")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Selexipag in the treatment of pulmonary arterial hypertension: design, development, and therapy
Received 30 July 2016
Accepted for publication 28 September 2016
Published 15 November 2016 Volume 2016:10 Pages 3747—3754
DOI https://doi.org/10.2147/DDDT.S103534
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Elizabeth Ashley Hardin,1 Kelly M Chin2
1Department of Internal Medicine, Division of Cardiology, 2Department of Internal Medicine, Division of Pulmonary and Critical Care Medicine, University of Texas Southwestern Medical Center, Dallas, TX, USA
Abstract: Pulmonary arterial hypertension is characterized by abnormalities in the small pulmonary arteries including increased vasoconstriction, vascular remodeling, proliferation of smooth muscle cells, and in situ thrombosis. Selexipag, a novel, oral prostacyclin receptor agonist, has been shown to improve hemodynamics in a phase II clinical trial and reduce clinical worsening in a large phase III clinical trial involving patients with pulmonary arterial hypertension. In this paper, we describe the prostacyclin signaling pathway, currently available oral prostanoid medications, and the development and clinical use of selexipag.
Keywords: selexipag, pulmonary arterial hypertension, prostacyclin
Overview
Pulmonary arterial hypertension (PAH) is a life-threatening disease associated with significant morbidity and mortality despite therapeutic advances in the modern era. Abnormalities in the small pulmonary arteries include increased vasoconstriction, vascular remodeling, proliferation of smooth muscle cells, and in situ thrombosis.1–3 The pathogenesis of PAH is thought to result from an imbalance in the amount and ratio of vasoactive substances including prostacyclin, nitric oxide, and endothelin-1. Reduction in the levels of prostacyclin relative to vasoconstrictive substances in the pulmonary vasculature has been implicated in the pathophysiology of PAH.4,5 Augmenting prostacyclin pathway activation is one of the major therapeutic strategies in the treatment of PAH. Selexipag, a novel, oral prostacyclin (IP) receptor agonist, has been shown to improve hemodynamics in a phase II clinical trial and to lead to a reduction in clinical worsening in a large phase III clinical trial involving patients with PAH. In this paper, we describe the prostacyclin signaling pathway, currently available oral prostanoid medications, and the development and clinical use of selexipag.
Prostacyclin
Prostacyclin, also known as prostaglandin I2, is an arachidonic acid derivative produced primarily by vascular endothelial cells. Prostacyclin acts predominantly through the G-protein coupled IP receptor leading to increases in cyclic adenosine monophosphate.6–9 Downstream effects include vasodilation, particularly under conditions of increased pulmonary vascular tone, inhibition of vascular smooth muscle cell proliferation, and of platelet aggregation.1–4
The importance of the IP receptor and prostacyclin signaling in the pulmonary vasculature has been shown in both basic science and clinical studies. In animal studies, mice with IP receptor knockout mutations develop much more severe PAH under hypoxic conditions compared with wild-type mice.5 Furthermore, transfer of the human prostacyclin synthase gene ameliorates the severity of monocrotaline-induced PAH in rats.6 Patients with PAH also have reduced IP receptor expression, reduced prostacyclin synthase expression, and reduced prostacyclin production.7–10
Epoprostenol, as prostacyclin is called when used therapeutically, was the first medication approved for the treatment of PAH. In clinical trials, epoprostenol led to improvement in functional class, quality of life, hemodynamics, exercise capacity, and survival, and it remains the only therapy to have shown a mortality benefit in randomized clinical trials in PAH.11 Administration requires an indwelling central catheter and continuous infusion pump due to the short half-life of epoprostenol and its limited stability at room temperature. Despite the complexities involved in its administration, it remains the therapy of choice for severe pulmonary hypertension.
Oral prostanoids in clinical use: beraprost and treprostinil
The significant hemodynamic and clinical benefits seen with the intravenous prostacyclins led to considerable interest in the development of oral prostanoids. Two are currently available for clinical use: beraprost, approved in Japan and South Korea and oral treprostinil, approved for use in the US. Clinical trial results with both medications have been mixed, likely in part due to difficulty in achieving therapeutic doses in some patients because of dose-limiting prostacyclin type side effects.
Beraprost was initially evaluated in a 12-week clinical trial with a primary endpoint of change in 6-minute walk distance (6MWD).12 A statistically significant improvement in 6MWD relative to placebo was seen in the 12-week study (mean change 25.1 m, P=0.04). However, a subsequent 12-month study found no difference between the beraprost and placebo groups in disease progression, the primary endpoint, and no difference in change in 6MWD compared with placebo at the 12-month time point.13 Possible explanations for the negative longer term study include beraprost’s short half-life of approximately 1 hour,14 difficulty in up-titration due to side effects, and potentially, the development of tolerance with long-term administration.
