulation was done by phase contrast microscopy,39 details of which are provided in the Supplementary materials section.
Scanning electron microscopy studies of C. albicans biofilm
An already published protocol for scanning electron microscopy (SEM) of C. albicans biofilm was followed.38 C. albicans biofilms were formed on plastic Thermanox coverslip disks (15 mm diameter; NalgeNunc International) in six-well cell culture plates (Corning Incorporated) by dispensing standardized cell suspensions (4 mL of a suspension containing 1.0×106 cells/mL of RPMI 1640 medium) onto the appropriate disks and incubating the plates at 37°C for 24 hours. After washing with PBS, a AmB-PGA formulation (5 μg/mL) was added to the preformed biofilms and the plates were incubated for a further 48 hours at 37°C. Control biofilms were incubated in RPMI 1640 medium only. After incubation, the biofilms were washed with PBS and placed in fixative (4% formaldehyde [v/v] and 1% glutaraldehyde [v/v] in PBS) overnight. The samples were rinsed in 0.1 M phosphate buffer (twice for 3 minutes each time) and then placed in 1% Zetterquist’s osmium for 30 minutes. The samples were subsequently dehydrated in a series of ethanol washes (70% ethanol for 10 minutes, 95% ethanol for 10 minutes, 100% ethanol for 20 minutes), and then treated (twice for 5 minutes each time) with hexamethyldisilazane (Polysciences Inc, Warrington, PA, USA) and finally air-dried in a desiccator. The specimens were coated with 40% gold–60% palladium. The samples were then observed by SEM (JSM-6510LV, JEOL, Tokyo, Japan) at 15 kV.
Biofilm quantitation by XTT assay
A semiquantitative measure of the antibiofilm activity of the PGA nanoparticles was performed by XTT (2, 3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino) carbonyl]-2H-tetrazolium hydroxide) reduction assay as described elsewhere.38 Briefly, XTT solution was prepared by mixing 0.5 mg/mL XTT in Ringer’s lactate. This solution was filter-sterilized and stored at −70°C. Prior to each assay, an aliquot of stock XTT was thawed, and menadione (10 mM prepared in acetone; Sigma-Aldrich) was added to obtain a final concentration of 1 μM. The solution was filter-sterilized using a 0.22 μm syringe filter (Millipore). The biofilms (developed as above) were then incubated in the presence of the different concentrations of AmB-PGA formulation (final concentrations from 0.195 to 100 μg/mL) for 48 hours. After treatment with the AmB-PGA formulation, 100 μL of XTT was added to each prewashed biofilm (to remove nonadhered cells). The plates were then incubated in the dark for up to 2 hours at 37°C. A colorimetric change in the XTT reduction assay, a direct correlation of the metabolic activity of the biofilm, was then measured in a microplate reader (Bio-Rad, Hercules, CA, USA) at 450 nm and expressed as percent viability in comparison with control cells. Untreated biofilms containing RPMI 1640 medium were included as positive controls. AmB-D and AmB-L were also included in the study as controls. Wells without biofilms served as a blank. Four replicate biofilms were included for each experiment.
|
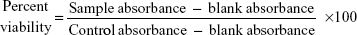
where blank absorbance is the medium alone and control absorbance is fungal cells with medium alone.
Biofilm time-kill curves
Biofilms were grown under shaking (100 rpm) for 24 or 48 hours at 37°C. Various AmB formulations were then added to the preformed biofilms, and the plates were incubated for selected time periods (2, 4, 8, 12, 24, and 48 hours). Antifungal effects were monitored by a metabolic assay based on the reduction of XTT as described above.38 Details of the experiment are provided in the Supplementary materials section.
Statistical analysis
In order to assess the statistical differences among various groups, the data were analyzed using the Student’s t-test and one way analysis of variance (Holm-Sidak method), using Sigma-Plot version 10 software. P-values <0.05 were considered to be statistically significant.
Results and discussion
Despite a worldwide search during the last three decades for a broad-spectrum, nontoxic, and well tolerated antifungal drug, AmB remains an ideal therapeutic agent for patients with unidentified invasive fungal infections. In the present scenario, a cheap, nontoxic, effective, and highly stable delivery system for AmB is needed to address the limitations of its existing delivery systems. Investigators have employed different water-soluble polymers to develop encapsulated, conjugated AmB-based delivery systems, but have met with little success.40–48 In some of the earlier efforts, polymers synergized the toxicity of AmB,49 while in others the AmB was unable to retain the beneficial physicochemical properties associated with its liposomal form (AmB-L).50 Although, AmB–cyclodextrin complexes showed improved water solubility,49,51 they were found to be toxic to human red blood cells.52,53 On the other hand, polyvinylpyrrolidone has been shown to complex AmB, but suffers from very low (0.249% w/w) AmB loading.54 Polymeric micelle systems have also been developed; however, encapsulation in the polymeric micelles decreased the in vitro antifungal activity of the drug.40 Moreover, these micelles have been shown to be unstable in the presence of serum due to disruption of their integrity,55 resulting in rapid and near-complete release of core-loaded molecules from circulating micelles in an uncontrolled fashion.56 Herein, we aimed to develop a stable, easy-to-prepare, nontoxic preparation of AmB with physicochemical characteristics comparable with those shown by AmB-L. Replacing the lipid moiety with a water-soluble homopolymer could reduce the costs and stability issues associated with the lipid components of AmB-L. In addition, an amphiphilic PGA-based delivery system may have some advantages, including biocompatibility, biodegradability,27,57 negligible toxicity, and cytoplasmic delivery of the entrapped molecules. The homopolymer contains carboxylic groups that can form ionic complexes with a charged compound such as AmB. Furthermore, PGA has the potential to encapsulate or immobilize a variety of molecules.26,27,57 For example, PGA has been used in clinical trials in the form of a covalently bound conjugate with paclitaxel (Opaxio, Cell Therapeutics, Seattle, WA, USA).29,30
The low aqueous solubility of amphiphilic AmB (<1 μg/mL)58 makes it a suitable candidate for association with an amphiphilic PGA homopolymer. The initial reaction of AmB with PGA was carried out in the presence of dimethyl sulfoxide, a water-miscible organic solvent. Next, an aqueous solution of PVA (a base) was added followed by extensive dialysis against water. In high abundance of an aqueous environment, AmB can either choose to associate with the PGA polymer or can form precipitates. The amphiphilic nature of both AmB and the PGA polymer enables them to remain in association, resulting in spontaneous formation of nanoparticles. The base is added to increase the solubility of the PGA, which ensures that the polymer remains in solution. The nanoparticles show unusually high stability in the presence of PVA.59,60 The simple and straightforward preparation protocol makes development of a AmB-PGA formulation an excellent strategy. The AmB-PGA formulation was found to be readily and rapidly soluble in water (Table 1). The EE was found to be around 20.5% and 45% for the two polymers in the 20–40 kDa and 50–70 kDa molecular weight range, respectively (Table 1). Since high AmB loading (>40%) ensures applicability in the clinical setting, the PGA nanoparticle having a molecular weight of 50–70 kDa (with high complexation efficiency and aqueous solubility) was used in the subsequent studies.
 | Table 1 Entrapment efficiency, particle size, and zeta potential of various amphotericin B formulations |
Characterization of AmB–PGA nanoparticles
The AmB-PGA nanoparticles prepared in-house were characterized for their size, shape, and zeta potential. The average zeta diameter and zeta potential of the AmB-PGA nanoparticle (molecular weight 50–70 kDa) were found to be 102±7 nm and −35.2±0.73 mV, respectively (Table 1). Generally, a high zeta potential (either positive or negative), more than 30 mV, maintains a stable system.61,62 Extremely positive or negative zeta potential values cause larger repulsive forces; this repulsion between similarly charged particles prevents aggregation of the particles and thus ensures easy redispersion.63,64 Thus, it can be said that AmB-PGA nanoparticles prepared using a 50–70 kDa molecular weight polymer are more stable than nanoparticles prepared with PGA having a molecular weight of 20–40 kDa (Table 1).
The mean size of the AmB-PGA nanoparticle was 107±0.7 nm as measured by dynamic light scattering (Figure 1A). A population of nearly spherical PGA nanoparticles with an average size of 98±2 nm was observed by transmission electron microscopy (Figure 1B). This is in agreement with the sizes determined by dynamic light scattering analysis (average diameter 107±0.7 nm, Figure 1A) and nanosizer (102±7 nm, Figure 1C).
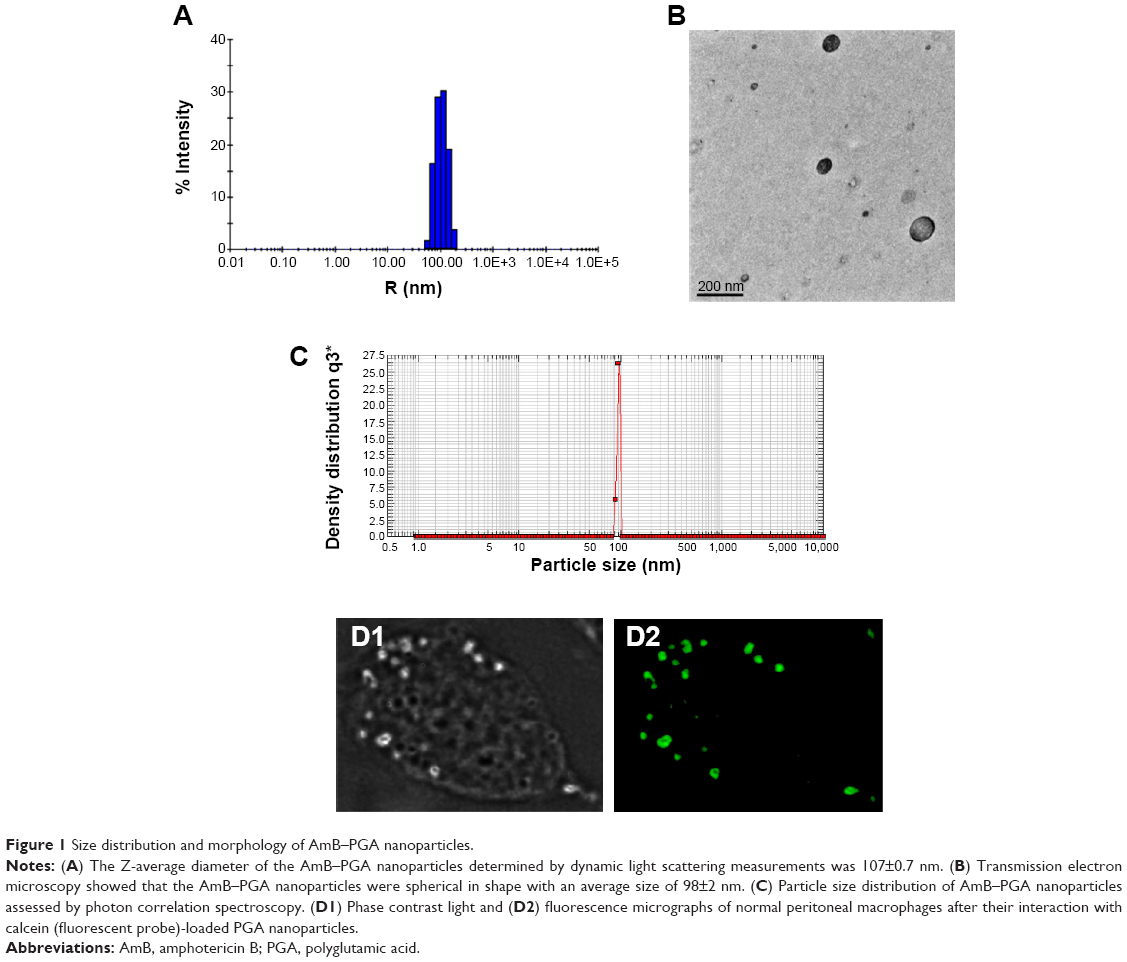 | Figure 1 Size distribution and morphology of AmB–PGA nanoparticles. |
To ascertain their fate after uptake by macrophages, calcein-loaded PGA nanoparticles were incubated with mouse macrophage cells (thioglycollate elicited). Incubation of the PGA nanoparticles resulted in a punctate fluorescence pattern owing to their active endocytic uptake by macrophages (Figure 1D). Commonly, drugs associated with colloidal carriers enable higher accumulation of drug inside macrophages and a small size enables them to be taken up through endocytosis via clathrin, caveolae, or specific independent receptors.65,66 The particle-loaded macrophages act as a secondary depot for the drug. Once internalized, a direct interaction of AmB with the intracellular yeast seems to be plausible. The released AmB can then readily act upon the C. albicans cells, resulting in effective killing.