Oral treprostinil, approved in the US in 2013, has been evaluated in three 12- to 16-week randomized controlled clinical trials. All three studies used change in 6MWD as the primary endpoint. The treatment effect (change in 6MWD in treprostinil-treated patients minus change in 6MWD in placebo patients) was statistically significant in FREEDOM-M (23 m, P=0.01), but only approached statistical significance in FREEDOM-C and FREEDOM-C2 (11 m, P=0.07 and 10 m, P=0.09, respectively).15–17 Differences across the three studies include a 16-week duration for FREEDOM-C and C2, compared with 12 weeks for FREEDOM-M, and importantly, the prerequisite for background PAH therapy in FREEDOM-C and C2 compared with treatment naïve patients in FREEDOM-M. No significant improvement was seen in the secondary endpoints of World Health Organization functional class or clinical worsening in any of the three studies. Prostacyclin-type side effects such as headache, diarrhea, jaw pain, nausea, and vomiting were common.18–20
There are several ongoing studies of oral beraprost and treprostinil, including a trial investigating modified release oral beraprost taken four times daily in combination with inhaled treprostinil (NCT01908699)21 and a trial of oral treprostinil administered three times daily (NCT01560624).22
Selexipag and ACT-333679
Limitations in the half-life and bioavailability of the oral prostanoids led to a broader search for compounds with activity at the IP receptor. Selexipag, whose synthesis was first reported in 2007, is the first nonprostanoid IP receptor agonist approved for clinical use. When taken orally, selexipag is rapidly absorbed and subsequently hydrolyzed by liver carboxylesterase to its more active metabolite, ACT-333679.23 Selexipag and ACT-333679 are both IP receptor agonists with a high degree of selectivity versus members of the prostaglandin family, but ACT-333679 is approximately 37-fold more potent in activating the IP receptor and is considered to be the major contributor to the efficacy of selexipag.24 Both compounds have been shown to lead to pulmonary artery vasodilation ex vivo.25–27 In a monocrotaline-induced PAH animal model, selexipag was shown to lower right ventricular systolic pressures, reduce pulmonary artery wall thickness and right ventricular hypertrophy, and improve survival.25
Phase II trial
Selexipag was first evaluated in patients with PAH in a multicenter, phase II study conducted in Europe.28 Simonneau et al enrolled 43 patients into a 17-week long randomized controlled clinical trial of selexipag vs placebo. Patients were required to be on a stable background therapy regimen consisting of an endothelin receptor antagonist, a phosphodiesterase-5 inhibitor, or both, and to have a pulmonary vascular resistance (PVR) >5 Wood units at study entry. The primary endpoint was a change in PVR as expressed as a percentage of the baseline value. This was analyzed both in a per-protocol analysis (all treated patients who did not violate the protocol) and in an all-treated set (all patients who received at least one dose of the study drug). The PVR declined in treated patients to 81% of baseline as compared with an increase in placebo patients to 116% of baseline (Figure 1). This difference was statistically significant with a reduction in the primary endpoint of 30.3% (selexipag vs placebo, P<0.01). Improvement compared with placebo was also seen in the cardiac index (mean 0.5 L, P<0.05). Typical prostacyclin-associated side effects were reported in a majority of patients in the selexipag group including headache (most common), jaw pain, and nausea.
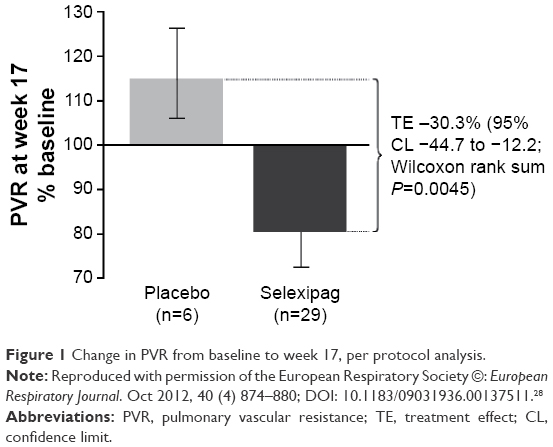 | Figure 1 Change in PVR from baseline to week 17, per protocol analysis. |
Phase III trial
The GRIPHON clinical trial enrolled 1,156 patients with PAH into a long-term time-to-event study with a primary endpoint of morbidity and mortality.29 The main finding was a significant reduction in the composite of death from any cause or a complication of PAH (41.6% in the placebo group and 27% in the selexipag group; hazard ratio 0.60, P≤0.001) (Figure 2). Improvement was also seen in the secondary endpoint of change in 6MWD at week 26 (12 m improvement vs placebo, P=0.003) and in the exploratory endpoint of change in N-terminal probrain natriuretic peptide level at 26 weeks (treatment effect −123 ng/L, P<0.001).
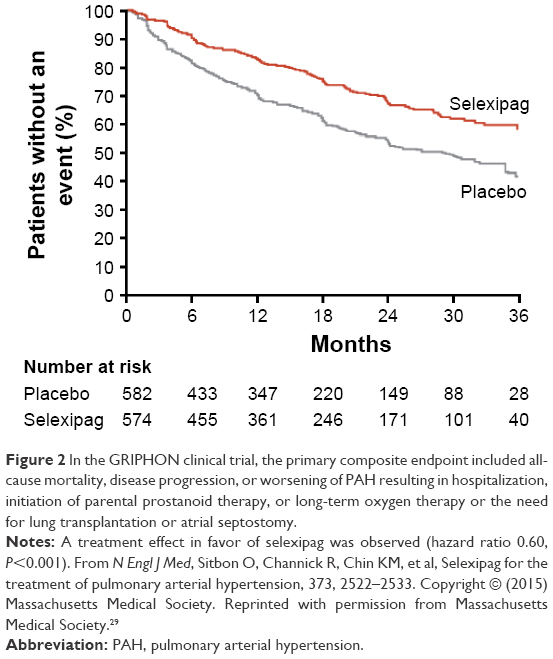 | Figure 2 In the GRIPHON clinical trial, the primary composite endpoint included all-cause mortality, disease progression, or worsening of PAH resulting in hospitalization, initiation of parental prostanoid therapy, or long-term oxygen therapy or the need for lung transplantation or atrial septostomy. |
Enrolled patients were allowed to be treatment naïve, in the event that no approved therapies were an option, or could be receiving an endothelin receptor antagonist, phosphodiesterase-5 inhibitor, or both. Approximately 80% of enrolled patients were on one or two background PAH therapies. All patients enrolled were required to have a formal diagnosis of PAH with a mean pulmonary arterial pressure >25 mmHg, PVR >5 Wood units, and a pulmonary capillary wedge pressure or left ventricular end diastolic pressure ≤15 mmHg upon cardiac catheterization. Patients were initiated on selexipag 200 μg twice daily with dose increases of 200 μg twice daily each week until the maximum tolerated dose was achieved over a 12-week titration phase. Importantly, the median length of treatment in the placebo and selexipag groups was 63.7 and 70.7 weeks, respectively.
The 40% reduction in the composite primary endpoint was driven largely by differences in clinical worsening and hospitalizations related to PAH, as there was no significant difference in mortality between the two study groups. In contrast to earlier studies in PAH, where greater benefit was reported in treatment-naïve patients,15,17,30 the treatment effect (primary endpoint) of selexipag was similar between treatment-naïve patients and patients on one or two background therapies. The treatment effect was also consistent across other prespecified subgroups including functional class, gender, age, PAH etiology, and geographic region (interaction P-value was not significant).
Prostacyclin-type adverse events were common in the selexipag group (Table 1), and adverse events leading to study discontinuation were seen in twice as many patients receiving selexipag as compared with placebo (14.3% vs 7.1%, respectively). Multiple sensitivity analyses were conducted in order to evaluate the potential effect of early study exit and of missing data on the primary endpoint, the results of which were consistent with the primary analysis. Based on this trial, the US Food and Drug Administration granted approval of this medication for the treatment of PAH in the US in December 2015, and subsequent approval was granted in the European Union in May 2016.