Spectroscopic studies
Earlier studies have shown that the degree of aggregation of AmB is directly correlated with its toxicity,33,67,68 antimicrobial activity,67,69,70 and stability.71,72 In aqueous dispersions, the antifungal compound AmB has a tendency to exist in three different forms, ie, a monomer state, a soluble state, and an insoluble aggregate state.3,73 Small aggregates or soluble oligomers are responsible for the toxic reactions in mammalian cells and are usually the predominant species present in AmB-D solutions.3,74 Association of monomers with ergosterol leads to pore formation in the fungal membrane, while the oligomeric form of the drug complexes with cell membrane sterols in mammalian cells, which facilitates leakage of electrolytes.3,74 This drug–sterol interaction can lead to systemic toxicity, including adverse renal, hepatic, and cardiac effects, along with blood dyscrasias and death.5 The oligomers also seem to be susceptible to oxidation, which may enhance their toxicity.71 However, when AmB in solution is below the critical concentration (1 μM) required for self-association, toxicity against fungi is still observed, while toxic effects on mammalian cells are minimized.75,76
The ultraviolet-visible spectra of polyenes (such as AmB) are very sensitive to conformational changes induced by different molecular interactions, including aggregation. It has been reported that the monomeric form of AmB absorbs maximally at about 405–409 nm (λmax),77 while soluble self-aggregates display an absorption with a λmax of about 328–340 nm.73 In higher order aggregates, the maximum absorption shifts to wavelengths lower than 322 nm. Nonetheless, the optimum aggregation state is not simple, and cannot be defined using a single measure.
In order to assess the aggregation state of AmB inside PGA nanoparticles, we performed ultraviolet-visible spectroscopy. When AmB-PGA nanoparticles were suspended in water, shifting of λmax toward a shorter wavelength was observed (ie, a hypsochromic effect), suggesting the presence of aggregated AmB. The principal absorption was found to be at around 345 nm, with AmB showing a minimal peak at 410 nm (Figure 2A). As AmB is known to exist in monomeric form in organic solvents, we also determined its absorption spectrum in dimethyl sulfoxide. AmB showed an absorption pattern characteristic of a heptaene chromophore, with the lowest peak at around 355 nm and the highest peak at 415 nm (Figure 2B). The spectral modifications induced by aggregation may be represented by the ratio of absorbance at two different peaks (A340/A410), ie, 340 nm (peak I) and 410 nm (peak IV). The degree of aggregation of AmB can be easily monitored using this ratio.33 The higher the aggregation ratio, the lower the amount of monomeric AmB present. When the PGA nanoparticles were dispersed in water, this ratio was found to be 2.39, whereas when dimethyl sulfoxide was used as the solvent, this ratio decreased to 0.24.
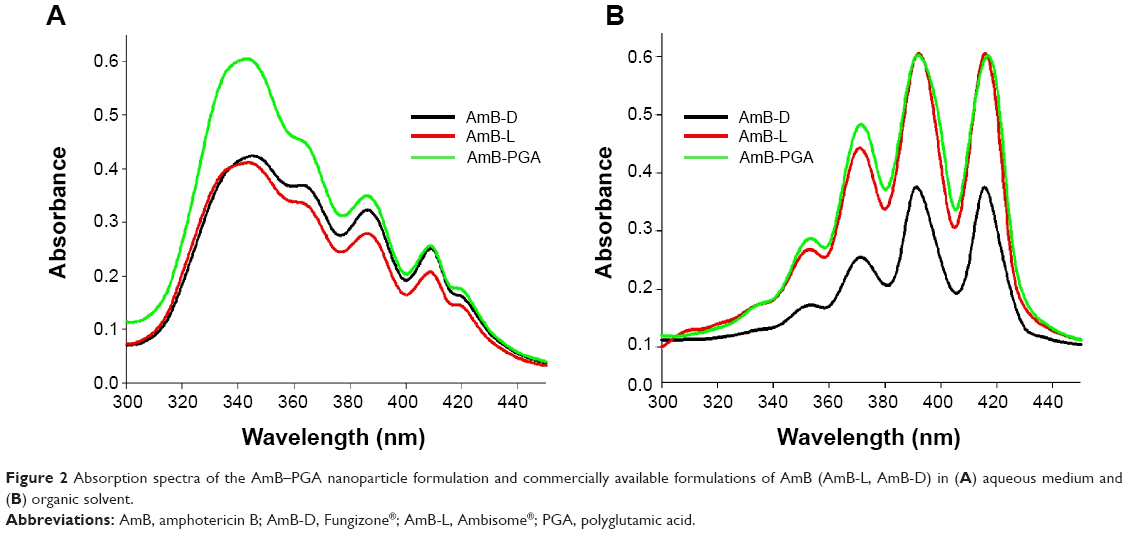 | Figure 2 Absorption spectra of the AmB–PGA nanoparticle formulation and commercially available formulations of AmB (AmB-L, AmB-D) in (A) aqueous medium and (B) organic solvent. |
Absorption peaks at a lower wavelength are generally observed when aggregated AmB is complexed with deoxycholate77 or upon formation of superaggregates by heat treatment of AmB alone.73 Taken together, these results suggest that AmB is present in a highly aggregated form inside PGA nanoparticles. Interestingly, it has been shown that AmB exists in a highly aggregated form in both AmB-D and AmB-L formulations. However, in AmB-L, there is an hypsochromic (blue) shift of 7-9 nm at λmax.78 This shift could be attributed to the close association between AmB and the lipid bilayer in AmB-L.79 Interaction of AmB with the PGA polymer in an aqueous environment results in formation of AmB aggregates that become entrapped inside the self-assembled PGA nanoparticles during their formation.
Release profile for AmB nanoparticle formulation
The PGA nanoformulation developed in the present study was assessed for its degradability under simulated physiological conditions (in PBS and in serum). The AmB-PGA nanoparticle was found to be stable in PBS, with 2.30%±0.09% of the total AmB being released into PBS after 24 hours of incubation (Figure 3A). This was followed by sustained release that continued for 240 hours; 13.6%±0.21% of the drug was released over this period of time. A latency period characterized by no drug release is observed in the initial phase. The occurrence of a lag time in the release process indicates that the polymer has to be degraded to a considerable extent before the drug can leave the nanoparticle.
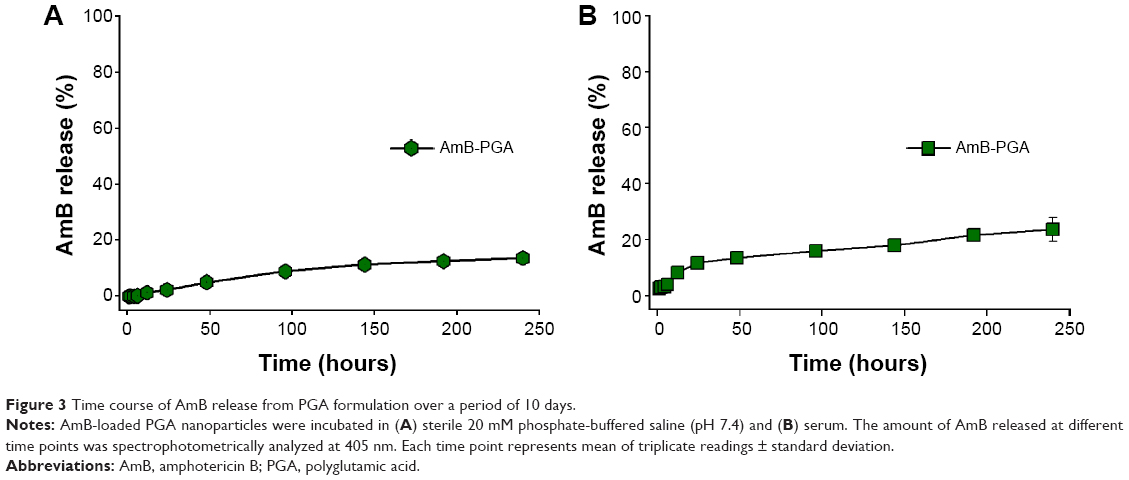 | Figure 3 Time course of AmB release from PGA formulation over a period of 10 days. |
In order to assess the fate of AmB-PGA nanoparticles in the plasma circulation, a simulation study involving incubation in mouse serum at 37°C was performed. The AmB-PGA nanoparticles showed high stability in serum, with only 20.9%±4.21% of AmB released from the nanoparticles after 240 hours (Figure 3B). Sustained stability in serum ensures that AmB does not bind to low-density plasma lipoproteins.76
The stability of encapsulated AmB in serum has been extensively studied in other systems, including ones derived from polymers.80,81 The AmB-PGA formulation showed better stability in PBS (pH 7.4) than AmB polymeric micelle systems, such as AmB-PEG-PLA (~45% of AmB was released after 24 hours),82 AmB-poly-lactic glycolic acid/dextran micelles (~20% AmB released after 24 hours), AmB-PEG-PLA polymersomes (~50% AmB released after 24 hours),83 and AmB-poly(2-ethyl-2-oxazoline)-block-poly-(aspartic acid) polyion complex micelles (~40% AmB released after 24 hours).84 Furthermore, the stability of these polymeric micelle systems was only examined in PBS, and there have been no data available for serum. The release pattern of the AmB-PGA nanoparticles in PBS and serum was similar to the stability data of AmBisome.The high stability of the AmB-PGA formulation could be attributed (in part) to the stabilizing properties of the PVA59,60 used in the preparatory cocktail during formation of the nanoparticles.