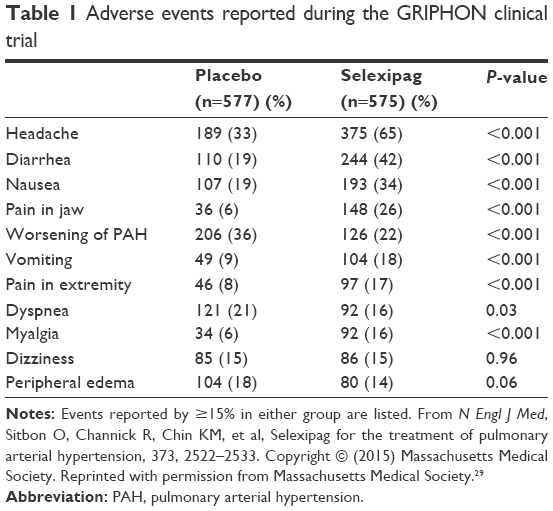 | Table 1 Adverse events reported during the GRIPHON clinical trial |
Dosing and adverse reactions
Selexipag is initiated at 200 μg twice daily and increased weekly until prostacyclin-associated side effects cannot be managed or to a maximum of 1,600 μg twice daily. Prostacyclin-type side effects including headache, diarrhea, jaw pain, and nausea are common, particularly during the up-titration phase. Improvement is often seen during the maintenance phase, particularly as those who have continued symptoms can be down-titrated. Published guidelines on symptom management are lacking. Based on the common side effect profile of prostacyclin therapy, our center provides patients with recommendations for the use of acetaminophen, ondansetron, and loperamide as needed at initiation of therapy, with a low threshold for the addition of tramadol for pain. We also recommend taking selexipag with food, as tolerability may be improved, but this is not required.
In the GRIPHON trial, the distribution of the final attained dosing range was 200–400 μg twice daily in 23%, 600–1,000 μg twice daily in 31%, and 1,200–1,600 μg twice daily in 43% of patients. Interestingly, in a prespecified analysis, the efficacy of selexipag was similar in patients regardless of their stratification based on dose achieved. The authors note, however, that this study was not conducted in a way to evaluate alternative dosing strategies, and recommended treatment with the highest dose at which side effects were manageable.
Several other adverse events of special interest were also reported in the phase III clinical trial. New onset hyperthyroidism was reported in eight patients in the selexipag group (1%) and none in the placebo group (P=0.004), while anemia was reported in 8% of the selexipag group and 5% of the placebo group (P=0.05). Serial laboratory follow-up of both measures (among others) are recommended for PAH in general,31 and no additional monitoring for patients receiving selexipag has been recommended.
Pharmacokinetics
The safety, tolerability, and pharmacokinetics of selexipag were initially evaluated in several dose ranging studies in healthy, normal volunteers.23 Selexipag has a half-life of 0.8–2.5 hours and is hydrolyzed to its pharmacologically active metabolite ACT-333679 with a half-life of 6.2–13.5 hours (Figure 3).24,32 It is rapidly absorbed after oral administration and may be taken with or without food. Single doses above 400 μg led to an increased incidence of headache, nausea, dizziness, and vomiting in normal volunteers. Gradual up-titration improves tolerability, and the maximum tolerated strength with repeated dosing in normal volunteers was 1,600 μg.32 In the presence of food, the absorption of selexipag was prolonged resulting in a delayed time to peak concentration (Tmax) and approximately 30% lower peak plasma concentration (Cmax). The exposure to selexipag and the active metabolite (area under the curve [AUC]) did not significantly change in the presence of food.24