In vitro toxicity tests
Before introducing the novel formulation of AmB-PGA as a potential antifungal therapy approach, we investigated the associated intrinsic toxicity issues both in vitro and in vivo. Given that hemolysis is a serious toxic manifestation of AmB-D,85 we tested for acute toxicity using an in vitro red blood cell toxicity test. The association of AmB with polymers resulted in a large decrease in their hemolytic activity. At a lower concentration (1 μg/mL) and shorter duration (1 hour), no significant hemolysis was observed with any of the formulations (Figure 4A). In fact, no hemolysis was observed with the PGA formulation across the whole range of tested concentrations, ie, 1–100 μg/mL (maximum leakage was 4.0%±0.9%; P<0.001). AmB-L, used as a control, also showed negligible hemolysis (2.60%±0.7%). However, the positive control, AmB-D, showed a sharp increase in toxicity as the concentration was increased from 5 μg/mL to 10 μg/mL (20.0%±5.0% and 90.0%±7.3% respectively); with complete hemolysis at the 50 μg/mL concentration of AmB (Figure 4A).
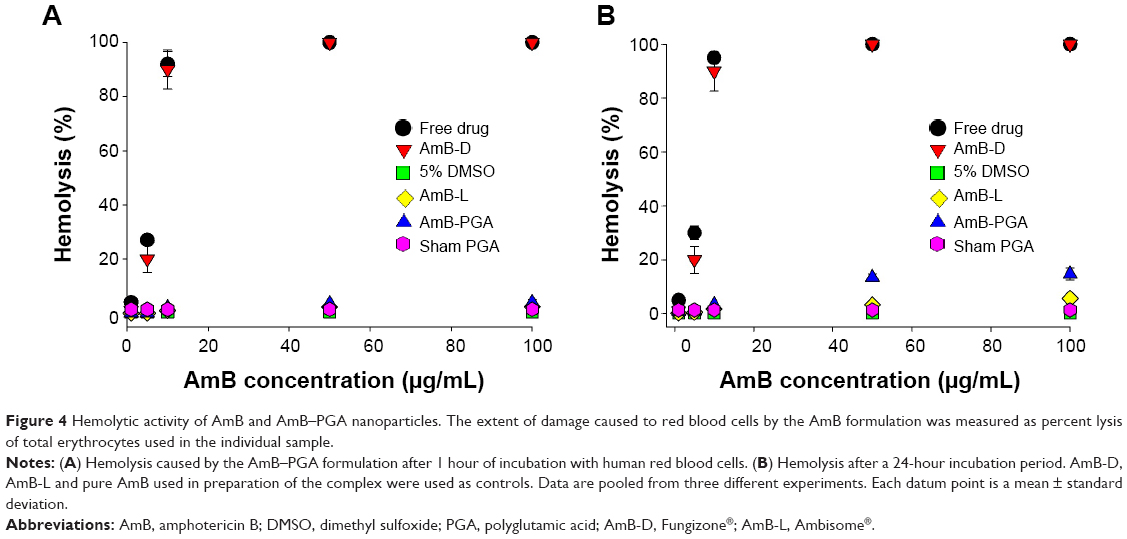 | Figure 4 Hemolytic activity of AmB and AmB–PGA nanoparticles. The extent of damage caused to red blood cells by the AmB formulation was measured as percent lysis of total erythrocytes used in the individual sample. |
Incubation for longer durations (24 hours) at 37°C did not affect the toxic properties of AmB-PGA nanoparticles and did not induce significant hemolysis (Figure 4B). Only 14.7%±2.2% hemolysis at 100 μg/mL (AmB equivalents) was observed (Figure 4B). AmB-D showed 100% hemolysis at an AmB equivalent concentration of 50 μg/mL. Empty nanoparticles (sham group) showed no signs of toxicity (1.40%±0.50%) and AmB-L displayed negligible hemolysis (5.6%±0.3%).
The low toxicity of AmB when formulated in AmB-PGA nanoparticles can be attributed to the shielding effect of the PGA nanoparticles resulting from association of AmB with PGA. In the case of AmB-D, the drug is available in total at a given time point, so has a direct adverse effect on the membrane of the red blood cell, resulting in leakage of hemoglobin and ions. On the other hand, the PGA formulation regulates drug release, so that the drug is not readily available to bind to the red blood cell membrane, thereby reducing toxicity. Moreover, the slow release of the drug from the PGA formulation ensures that the drug remains as a monomer and does not form oligomer aggregates, which is the species mainly responsible for drug toxicity.
The cytotoxic effect of the AmB-PGA formulation was also assessed in mammalian cells. The AmB-PGA nanoparticle was not toxic to mammalian cells (KB and RAW 264.7), whereas AmB-D showed dose-dependent toxicity (Figure 5A and B). A 10 μg/mL concentration of free drug (pure AmB) allowed survival of only 55.4%±7.6% of the cells. On the other hand, our PGA formulation prepared in-house had much less toxicity at the same concentration (86.3%±3.4% cell survival), comparable with that of AmB-L (90.01%±9.75%). A cell survival rate of 79.2%±6.7% was observed for PGA nanoparticles at an AmB concentration of 50 μg/mL, whereas AmB-D was completely cytotoxic to KB cells at the same AmB concentration (Figure 5A). At a concentration of 200 μg/mL AmB equivalents, the viability of KB cells was maintained at 75.7%±3.2% when treated with the AmB-PGA formulation. AmB-L maintained a cell survival of 84.5%±9.7%; whereas the pure drug was 100% lethal at the same concentration (200 μg/mL AmB equivalents). The MTT assay performed on RAW 264.7 cells treated with AmB-PGA nanoparticles showed similar results. However, the survival rate was much higher (~93% at 200 μg/mL AmB equivalents) than that observed with KB cells (Figure 5B), equivalent to the cell survival rate seen with AmB-L. The sodium salt of PGA alone did not display toxicity toward KB cells or RAW 264.7 cells at much higher doses (data not shown).
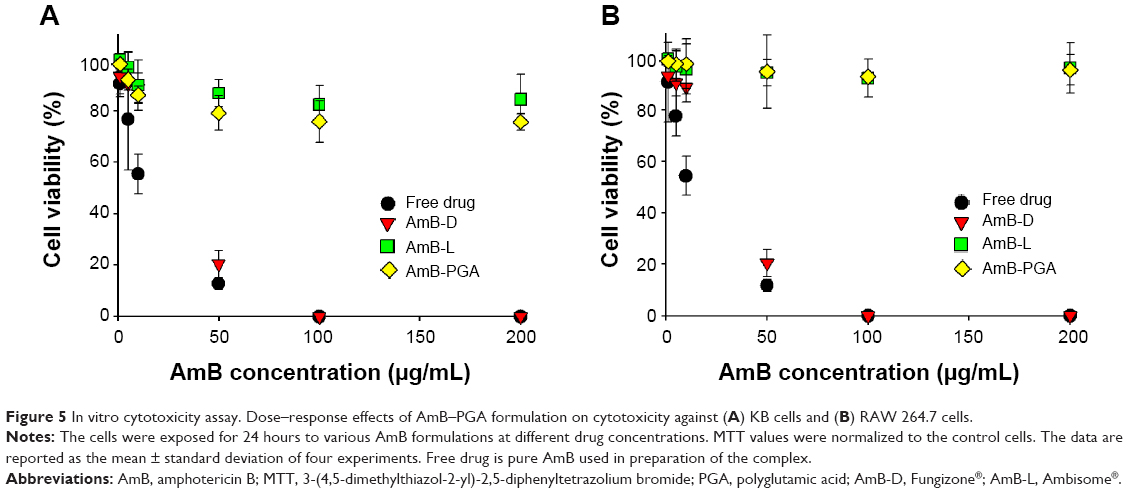 | Figure 5 In vitrocytotoxicity assay. Dose–response effects of AmB–PGA formulation on cytotoxicity against (A) KB cells and (B) RAW 264.7 cells. |
In vivotoxicity of AmB–PGA formulation
Next, we evaluated the toxic manifestations (if any) of the AmB-PGA nanoformulation in in vivo models treated with three different doses (5, 10, and 15 mg/kg body weight). AmB-D was administered at a dose of 5 mg/kg body weight. Liver and renal function tests (Table 2) suggest that while AmB-D imparts significant toxicity to the host liver and kidney, the AmB-PGA formulation was not toxic even at a dose of 15 mg/kg body weight and did not significantly alter serum biochemical parameters for renal and hepatic function. There was no significant elevation in serum alanine transaminase levels, while aspartate transaminase levels were slightly elevated. However, animals treated with AmB-D exhibited significantly elevated levels of both alanine aminotransferase and aspartate aminotransferase. Creatinine concentrations were within the normal range for mouse serum in the AmB-PGA-treated groups, whereas mice treated with AmB-D showed relatively higher levels of creatinine (Table 2). The AmB-PGA nanoparticle was well tolerated by the experimental animals up to a dose of 15 mg/kg body weight, with no signs of illness or loss of body weight (data not shown).
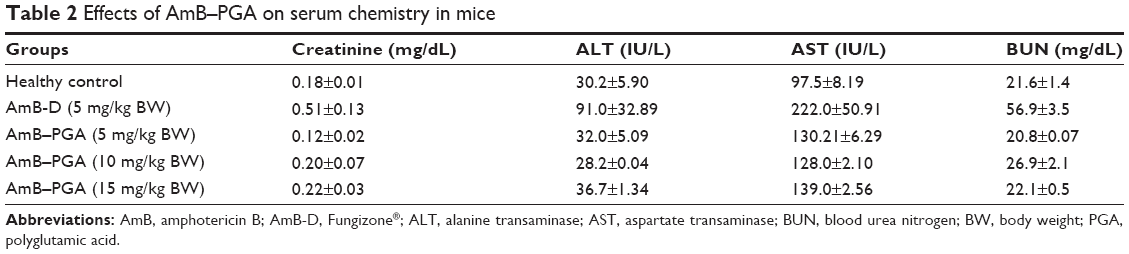 | Table 2 Effects of AmB–PGA on serum chemistry in mice |
The possible toxicity issues related to the formulation were further ruled out by histopathological examination of the kidneys from treated animals. As observed under light microscopy, features of the renal parenchyma from animals treated with the AmB-PGA nanoparticles (Figure S1C) were very much comparable with those of healthy controls (Figure S1D) as well as AmB-L (Figure S1B). There were no changes in the normal morphology of the kidneys, which showed normal renal corpuscles in terms of glomerular mass, cellularity, and urinary space. Intact renal tubules with characteristic cell linings as well as interstitial space could be identified. Both the proximal and distal convoluted tubules appear normal in architecture. AmB-D showed toxic effects on the kidney, with varying degrees of tissue necrosis (Figure S1A). These results are consistent with preclinical reports by other investigators that AmB-D is more toxic than AmB-L and ABLC (amphotericin B lipid complex) at the cellular level86,87 and more toxic than AmB-L and AmB colloidal dispersion (Amphotec®) in organs.88,89
In vitro antifungal activity
The antifungal activity of the AmB-PGA formulation was evaluated over a final concentration range of 0.015 μg/mL to 8 μg/mL using a broth microdilution method. The relative potency of the AmB-PGA complexes in vitro was similar to that of AmB-D (Table 3). The MIC of PGA-formulated AmB as well as AmB-D was 0.125 μg/mL, while the MIC of AmB-L against the C. albicans strain was found to be 0.5 μg/mL. In the case of C. glabrata, AmB-PGA nanoparticles (MIC 0.25 μg/mL) were found to be more potent than either AmB-D (MIC 0.5 μg/mL) or AmB-L (MIC 1.0 μg/mL). Fungicidal activity has been routinely expressed as a 99.9% reduction in CFU over a fixed sampling period. MFC values were found to be 0.5 μg/mL for the AmB-PGA formulation, while AmB-D showed an MFC of 1.0 μg/mL. The MIC and MFC of the AmB-PGA nanoparticle and of AmB-D against the C. albicans quality control strain (ATCC 10231) was found to be 0.25 μg/mL and 0.5 μg/mL, respectively (Table 3). When the different AmB formulations were tested at 4× MIC, the AmB-PGA nanoparticle as well as AmB-D caused a 99% loss of yeast viability after 4 hours of incubation (Figure 6). The PGA polymer was inactive against the pathogen up to a concentration of 500 μg/mL (data not shown).