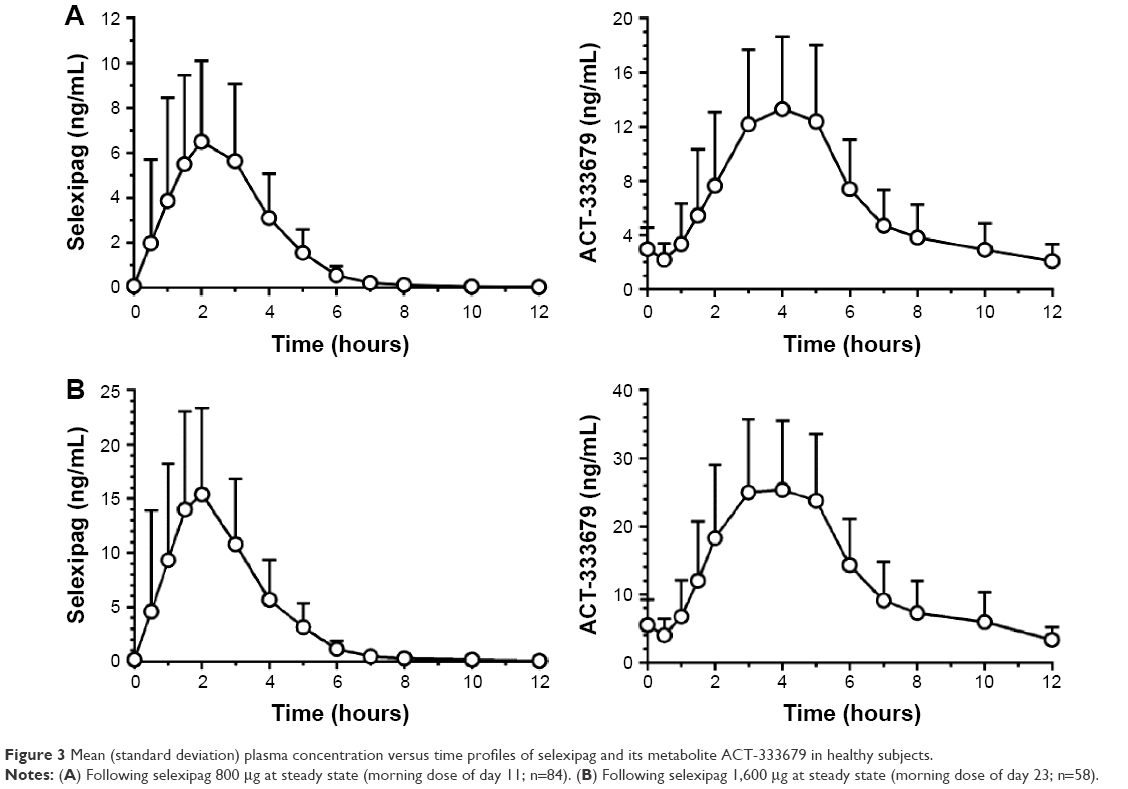 | Figure 3 Mean (standard deviation) plasma concentration versus time profiles of selexipag and its metabolite ACT-333679 in healthy subjects. |
Steady state is achieved within 3 days of twice daily dosing. Elimination is mainly via the hepatobiliary route,33 and renal and hepatic impairment increase exposure to both selexipag and its metabolite ACT-333679. Greater caution is recommended during up-titration in individuals with liver or renal impairment, and once daily administration is recommended for patients with moderate liver impairment.24 Use of selexipag should be avoided in those with severe hepatic impairment.34 The initiation of selexipag in patients with impaired renal function is similar to patients with normal renal function, however, caution during titration is advised. There is a 40%–70% increase in exposure of selexipag and its active metabolite in patients with estimated glomerular filtration rates of 15–30 mL/minute/1.73 m2. Of note, there have been no studies involving patients with severely impaired renal function (estimated glomerular filtration rate <15 mL/minute/1.73 m2).24
Selectivity and tachyphylaxis: selexipag vs prostacyclin analogs
Downstream effects mediated via the IP receptor are thought to be responsible for the benefits of prostacyclin therapy in PAH. Selexipag and ACT-333679 act selectively at the IP receptor, in contrast to epoprostenol and the synthetic prostacyclin analogs.25,35–40 Nonselective prostanoids have a shared affinity for a variety of other prostanoid receptors, some of which promote vasoconstriction or alter gastric motility.25,26,37,41 These data suggest that selectivity at the IP receptor could be advantageous.25,26,37 In contrast, other studies suggest that prostanoid activity at other vasodilatory cell surface receptors, or nuclear receptors involved in vascular cell proliferation, might be beneficial.9,42 Characterization of receptor profiles has been largely based on in vitro studies, thus the clinical consequences of drug actions at different receptors is unclear. This is particularly apparent in the context of pivotal placebo controlled clinical trials demonstrating strong evidence of benefit for both selective and nonselective therapies (Table 2). Nevertheless, the variability in prostanoid receptor expression across different experimental conditions26 and disease states43 does raise the possibility that some patients, or groups of patients, might differ in clinical response or side effects with different prostacyclin pathway medications.