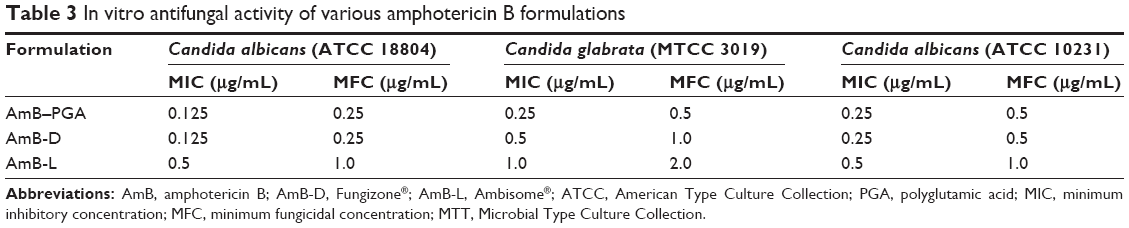 | Table 3 In vitro antifungal activity of various amphotericin B formulations |
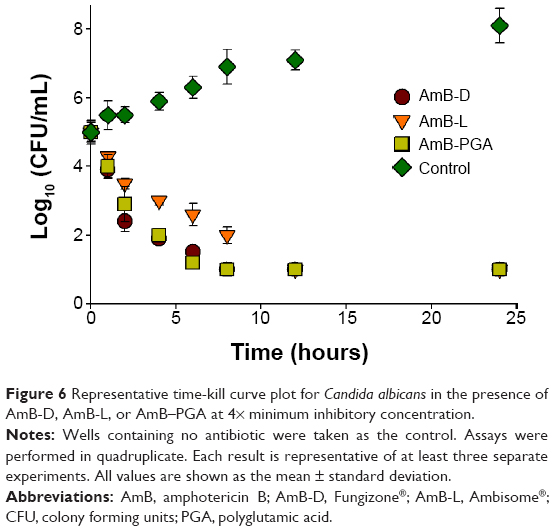 | Figure 6 Representative time-kill curve plot for Candida albicans in the presence of AmB-D, AmB-L, or AmB–PGA at 4× minimum inhibitory concentration. |
Antifungal activity of AmB–PGA nanoparticles against intracellular C. albicans
Occasionally, the dimorphic C. albicans may opt for intracellular life cycle inside the macrophages in order to avoid antibody onslaught. This intracellular abode also offers protection against the circulating antibiotics. Taking this fact into consideration, we assessed the antifungal potential of AmB entrapped in PGA nanoparticles against macrophages harboring intracellular C. albicans. It should be noted that no complete eradication of C. albicans was observed up to a drug concentration of 50 μg/mL (Figure 7). Overall, AmB-L was found to be less effective for killing intracellular C. albicans than either of the formulations tested (ie AmB–PGA and AmB-D). At a dose of 5 μg/mL, AmB-D showed higher intracellular antifungal activity than AmB-L (log 2.5 versus log 3.9, respectively). For AmB-D, doses higher than 10 μg/mL were not studied because of their detrimental effects toward the host cell. The antifungal activity of AmB-PGA was comparable with that of AmB-D (log 1.6 versus log 1.5, respectively) at a concentration of 10 μg/mL AmB equivalent; with a log 2 decrease in CFU (Figure 7). A further increase in AmB dose (to 50 μg/mL) caused a 1 log decrease in CFU.
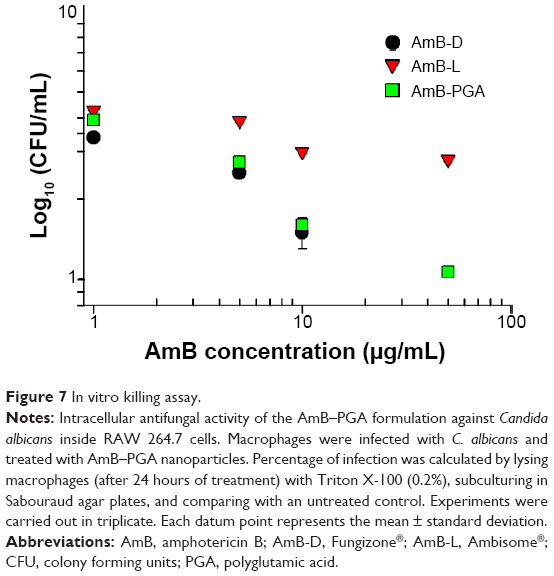 | Figure 7 In vitrokilling assay. |
The reduced toxicity of AmB following liposomal encapsulation (ie, AmBisome) seemed to be associated with a substantial reduction of its direct antifungal activity. Low antifungal activity of AmB-L against intracellular C. albicans has also been reported by other investigators.37 This may be accounted for by the low uptake of AmBisome (demonstrated in the J774 macrophage-like cell line) in comparison with other lipid based AmB formulations.90 A higher level of uptake of AmB-PGA nanoparticles than of AmBisome in murine peritoneal macrophages and RAW 264.7 cells (our unpublished data) seems to be responsible for the observed high antifungal activity of the AmB-PGA formulation. The particulate nature of PGA nanoparticles allows them to be efficiently taken up by these cells, giving rise to a higher concentration of drug. Simultaneous endocytosis of the AmB-PGA complex and C. albicans brings them into close proximity, which favors more pronounced killing, presumably by apoptosis of target cells.91
Regression of C. albicans biofilm induced by AmB–PGA
In order to determine the potential of AmB-PGA nanoparticles to interfere with formationof C. albicans biofilm, we studied their antibiofilm activity. Preliminary experiments on the antibiofilm activity of the nanoparticles were performed using phase contrast microscopy. Surprisingly, the AmB-PGA formulation prevented formation of biofilm to a considerable extent (Figure S2). This effect was unexpected, given the fact that the AmB-PGA nanoparticles would only reduce cell growth and not completely inhibit it. These encouraging results prompted us to examine its antibiofilm activity employing SEM and XTT studies.
SEM studies revealed that complete eradication of the biofilms was not achieved by treatment with AmB-PGA nanoparticles, and fungal cells adhering to the biomaterial were still visible (Figure 8). However, compared with control (untreated) biofilms, AmB-PGA-treated biofilms were less hyphal and some of the cells within the biofilms showed aberrant morphology. The XTT assay was also performed to quantitatively assess the potential of the AmB-PGA formulation to inhibit C. albicans biofilm (Figure 9). Substantial inhibition of biofilm growth was observed upon exposure to the AmB-PGA formulation. The AmB-PGA nanoparticles showed concentration-dependent activity on C. albicans biofilm. Approximately 30%–35% of viable biofilm was observed in fully developed film at a concentration of 100 μg/mL. AmB-L was the least effective against Candida biofilm, with a higher cell survival percentage (55.0%±2.8%). On the other hand, AmB-D showed viability of 43.5%±2.4% (Figure 9).
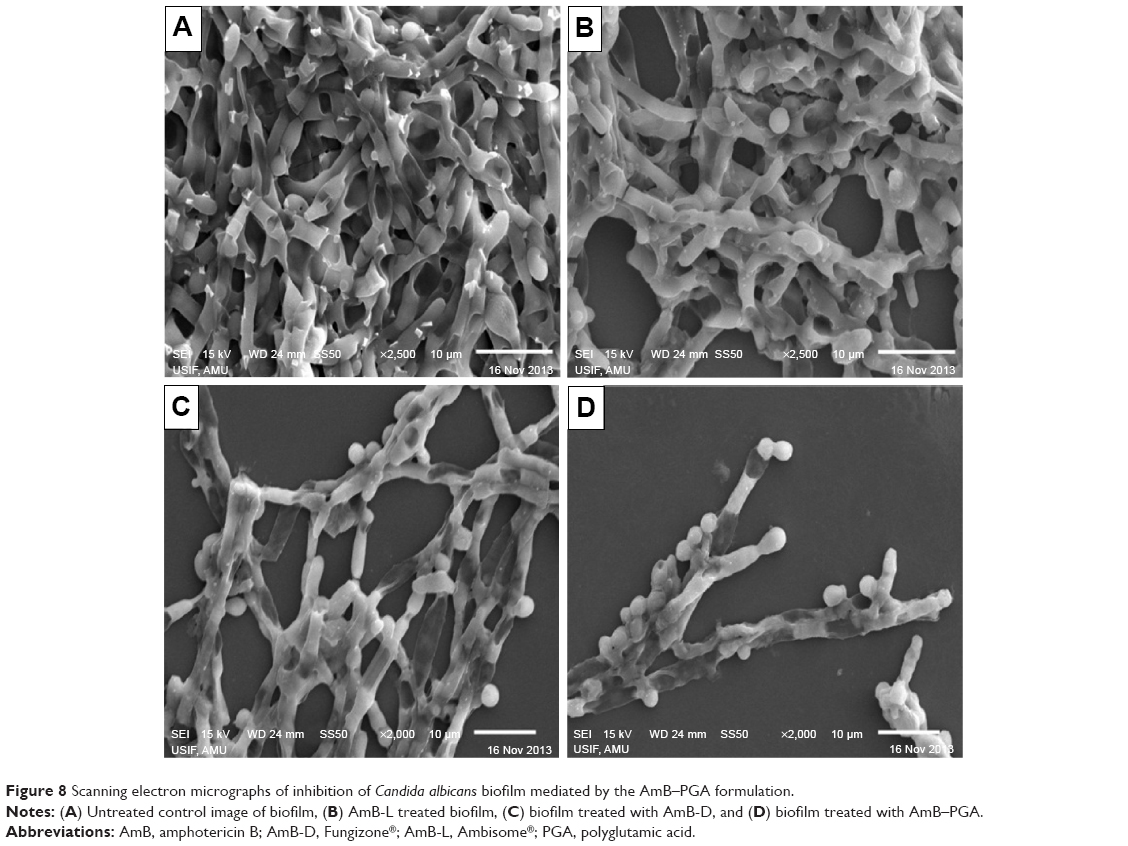 | Figure 8 Scanning electron micrographs of inhibition of Candida albicans biofilm mediated by the AmB–PGA formulation. |
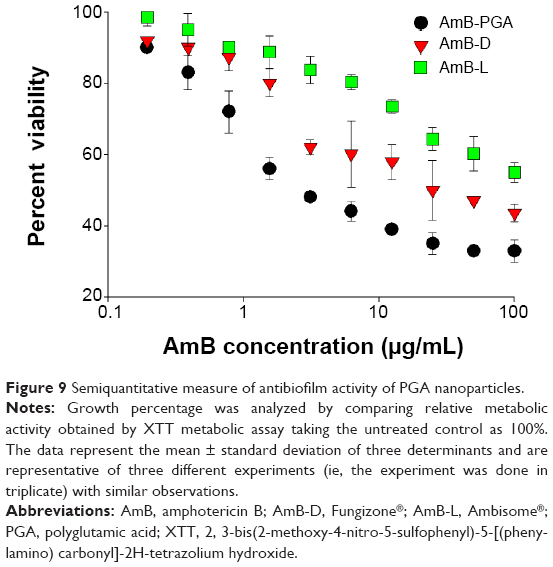 | Figure 9 Semiquantitative measure of antibiofilm activity of PGA nanoparticles. |
The antifungal susceptibility of C. albicans biofilm was tested using highly developed biofilms of intermediate phase (24 hours) and maturation phase (48 hours).92 All three antifungal formulations (AmB-D, AmB-L, and AmB-PGA) were found to be more effective against 24-hour biofilm than 48-hour biofilm. AmB-PGA nanoparticles demonstrated slightly higher activity than AmB-D for both 24-hour and 48-hour biofilm (Figure S3), with AmB-L being the least effective. Despite the improved activity of the AmB-PGA formulation, complete eradication of sessile organisms within mature biofilms was not observed (Figure S3). For 24-hour preformed biofilm, AmB-PGA reduced the optical density of the biofilm by 50% (P<0.001) after 4 hours of incubation time. In the case of 48-hour biofilm, the AmB-PGA formulation achieved a 50% reduction in biofilm only after 24 hours. This decrease in antibiofilm activity as the biofilm matures is a common phenomenon with antifungal agents.92 Similar results were obtained for AmB-D.
Consistent with our results (Figures 9 and S3), other researchers have shown that the antifungal killing effects of AmB against biofilms is generally more pronounced at the highest concentration (64 μg/mL) but not at therapeutic concentrations (0.125 and 0.5 μg/mL).93 Researchers have observed that 20 times the MIC of commonly used antifungals was required to achieve a significant reduction in cell numbers.94 Thus, higher levels of AmB-D are required to manage biofilm-related infection in the clinic, and this is not advisable because of the toxic behavior of AmB. We have shown here that our AmB-PGA formulation prepared in-house is well tolerated even at high concentration and has little or no toxicity (Figures 4 and 5, Table 2). Therefore, it can be said that this AmB-PGA formulation represents a good alternative to improve the management of Candida biofilm-associated infection, to disrupt biofilms, and to prevent the emergence of resistance.
C. albicans biofilm, like many other microorganisms, arises from microcolonies embedded in an extracellular matrix.95 The prominence of hyphal filaments is the hallmark of C. albicans biofilm maturation.92 In addition to assisting evasion of host defense mechanisms, hyphal transformants are lethal to macrophages and endothelial cells.96,97 A reduction in hyphal content as well as lower fungal viability (Figures 8 and 9) makes the AmB-PGA nanoformulation a promising strategy for dealing with biofilms. The mechanism behind the unique antibiofilm activity of our formulation prepared in-house is unknown. Given the size of the formulation, it is somewhat surprising that it can penetrate the extracellular matrix to target fungal cells; however, it can be speculated that the dispersion of AmB in the polymer may in fact facilitate passage through the charged polysaccharide extracellular matrix.
More elaborate studies involving confocal laser scanning microscopy are required to ascertain the potent antibiofilm activity of AmB–PGA nanoparticles. We are presently undertaking studies to evaluate the effect of AmB-PGA nanoparticles on the morphogenesis of C. albicans during biofilm formation by means of real-time polymerase chain reaction and other techniques. Further, the potential of the AmB-PGA formulation in the treatment of oral and systemic C. albicans infection is also currently under investigation in a mouse model.
Conclusion
In summary, the results of the present study indicate that encapsulation with PGA markedly improves the antifungal activity of AmB, without signs of drug-induced toxicity. A PGA homopolymer can associate with AmB to produce a formulation that is water-soluble, efficacious, well tolerated, and highly stable, and can be used as an alternative to the presently available AmB formulations. PGA-based AmB nanoparticles offer a safe and efficacious drug delivery vehicle with potential for future use as a nanomedicine in the clinical setting.
Disclosure
The authors report no conflicts of interest in this work.
References
Barratt G, Bretagne S. Optimizing efficacy of amphotericin B through nanomodification. Int J Nanomedicine. 2007;2:301–313. | ||
Torrado JJ, Espada R, Ballesteros MP, Torrado-Santiago S. Amphotericin B formulations and drug targeting. J Pharm Sci. 2008;97:2405–2425. | ||
Kleinberg M. What is the current and future status of conventional amphotericin B? Int J Antimicrob Agents. 2006;27(S1):12–16. | ||
Ha YE, Peck KR, Joo EJ, et al. Impact of first-line antifungal agents on the outcomes and costs of candidemia. Antimicrob Agents Chemother. 2012;56:3950–3956. | ||
Laniado-Laborin R, Cabrales-Vargas MN. Amphotericin B: side effects and toxicity. Rev Iberoam Micol. 2009;26:223–227. | ||
Gupta S, Pal A, Vyas SP. Drug delivery strategies for therapy of visceral leishmaniasis. Expert Opin Drug Deliv. 2010;7:371–402. | ||
Deray G. Amphotericin B nephrotoxicity. J Antimicrob Chemother. 2002;49(S1):37–41. | ||
Sabra R, Branch RA. Amphotericin B nephrotoxicity. Drug Saf. 1990;5:94–108. | ||
Dupont B. Overview of the lipid formulations of amphotericin B. J Antimicrob Chemother. 2002;49(S1):31–36. | ||
Herbrecht R, Natarajan-Ame S, Nivoix Y, Letscher-Bru V. The lipid formulations of amphotericin B. Expert Opin Pharmacother. 2003;4:1277–1287. | ||
Antoniadou A, Dupont B. Lipid formulations of amphotericin B: where are we today? J Med Mycol. 2005;15:230–238. | ||
Adler-Moore PJ, Proffitt TR. Development, characterization, efficacy and mode of action of Ambisome, a unilamellar liposomal formulation of Amphotericin B. J Liposome Res. 1993;3:429–450. | ||
Adler-Moore J, Proffitt RT. AmBisome: liposomal formulation, structure, mechanism of action and pre-clinical experience. J Antimicrob Chemother. 2002;49:21–30. | ||
Bruynesteyn K, Gant V, McKenzie C, et al. A cost-effectiveness analysis of caspofungin vs liposomal amphotericin B for treatment of suspected fungal infections in the UK. Eur J Haematol. 2007;78:532–539. | ||
Olliaro P, Darley S, Laxminarayan R, Sundar S. Cost-effectiveness projections of single and combination therapies for visceral leishmaniasis in Bihar, India. Trop Med Int Health. 2009;14:918–925. | ||
Adler-Moore JP, Proffitt RT. Amphotericin B lipid preparations: what are the differences? Clin Microbiol Infect. 2008;14(S4):25–36. | ||
Olson JA, Adler-Moore JP, Jensen GM, et al. Comparison of the physicochemical, antifungal, and toxic properties of two liposomal amphotericin B products. Antimicrob Agents Chemother. 2008;52:259–268. | ||
Cifani C, Costantino S, Massi M, Berrino L. Commercially available lipid formulations of amphotericin B: are they bioequivalent and therapeutically equivalent? Acta Biomed. 2012;83:154–163. | ||
Patel GP, Crank CW, Leikin JB. An evaluation of hepatotoxicity and nephrotoxicity of liposomal amphotericin B (L-AMB). J Med Toxicol. 2011;7:12–15. | ||
Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75:1–18. | ||
Saffie-Siebert R, Ogden J, Parry-Billings M. Nanotechnology approaches to solving the problems of poorly water-soluble drugs. Drug Discovery World. 2005;3:71–76. | ||
Yang D, Yu L, Van S. Clinically relevant anticancer polymer paclitaxel therapeutics. Cancers (Basel). 2011;3:17–42. | ||
Kopecek J, Kopecková P, Minko T, Lu ZR, Peterson CM. Water soluble polymers in tumor targeted delivery. J Control Release. 2001;74:147–158. | ||
Farazuddin M, Sharma B, Khan AA, Joshi B, Owais M. Anticancer efficacy of perillyl alcohol-bearing PLGA microparticles. Int J Nanomedicine. 2012;7:35–47. | ||
Farazuddin M, Chauhan A, Khan RM, Owais M. Amoxicillin-bearing microparticles: potential in the treatment of Listeria monocytogenes infection in Swiss albino mice. Biosci Rep. 2011;31:265–272. | ||
Wang C, Feng M, Deng J, et al. Poly(alpha-glutamic acid) combined with polycation as serum-resistant carriers for gene delivery. Int J Pharm. 2010;398:237–245. | ||
Li C. Poly(L-glutamic acid)-anticancer drug conjugates. Adv Drug Deliv Rev. 2002;54:695–713. | ||
Li C, Wallace S. Polymer-drug conjugates: recent development in clinical oncology. Adv Drug Deliv Rev. 2008;60:886–898. | ||
Paz-Ares L, Ross H, O’Brien M, et al. Phase III trial comparing paclitaxel poliglumex vs docetaxel in the second-line treatment of non-small-cell lung cancer. Br J Cancer. 2008;98:1608–1613. | ||
Sabbatini P, Aghajanian C, Dizon D, et al. Phase II study of CT-2103 in patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. J Clin Oncol. 2004;22:4523–4531. | ||
Cho CS, Kobayashi A, Takei R, et al. Receptor-mediated cell modulator delivery to hepatocyte using nanoparticles coated with carbohydrate-carrying polymers. Biomaterials. 2001;22:45–51. | ||
Nilsson-Ehle I, Yoshikawa TT, Edwards JE, Schotz MC, Guze LB. Quantitation of amphotericin B with use of high-pressure liquid chromatography. J Infect Dis. 1977;135:414–422. | ||
Barwicz J, Christian S, Gruda I. Effects of the aggregation state of amphotericin B in its toxicity to mice. Antimicrob Agents Chemother. 1992;36:2310–2315. | ||