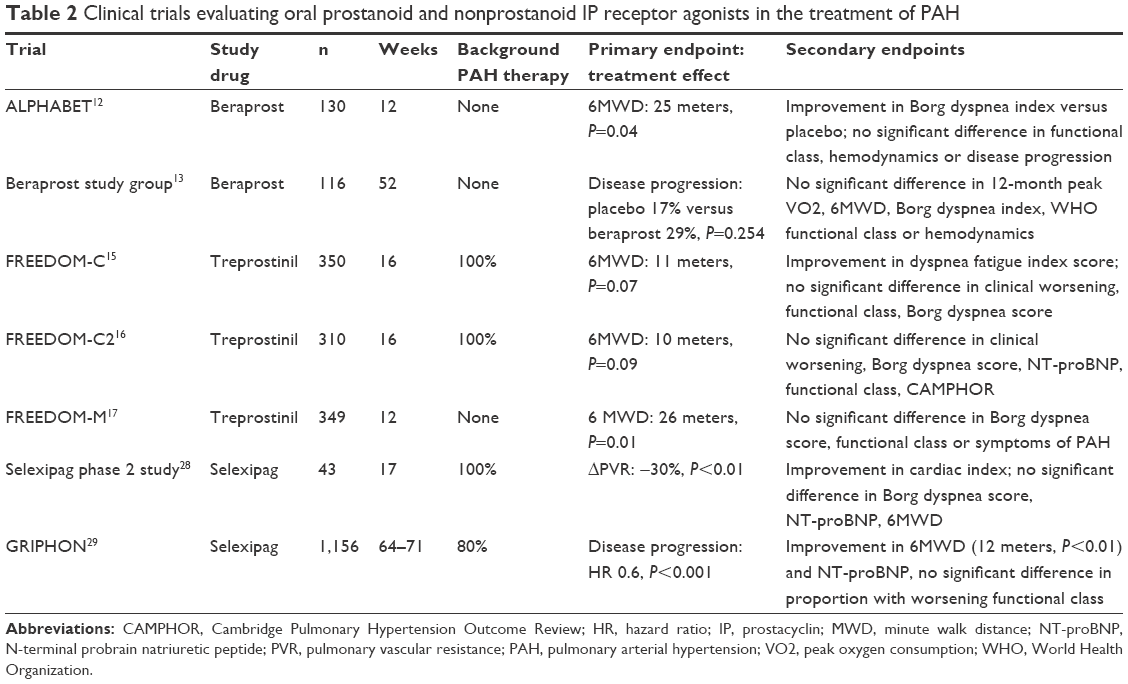 | Table 2 Clinical trials evaluating oral prostanoid and nonprostanoid IP receptor agonists in the treatment of PAH |
Another important difference between selexipag and the prostacyclin analogs is that selexipag does not appear to lead to IP receptor downregulation over time. Continuous infusion of the prostacyclin analogs, but not selexipag, leads to receptor internalization and tachyphylaxis in experimental conditions.44–47 The degree to which tachyphylaxis contributes to the need for late uptitration of epoprostenol in some patients during long-term infusion is unknown;48 however, the lack of receptor downregulation with selexipag treatment could potentially reduce the need for late uptitration. The mechanism of reduced IP receptor downregulation with selexipag and its active metabolite is thought to be due to partial antagonism at the IP receptor. This partial antagonism leads to decreased recruitment of beta arrestin, a protein involved in the first step of internalization of the IP receptor, which is likely due to its nonprostanoid structure.47
Selexipag for the treatment of PAH
As the treatment of PAH continues to evolve, there has been a shift toward earlier use of combination therapy based on the paradigm that derangements in three distinct signaling pathways underlie the development of PAH. This includes both upfront combination therapy, as studied in the AMBITION clinical trial (tadalafil and ambrisentan), and earlier use of sequential combination therapy.49 Current guidelines also recommend consideration of triple combination therapy in patients with an inadequate clinical response,50 supported by results of both the GRIPHON clinical trial, where 33% of patients were on combination background therapy, and a small pilot study of upfront triple combination therapy in patients presenting with advanced, severe PAH.51 Strengths of the GRIPHON trial include the enrollment of an unprecedented number of patients, long trial duration, and use of background therapy in a majority of patients.
The approval of selexipag for the treatment of PAH will likely allow for earlier treatment of a larger number of patients with an oral drug that targets the prostacyclin pathway24 thought to play a key role in disease pathophysiology. A number of clinical trials studying selexipag are ongoing in order to improve our understanding of its use in the treatment of PAH in the modern era. One such trial is the TRITON study (NCT02558231) which aims to evaluate initial triple vs dual oral combination therapy in patients with newly diagnosed PAH.21
Conclusion
Selexipag, the first highly selective, nonprostanoid IP receptor agonist to be approved for PAH, significantly reduced the likelihood of clinical worsening in a long-term randomized controlled clinical trial. Beneficial effects were observed both in treatment-naïve patients as well as in patients with one or two background PAH therapies.
Disclosure
Elizabeth Ashley Hardin reports no conflict of interest in this work. Dr Kelly M Chin has received fees for consulting work with Actelion and United Therapeutics and has received institutional support for pulmonary hypertension research from the Actelion, Bayer, GeNO, Gilead, Pfizer, Reata, United Therapeutics, and the NIH.