Nahar M, Dubey V, Mishra D, Mishra P, Dube A, Jain NK. In vitro evaluation of surface functionalized gelatin nanoparticles for macrophage targeting in the therapy of visceral leishmaniasis. J Drug Target. 2010;18:93–105. | ||
Olson JA, Adler-Moore JP, Schwartz J, Jensen GM, Proffitt RT. Comparative efficacies, toxicities, and tissue concentrations of amphotericin B lipid formulations in a murine pulmonary aspergillosis model. Antimicrob Agents Chemother. 2006;50:2122–2131. | ||
Clinical and Laboratory Standards Institute. M27-A2. Vol 22 No 15. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard method.2nd ed. (ISBN 1-56238-469-4). Wayne, PA, USA: Clinical and Laboratory Standards Institute; 2002. | ||
van Etten EW, van Vianen W, Hak J, Bakker-Woudenberg IA. Activity of liposomal amphotericin B with prolonged circulation in blood versus those of AmBisome and Fungizone against intracellular Candida albicans in murine peritoneal macrophages. Antimicrob Agents Chemother. 1998;42:2437–2439. | ||
Bachmann SP, Van de Walle K, Ramage G, et al. In vitro activity of caspofungin against Candida albicans biofilms. Antimicrob Agents Chemother. 2002;46:3591–3596. | ||
Kuhn DM, George T, Chandra J, Mukherjee PK, Ghannoum MA. Antifungal susceptibility of Candida biofilms: unique efficacy of amphotericin B lipid formulations and echinocandins. Antimicrob Agents Chemother. 2002;46:1773–1780. | ||
Vandermeulen G, Rouxhet L, Arien A, Brewster ME, Preat V. Encapsulation of amphotericin B in poly(ethylene glycol)-block-poly(ε-caprolactone-co-trimethylene carbonate) polymeric micelles. Int J Pharm. 2006;309:234–240. | ||
Choi KC, Bang JY, Kim PI, Kim C, Song CE. Amphotericin B-incorporated polymeric micelles composed of poly-(d,l-lactide-co-glycolide)/dextran graft copolymer. Int J Pharm. 2008;355:224–230. | ||
Adams ML, Andes RD, Kwon SG. Amphotericin B encapsulated in micelles based on poly(ethylene oxide)-block–poly(L-amino acid) derivatives exerts reduced in vitro haemolysis but maintains potent in vivo antifungal activity. Biomacromolecules. 2003;4:750–757. | ||
Falamarzian A, Lavasanifar A. Optimization of the hydrophobic domain in poly(ethylene oxide)-poly(varepsilon-caprolactone) based nano-carriers for the solubilization and delivery of amphotericin B. Colloids Surf Biointerfaces. 2010;81:313–320. | ||
Yoo BK, Jalil Miah MA, Lee ES, Han K. Reduced renal toxicity of nanoparticular amphotericin B micelles prepared with partially benzylated poly-L-aspartic acid. Biol Pharm Bull. 2006;29:1700–1705. | ||
Nishi KK, Antony M, Mohanan PV, Anilkumar TV, Loiseau PM, Jayakrishnan A. Amphotericin B-gum arabic conjugates: synthesis, toxicity, bioavailability, and activities against Leishmania and fungi. Pharm Res. 2007;24:971–980. | ||
Conover DC, Zahao H, Longley BC, Shum LK, Greenwald BR. Utility of poly(ethylene glycol) conjugation to create prodrugs of amphotericin B. Bioconjug Chem. 2003;14:661–666. | ||
Nicoletti S, Seifert K, Gilbert IH. N-(2-hydroxypropyl)-methacrylamide-amphotericin B (HPMA-AmB) copolymer conjugates as antileishmanial agents. Int J Antimicrob Agents. 2009;33:441–448. | ||
Ehrenfreund-Kleinman T, Azzam T, Falk R, Polacheck I, Golenser J, Domb JA. Synthesis and characterization of novel water soluble amphotericin B-arabinogalactan conjugates. Biomaterials. 2002;23:1327–1335. | ||
Jansook P, Loftsson T. CDs as solubilizers: effects of excipients and competing drugs. Int J Pharm. 2009;379:32–40. | ||
Corware KF, Harris DF, Teo IF, et al. Accelerated healing of cutaneous leishmaniasis in non-healing BALB/c mice using water soluble amphotericin B-polymethacrylic acid. Biomaterials. 2011;32:8029–8039. | ||
Kajtar M, Vikmon M, Morlin E, Szejtli J. Aggregation of amphotericin B in the presence of gamma-cyclodextrin. Biopolymers. 1989;28:1585–1596. | ||
Ohtani Y, Irie T, Uekama K, Fukunaga K, Pitha J. Differential effects of alpha-, beta- and gamma-cyclodextrins on human erythrocytes. Eur J Biochem. 1989;186:17–22. | ||
Donaubauer HH, Fuchs H, Langer KH, Bar A. Subchronic intravenous toxicity studies with gamma-cyclodextrin in rats. Regul Toxicol Pharmacol. 1998;27:189–198. | ||
Charvalos E, Tzatzarakis MN, Van Bambeke F, et al. Water soluble amphotericin B-polyvinylpyrrolidone complexes with maintained antifungal activity against Candida spp. and Aspergillus spp. and reduced haemolytic and cytotoxic effects. J Antimicrob Chemother. 2006;57:236–244. | ||
Savic R, Azzam T, Eisenberg A, Maysinger D. Assessment of the integrity of poly(caprolactone)-b-poly(ethylene oxide) micelles under biological conditions: a fluorogenic-based approach. Langmuir. 2006;22:3570–3578. | ||
Chen H, Kim S, He W, et al. Fast release of lipophilic agents from circulating PEG-PDLLA micelles revealed by in vivoForster resonance energy transfer imaging. Langmuir. 2008;24:5213–5217. | ||
Akagi T, Kaneko T, Kida T, Akashi M. Multifunctional conjugation of proteins on/into bio-nanoparticles prepared by amphiphilic poly(gamma-glutamic acid). J Biomater Sci Polym Ed. 2006;17:875–892. | ||
Lemke A, Kiderlen A, Kayser O, Amphotericin B. Appl Microbiol Biotechnol. 2005;68:151–162. | ||
Pimpang P, Choopun S. Monodispersity and stability of gold nanoparticles stabilized by using polyvinyl alcohol. Chiang Mai J Sci. 2011;38:31–38. | ||
Rosenblatt KM, Bunjes H. Poly(vinyl alcohol) as emulsifier stabilizes solid triglyceride drug carrier nanoparticles in the alpha-modification. Mol Pharm. 2009;6:105–120. | ||
Prokop A, Kozlov E, Carlesso J, Davidson M. Hydrogel based colloidal polymeric system for protein and drug delivery: physical and chemical characterization permeability control and applications. Advances in Polymer Sciences. 2002;160:119–173. | ||
Beck-Broichsitter M, Ruppert C, Schmehl T, et al. Biophysical investigation of pulmonary surfactant surface properties upon contact with polymeric nanoparticles in vitro. Nanomedicine. 2011;7:341–350. | ||
Patila S, Sandberg A, Heckert E, Self W, Sea S. Protein adsorption and cellular uptake of cerium oxide nanoparticles as a function of zeta potential. Biomaterials. 2007;28:4600–4607. | ||
Puttipipatkhachorn S, Nunthanid J, Yamamotoc K, Peck GE. Drug physical state and drug–polymer interaction on drug release from chitosan matrix films. J Control Release. 2001;75:143–153. | ||
Sutherland MD, Thorkildson P, Parks SD, Kozel TR. In vivo fate and distribution of poly-gamma-D-glutamic acid, the capsular antigen from Bacillus anthracis. Infect Immun. 2008;76:899–906. | ||
Pelkmans L. Secrets of caveolae- and lipid raft-mediated endocytosis revealed by mammalian viruses. Biochim Biophys Acta. 2005;1746:295–304. | ||
Espada R, Valdespina S, Dea MA, et al. In vivo distribution and therapeutic efficacy of a novel amphotericin B poly-aggregated formulation. J Antimicrob Chemother. 2008;61:1125–1131. | ||
Legrand P, Romero EA, Cohen BE, Bolard J. Effects of aggregation and solvent on the toxicity of amphotericin B to human erythrocytes. Antimicrob Agents Chemother. 1992;36:2518–2522. | ||
Sanchez-Brunete JA, Dea MA, Rama A, et al. Amphotericin B molecular organization as an essential factor to improve activity/toxicity ratio in the treatment of visceral leishmaniasis. J Drug Target. 2004;12:453–460. | ||
Mullen AB, Carter KC, Baillie AJ. Comparison of the efficacies of various formulations of amphotericin B against murine visceral leishmaniasis. Antimicrob Agents Chemother. 1997;41:2089–2092. | ||
Lamy-Freund MT, Ferreira VF, Faljoni-Alario A, Schreier S. Effect of aggregation on the kinetics of autoxidation of the polyene antibiotic amphotericin B. J Pharm Sci. 1993;82:162–166. | ||
Mazerski J, Borowski E. Molecular dynamics of amphotericin B. II Dimer in water. Biophys Chem. 1996;57:205–217. | ||
Gaboriau F, Cheron M, Leroy L, Bolard J. Physico-chemical properties of the heat-induced ‘superaggregates’ of amphotericin B. Biophys Chem. 1997;66:1–12. | ||
Egito LC, de Medeiros SR, Medeiros MG, Price JC, Egito ES. Evaluation of the relationship of the molecular aggregation state of amphotericin B in medium to its genotoxic potential. J Pharm Sci. 2004;93:1557–1565. | ||
Bolard J, Legrand P, Heitz F, Cybulska B. One-sided action of amphotericin-B on cholesterol-containing membranes is determined by its self-association in the medium. Biochemistry. 1991;30:5707–5715. | ||
Hartsel S, Bolard J. Amphotericin B: new life for an old drug. Trends Pharmacol Sci. 1996;17:445–449. | ||
Brajtburg J, Bolard J. Carrier effects on biological activity of amphotericin B. Clin Microbiol Rev. 1996;9:512–531. | ||
Espada R, Valdespina S, Alfonso C, Rivas G, Ballesteros MP, Torrado JJ. Effect of aggregation state on the toxicity of different amphotericin B preparations. Int J Pharm. 2008;361:64–69. | ||
Iman M, Huang Z, Szoka FC, Jaafari MR. Characterization of the colloidal properties, in vitro antifungal activity, antileishmanial activity and toxicity in mice of a di-stigma-steryl hemi-succinoyl-glycero-phosphocholine liposome-intercalated amphotericin B. Int J Pharm. 2011;408:163–172. | ||
Jee J, McCoy A, Mecozzi S. Encapsulation and release of amphotericin B from an ABC tri-block fluorous copolymer. Pharm Res. 2012;29:69–82. | ||
Diezi T, Bae Y, Kwon GS. Enhanced stability of PEG-block poly (N-hexyl stearate l-aspartamide) micelles in the presence of serum proteins. Mol Pharm. 2010;7:1355–1360. | ||
Yang ZL, Li XR, Yang KW, Liu Y. Amphotericin B loaded poly(ethylene glycol)-poly(lactide) micelles: preparation, freeze-drying, and in vitro release. J Biomed Mater Res A. 2008;85:539–546. | ||
Jain JP, Kumar N. Development of amphotericin B loaded polymersomes based on (PEG)(3)-PLA co-polymers: factors affecting size and in vitro evaluation. Eur J Pharm Sci. 2010;40:456–465. | ||
Wang CH, Wang WT, Hsiue GH. Development of polyion complex micelles for encapsulating and delivering amphotericin B. Biomaterials. 2009;30:3352–3358. | ||
Skrocka-Knopik A, Bielawski J. The mechanism of the haemolytic activity of polyene antibiotics. Cell Mol Biol Lett. 2002;7:31–48. | ||
Jensen GM, Skenes CR, Bunch TH, et al. Determination of the relative toxicity of amphotericin B formulations: a red blood cell potassium release assay. Drug Deliv. 1999;6:81–88. | ||
Sperry PJ, Cua DJ, Wetzel SA, Adler-Moore JP. Antimicrobial activity of AmBisome and non-liposomal amphotericin B following uptake of Candida glabrata by murine epidermal Langerhans cells. Med Mycol. 1998;36:135–141. | ||
Lee JW, Amantea MA, Francis PA, et al. Pharmacokinetics and safety of a unilamellar liposomal formulation of amphotericin B (AmBisome) in rabbits. Antimicrob Agents Chemother. 1994;38:713–718. | ||
Van Etten EW, van den Heuvel-de Groot C, Bakker-Woudenberg IA. Efficacies of amphotericin B-desoxycholate (Fungizone), liposomal amphotericin B (AmBisome) and fluconazole in the treatment of systemic candidosis in immunocompetent and leucopenic mice. J Antimicrob Chemother. 1993;32:723–739. | ||
Legrand P, Vertut-Do A, Bolard J. Comparative internalization and recycling of different amphotericin B formulations by a macrophage-like cell line. J Antimicrob Chemother. 1996;37:519–533. | ||
Al-Dhaheri RS, Douglas LJ. Apoptosis in Candida biofilms exposed to amphotericin B. J Med Microbiol. 2010;59:149–157. | ||
Chandra J, Kuhn DM, Mukherjee PK, Hoyer LL, McCormick T, Ghannoum MA. Biofilm formation by the fungal pathogen Candida albicans: development, architecture, and drug resistance. J Bacteriol. 2001;183:5385–5394. | ||
Ramage G, Vandewalle K, Bachmann SP, et al. In vitro pharmacodynamic properties of three antifungal agents against preformed Candida albicans biofilms determined by time-kill studies. Antimicrob Agents Chemother. 2002;46:3634–3636. | ||
Baillie GS, Douglas LJ. Effect of growth rate on resistance of Candida albicans biofilms to antifungal agents. Antimicrob Agents Chemother. 1998;42:1900–1905. | ||
Stoodley P, Sauer K, Davies DG, Costerton JW. Biofilms as complex differentiated communities. Annu Rev Microbiol. 2002;56:187–209. | ||
Hoyer LL, Payne TL, Bell M, et al. Relationship between Candida albicans virulence during experimental hematogenously disseminated infection and endothelial cell damage in vitro. Infect Immun. 2004;72:598–601. | ||
Lo HJ, Kohler JR, Di Domenico B, Loebenberg D, Caccia-Puoti A, Fink GR. Nonfilamentous C. albicans mutants are avirulent. Cell. 1997;90:939–949. |
Supplementary materials
Animals
Inbred female Balb/c mice (aged 6–8 weeks, mean weight 20±2 g) were obtained from the animal facility at the Indian Institute of Toxicological Research, Lucknow, India. After procurement, the animals were acclimatized for a few days under standard husbandry conditions (room temperature 22°C±3°C, relative humidity 65%±10%, and a 12-hour light/dark cycle). They had free access to a standard dry pellet diet and water ad libitum under strict hygienic conditions. Animals were anesthetized with ketamine 5 mg/kg and xylazine 4 mg/kg prior to sacrifice. Humane endpoints were necessary for mice that survived at the conclusion of the experiment. Mice were administered an anesthetic cocktail of ketamine and xylazine intraperitoneally and then euthanized by cervical dislocation. Experiments involving bleeding, injection, and sacrifice of animals were strictly performed following the mandates approved by the institutional animal ethics committee constituted as per the recommendations of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India (http://moef.nic.in/division/committee-purpose-control-and-supervision-experiments-animals-cpcsea-1). During the experimental procedures, all efforts were made to minimize pain and suffering.
Ethics statement
All animal experiments were reviewed and approved by the institutional animal ethics committee of the Interdisciplinary Biotechnology Unit, Aligarh Muslim University, India, and were performed according to the National Regulatory Guidelines issued by CPCSEA, Ministry of Environment and Forest, Government of India. Our approval identifier was 332/CPCSEA.
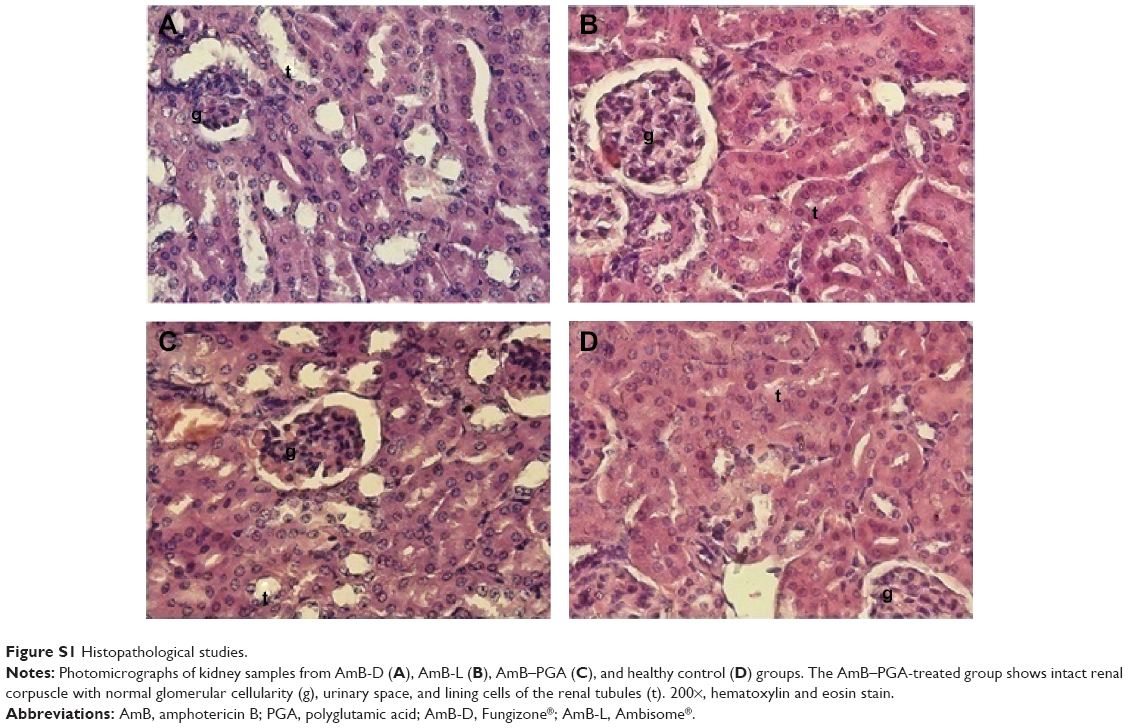 | Figure S1 Histopathological studies. |
Determination of particle size and zeta potential
The zeta potential of the nanoparticles was determined using DTS software (Malvern Instruments Limited, Malvern, UK) based on M3-PALS technology. Samples were filtered using a 0.22 μm polyethersulfone syringe filter (Millipore, Bangalore, India) to remove contamination with dust particles. The formulation was lyophilized in a 2.0 mL microfuge tube and the samples were reconstituted in 20 mM phosphate buffer (pH 7.4). This dispersion was then rapidly dispensed to an electrophoresis cell to measure the electrophoretic mobility, and zeta potential values were calculated. The concentration of the AmB-PGA formulation used was 10−4 M. The experiments were repeated three times and the average zeta potential and standard deviations were calculated. The particle size of the nanoparticles was determined by photon correlation spectroscopy (90Plus/BI-MAS, Brookhaven Instruments, Holtville, NY, USA). For size measurements, samples were diluted in Milli-Q water and measured for at least 120 seconds. The concentration of AmB-PGA formulation used was 10−4 M. All measurements were performed in triplicate.
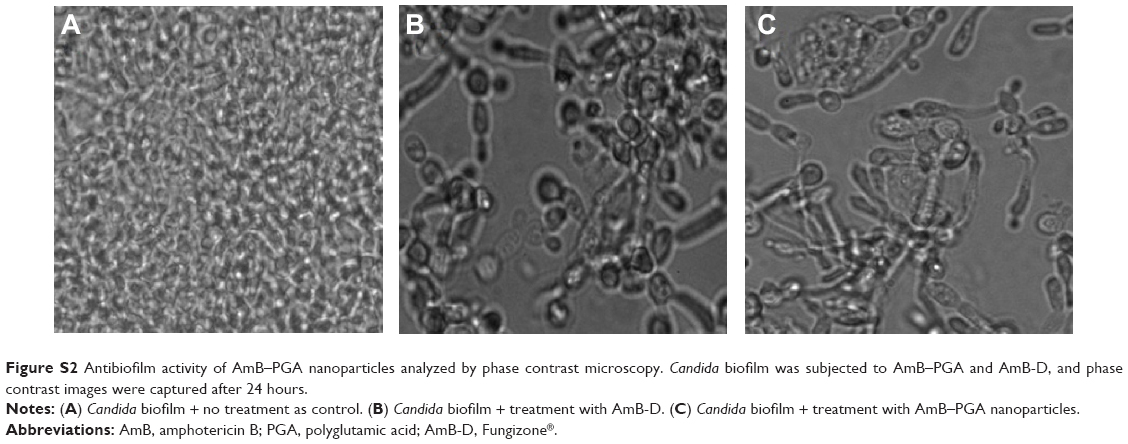 | Figure S2 Antibiofilm activity of AmB–PGA nanoparticles analyzed by phase contrast microscopy.Candida biofilm was subjected to AmB–PGA and AmB-D, and phase contrast images were captured after 24 hours. |
Transmission electron microscopy
Transmission electron microscopy was performed to characterize the size and surface morphology of AmB-loaded nanoparticles using an electron microscope (CM-10, Philips, Hamburg, Germany). For these studies, the AmB-PGA formulation was used at a concentration of 10 μg/mL AmB equivalents in double-distilled water. The lyophilized preparation of nanoparticles was suspended in 20 mM phosphate-buffered saline (pH 7.4) and a drop of the formulation was mounted on a clear glass stub, air-dried, and coated with gold-palladium alloy using a sputter coater. An accelerating voltage of 20 kV was used for imaging.