References
Clapp LH, Finney P, Turcato S, Tran S, Rubin LJ, Tinker A. Differential effects of stable prostacyclin analogs on smooth muscle proliferation and cyclic AMP generation in human pulmonary artery. Am J Respir Cell Mol Biol. 2002;26(2):194–201. | ||
Humbert M, Morrell NW, Archer SL, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):13s–24s. | ||
Oida H, Namba T, Sugimoto Y, et al. In situ hybridization studies of prostacyclin receptor mRNA expression in various mouse organs. Br J Pharmacol. 1995;116(7):2828–2837. | ||
Rubin LJ, Groves BM, Reeves JT, Frosolono M, Handel F, Cato AE. Prostacyclin-induced acute pulmonary vasodilation in primary pulmonary hypertension. Circulation. 1982;66(2):334–338. | ||
Hoshikawa Y, Voelkel NF, Gesell TL, et al. Prostacyclin receptor-dependent modulation of pulmonary vascular remodeling. Am J Respir Crit Care Med. 2001;164(2):314–318. | ||
Nagaya N, Yokoyama C, Kyotani S, et al. Gene transfer of human prostacyclin synthase ameliorates monocrotaline-induced pulmonary hypertension in rats. Circulation. 2000;102(16):2005–2010. | ||
Christman BW, McPherson CD, Newman JH, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(2):70–75. | ||
Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159(6):1925–1932. | ||
Falcetti E, Hall SM, Phillips PG, et al. Smooth muscle proliferation and role of the prostacyclin (IP) receptor in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182(9):1161–1170. | ||
Stearman RS, Cornelius AR, Lu X, et al. Functional prostacyclin synthase promoter polymorphisms. Impact in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;189(9):1110–1120. | ||
Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334(5):296–301. | ||
Galie N, Humbert M, Vachiery JL, et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2002;39(9):1496–1502. | ||
Barst RJ, McGoon M, McLaughlin V, et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2003;41(12):2119–2125. | ||
Demolis JL, Robert A, Mouren M, Funck-Brentano C, Jaillon P. Pharmacokinetics and platelet antiaggregating effects of beraprost, an oral stable prostacyclin analogue, in healthy volunteers. J Cardiovasc Pharmacol. 1993;22(5):711–716. | ||
Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest. 2012;142(6):1383–1390. | ||
Tapson VF, Jing ZC, Xu KF, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial. Chest. 2013;144(3):952–958. | ||
Jing ZC, Parikh K, Pulido T, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation. 2013;127(5):624–633. | ||
White RJ, Allen R, Torres F. Sustained plasma concentrations of treprostinil following chronic dosing of oral treprostinil in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2009; 179:A3360. | ||
Wade M, Baker FJ, Roscigno R, DellaMaestra W, Hunt TL, Lai AA. Absolute bioavailability and pharmacokinetics of treprostinil sodium administered by acute subcutaneous infusion. J Clin Pharmacol. 2004;44(1):83–88. | ||
Orinetram (Oral Treprostinil) Prescribing Information. Available from: https://www.orenitram.com/pdf/Orenitram_Full_Prescribing_Information.pdf. Accessed July 12, 2016. | ||
Beraprost 314d trial (BEAT). Available from: https://clinicaltrials.gov/ct2/show/NCT01908699. Accessed July 26, 2016. | ||
Freedom Ev Trial (Oral Treprostinil). Available from: https://clinicaltrials.gov/ct2/show/NCT01560624. Accessed July 29, 2016. | ||
Kaufmann P, Okubo K, Bruderer S, et al Pharmacokinetics and tolerability of the novel oral prostacyclin IP receptor agonist selexipag. Am J Cardiovasc Drugs. 2015;15(3):195–203. | ||
U.S Department of Health and Human Services, Food and Drug Administration. US Prescribing Information: Uptravi (Selexipag) Tables, for Oral Use; 2015;2016. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207947s000lbl.pdf. Accessed September 25, 2016. | ||
Kuwano K, Hashino A, Noda K, Kosugi K, Kuwabara K. A long-acting and highly selective prostacyclin receptor agonist prodrug, 2-{4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl)acetam ide (NS-304), ameliorates rat pulmonary hypertension with unique relaxant responses of its active form, {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}acetic acid (MRE-269), on rat pulmonary artery. J Pharmacol Exp Ther. 2008;326(3):691–699. | ||
Morrison K, Studer R, Ernst R, Haag F, Kauser K, Clozel M. Differential effects of selexipag [corrected] and prostacyclin analogs in rat pulmonary artery. J Pharmacol Exp Ther. 2012;343(3):547–555. | ||
Oka M, Fagan KA, Jones PL, McMurtry IF. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br J Pharmacol. 2008;155(4):444–454. | ||
Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40(4):874–880. | ||
Sitbon O, Channick R, Chin KM, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373(26):2522–2533. | ||
Barst RJ, Oudiz RJ, Beardsworth A, et al. Tadalafil monotherapy and as add-on to background bosentan in patients with pulmonary arterial hypertension. J Heart Lung Transplant. 2011;30(6):632–643. | ||
Galie N, Humbert M, Vachiery JL, et al. ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;46(4):903–975. | ||
Bruderer S, Hurst N, Kaufmann P, Dingemanse J. Multiple-dose up-titration study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of selexipag, an orally available selective prostacyclin receptor agonist, in healthy subjects. Pharmacology. 2014;94(3–4):148–156. | ||
Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med. 2010;104(1):9–21. | ||
Kaufmann P, Cruz HG, Krause A, Ulc I, Halabi A, Dingemanse J. Pharmacokinetics of the novel oral prostacyclin receptor agonist selexipag in subjects with hepatic or renal impairment. Br J Clin Pharmacol. 2016;82(2):369–379. | ||
Clapp LH, Gurung R. The mechanistic basis of prostacyclin and its stable analogues in pulmonary arterial hypertension: role of membrane versus nuclear receptors. Prostaglandins Other Lipid Mediat. 2015;120:56–71. | ||
Asaki T, Kuwano K, Morrison K, Gatfield J, Hamamoto T, Clozel M. Selexipag: an oral and selective IP prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. J Med Chem. 2015;58(18):7128–7137. | ||
Lu A, Zuo C, He Y, et al. EP3 receptor deficiency attenuates pulmonary hypertension through suppression of Rho/TGF-beta1 signaling. J Clin Invest. 2015;125(3):1228–1242. | ||
Qian YM, Jones RL, Chan KM, Stock AI, Ho JK. Potent contractile actions of prostanoid EP3-receptor agonists on human isolated pulmonary artery. Br J Pharmacol. 1994;113(2):369–374. | ||
Walch L, de Montpreville V, Brink C, Norel X. Prostanoid EP(1)- and TP-receptors involved in the contraction of human pulmonary veins. Br J Pharmacol. 2001;134(8):1671–1678. | ||
Whittle BJ, Silverstein AM, Mottola DM, Clapp LH. Binding and activity of the prostacyclin receptor (IP) agonists, treprostinil and iloprost, at human prostanoid receptors: treprostinil is a potent DP1 and EP2 agonist. Biochem Pharmacol. 2012;84(1):68–75. | ||
Morrison K, Ernst R, Hess P, Studer R, Clozel M. Selexipag: a selective prostacyclin receptor agonist that does not affect rat gastric function. J Pharmacol Exp Ther. 2010;335(1):249–255. | ||
Benyahia C, Boukais K, Gomez I, et al. A comparative study of PGI2 mimetics used clinically on the vasorelaxation of human pulmonary arteries and veins, role of the DP-receptor. Prostaglandins Other Lipid Mediat. 2013;107:48–55. | ||
Knebel SM, Sprague RS, Stephenson AH. Prostacyclin receptor expression on platelets of humans with type 2 diabetes is inversely correlated with hemoglobin A1c levels. Prostaglandins Other Lipid Mediat. 2015;116–117:131–135. | ||
Smyth EM, Austin SC, Reilly MP, FitzGerald GA. Internalization and sequestration of the human prostacyclin receptor. J Biol Chem. 2000;275(41):32037–32045. | ||
Kuwano K, Hashino A, Asaki T, et al. 2-[4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy]-N-(methylsulfonyl)acetam ide (NS-304), an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther. 2007;322(3):1181–1188. | ||
Morrison K, Wanner D, Gatfield J, Gnerre C, Hess P, Clozel M. Repeated oral administration of the selective prostacyclin receptor agonist selexipag does not cause tachyphylaxis in spontaneously hypertensive rats [Abstract]. J Am Coll Cardiol. 2015;65(10S):A1558. | ||
Gatfield J, Menyhart K, Morrison K, Nayler O. The non-prostanoid prostacyclin receptor agonist ACT-333679, the active metabolite of selexipag, is characterized by low beta-arrestin recruitment and receptor internalization activity [abstract]. J Am Coll Cardiol. 2015;65(10S):A1542. | ||
McLaughlin VV, Genthner DE, Panella MM, Rich S. Reduction in pulmonary vascular resistance with long-term epoprostenol (prostacyclin) therapy in primary pulmonary hypertension. N Engl J Med. 1998;338(5):273–277. | ||
Galie N, Barbera JA, Frost AE, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373(9):834–844. | ||
Galie N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D60–D72. | ||
Sitbon O, Jais X, Savale L, et al. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Respir J. 2014;43(6):1691–1697. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.