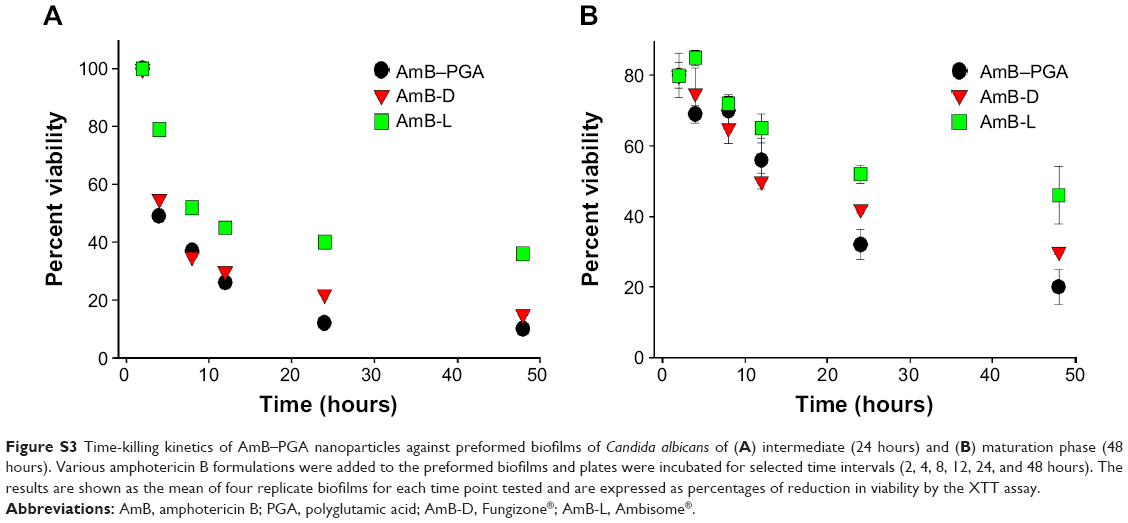 | Figure S3 Time-killing kinetics of AmB–PGA nanoparticles against preformed biofilms of Candida albicans of (A) intermediate (24 hours) and (B) maturation phase (48 hours). Various amphotericin B formulations were added to the preformed biofilms and plates were incubated for selected time intervals (2, 4, 8, 12, 24, and 48 hours). The results are shown as the mean of four replicate biofilms for each time point tested and are expressed as percentages of reduction in viability by the XTT assay. |
Dynamic light scattering measurements
Dynamic light scattering measurements were carried out using DynaPro-TC-04 equipment (Protein Solutions, Wyatt Technology, Santa Barbara, CA, USA) equipped with a temperature-controlled microsampler. The PGA nanoparticles (2 mg/mL) were filtered serially through 0.22 μm and 0.02 μm Whatman syringe filters directly into a 12 μL quartz cuvette. For each experiment, 20 measurements were taken. Mean hydrodynamic radius (Rh) was analyzed using Dynamics software at optimized resolution. The Rh was estimated on the basis of an autocorrelation analysis of scattered light intensity data based on a translation diffusion coefficient by Stokes–Einstein relationship:
|
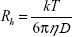
where Rh is the hydrodynamic radius, k is the Boltzmann constant, T is temperature, η is the viscosity of water, and D is the diffusion coefficient.
PGA nanoparticle-mediated transfer of entrapped molecules to macrophages
Mice were injected intraperitoneally with 2 mL of 4% thioglycolate, and peritoneal exudate cells were harvested 4 days later using cold Roswell Park Memorial Institute (RPMI) 1640 medium.1 Peritoneal macrophages (1×104 cells) were cultured on a glass coverslip using complete RPMI 1640 medium overnight in a CO2 incubator (with 5% CO2) at 37°C. The next morning, the cells were washed thoroughly with Hank’s Balanced Salt Solution and incubated with fluorescent dye (calcein)-loaded PGA nanoparticles for 30 minutes at 37°C. The calcein-loaded PGA nanoparticles were prepared using the same procedure as that used for AmB-PGA nanoparticles. The noninteracted PGA population was removed by thorough washing followed by mounting of the macrophage-harboring coverslip in glycerol. The cells were observed under a fluorescence microscope (Zeiss, Oberkochen, Germany). Phase contrast and fluorescent micrographs were recorded accordingly.
High-performance liquid chromatography analysis
Quantitation of AmB was performed by a slight modification of the method described by Nilsson-Ehle et al2 as standardized in our laboratory. High performance liquid chromatography was conducted using a MicroBondapak C-18 column (30 cm ×4 mm [diameter]; particle size 5 μm; Waters Associates, Milford, MA, USA) with a Waters high-performance liquid chromatography system. The mobile phase was a mixture of methanol and 0.005 M ethylenediaminetetraacetic acid (8:2 vol/vol) deaerated by vacuum. The injection volume was 20 μL, delivered at a rate of 1.0 mL/min. The eluent was monitored at 405 nm for AmB content with a run time of 15 minutes. The retention time of AmB was 8 minutes. The internal standard was 1-amino-4-nitronaphthalene. The limit of detection was 1 ng. The results were quantified by comparison of the AmB peak at 405 nm with a standard of known concentration eluted at the same retention time. A standard calibration curve of the drug was plotted at 405 nm by determining the area under the curve corresponding to the increasing amount of the drug (3.125–200 ng of AmB equivalent in 20 μL of sample injection volume). The highest AmB concentration was 10μg/mL (equivalent to 200 ng of AmB in 20 μL). Dilutions of AmB were performed in methanol to ensure that AmB remained in the monomeric form.
Histopathological analysis
Histopathological evaluation was also done on the kidneys of nonimmunosuppressed, uninfected mice following intravenous treatment with a single dose of 15 mg/kg AmB-PGA, AmB-L, or phosphate-buffered saline (control) following the published protocol.3 AmB-D was administered at a dose of 5 mg/kg body weight. The tissue sample was aseptically collected 24 hours after treatment. At necropsy, kidney samples were fixed in 10% neutral buffered formalin. Tissue blocks of kidney (1×0.2 cm) were processed routinely. They were dehydrated in alcohol, cleared in xylene, and infiltrated and embedded in paraffin. Sections (5 μm thick) were cut and mounted on glass slides. The slides were then rehydrated in water, stained with hematoxylin and eosin, dehydrated, cleared in xylene, mounted with a synthetic resin, and covered with a coverslip. All tissue sections were examined and photographed with a light microscope (BX45, Olympus, Tokyo, Japan) at ×200 by a board-certified pathologist form Jawaharlal Nehru Medical College, Aligarh Muslim University, Aligarh, India.
In vitro susceptibility testing
A concentrated (1,000 mg/L) stock solution of AmB (Sigma Aldrich, St Louis, MO, USA) was dissolved in dimethyl sulfoxide. The solution was diluted with dimethyl sulfoxide and then with the appropriate test medium. AmB was tested over a concentration range of 0.015–8 μg/mL. Candida albicans ATCC 10231 was used as a quality control strain.
The broth microdilution method was used as the reference method, as described by the Clinical and Laboratory Standards Institute (document M27-A24). The inoculum was prepared from Sabouraud dextrose agar subcultures incubated at 35°C for 24 hours and the resulting suspension was adjusted spectrophotometrically to a density equivalent to a 0.5 McFarland standard at 530 nm (1.5×106 colony forming units [CFU]/mL). A working suspension was made by 1:100 dilution of the suspension in RPMI 1640 medium with l-glutamine and without bicarbonate (Sigma-Aldrich), buffered to pH 7.0 with 0.165 M morpholinepropanesulfonic acid (Sigma-Aldrich). Next, 100 μL of the diluted inoculum was added to each well. The final inoculum size was (0.5–2.5) 103 CFU/mL. The broth dilution tests were incubated at 35°C, and minimum inhibitory concentrations (MICs) were determined after 48 hours of incubation by observation of the presence or absence of visible growth. For AmB, the MIC endpoint is the lowest concentration that inhibits visual growth or an endpoint score of 0 (100% inhibition).
After determination of the MICs, the entire contents (200 μL) of each well with drug concentrations above the MIC were plated onto two drug-free Sabouraud agar plates (100 μL aliquots/plate). Aliquots were placed in a single spot on the agar plate and, after drying, the cells were dispersed by streaking. Plates were incubated at 35°C for 48 hours; the minimum fungicidal concentrations (MFC) was the lowest drug concentration that showed either no growth or fewer than three colonies to obtain approximately 99%–99.5% killing activity. All assays were performed in duplicate and repeated at least twice.
Microscopic visualization of C. albicans biofilms
C. albicans biofilms were grown on sterile glass coverslips placed in six-well polystyrene plates.5 After 2 hours of incubation of the fungal cell suspension (at the density of 107 cells) on the glass coverslips at 37°C, the coverslips were washed three times to remove nonadhered cells. Further, the glass coverslips were incubated in RPMI 1640 medium for 48 hours at 37°C to allow development of biofilm. Following biofilm formation, the medium was aspirated and nonadherent cells were removed by thoroughly washing the biofilms three times in sterile phosphate-buffered saline. The AmB-PGA formulation was then added and the coverslips were incubated further at 37°C. After 24 hours, the coverslips were fixed in 2% paraformaldehyde followed by washing, and the activity of AmB-PGA nanoparticles against C. albicans biofilm was visualized using a Z1 Axio Inverted microscope (Carl Zeiss, Thornwood, NY, USA).
Time-kill kinetics of biofilm
Biofilms were grown under shaking (100 rpm) for 24 or 48 hours at 37°C. Various amphotericin B formulations were then added at a concentration of 8 μg/mL to the preformed biofilms and the plates were incubated for selected time intervals (2, 4, 8, 12, 24, and 48 hours). At each time interval, the antifungal effects were monitored by a metabolic assay based on the reduction of XTT as described previously.6 After treatment, 100 μL of the XTT/menadione solution, ie, 2, 3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide, was added to each prewashed biofilm and to the negative control wells. The plates were covered with aluminum foil and incubated in the dark for 2 hours at 37°C. The plates were then read in a microtiter plate reader at 490 nm. Untreated biofilms containing RPMI 1640 medium were included as positive controls. AmB-D and AmB-L were also included in the study as controls. Wells without biofilms served as a blank. Four replicate biofilms were included for each experiment.
|
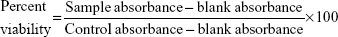
where blank absorbance is the medium alone and control absorbance is fungal cells with medium alone.
References
Alam M, Zubair S, Mohammad F, et al. Development, characterization and efficacy of niosomal diallyl disulphide in treatment of disseminated murine candidiasis. Nanomedicine. 2013;9:247–256. | ||
Nilsson-Ehle I, Yoshikawa TT, Edwards JE, Schotz MC, Guze LB. Quantitation of amphotericin B with use of high-pressure liquid chromatography. J Infect Dis. 1977;135:414–422. | ||
Olson JA, Adler-Moore JP, Schwartz J, Jensen GM, Proffitt RT. Comparative efficacies, toxicities, and tissue concentrations of amphotericin B lipid formulations in a murine pulmonary aspergillosis model. Antimicrob Agents Chemother. 2006;50:2122–2131. | ||
Clinical and Laboratory Standards Institute. M27-A2. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard-method. 2nd ed.Wayne, PA, USA: Clinical and Laboratory Standards Institute; 2002. | ||
Kuhn DM, George T, Chandra J, Mukherjee PK, Ghannoum MA. Antifungal susceptibility of Candida biofilms: unique efficacy of amphotericin B lipid formulations and echinocandins. Antimicrob Agents Chemother. 2002;46:1773–1780. | ||
Bachmann SP, Vande Walle K, Ramage G, et al. In vitro activity of caspofungin against Candida albicans biofilms. Antimicrob Agents Chemother. 2002;46:3591–3596. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.