")
Back to Journals » Journal of Inflammation Research » Volume 15
Secretory Autophagosomes from Alveolar Macrophages Exacerbate Acute Respiratory Distress Syndrome by Releasing IL-1β
Authors Xu X, Liu X, Dong X, Qiu H, Yang Y, Liu L
Received 22 October 2021
Accepted for publication 24 December 2021
Published 7 January 2022 Volume 2022:15 Pages 127—140
DOI https://doi.org/10.2147/JIR.S344857
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Xinyi Xu, Xu Liu, Xuecheng Dong, Haibo Qiu, Yi Yang, Ling Liu
Jiangsu Provincial Key Laboratory of Critical Care Medicine, Department of Critical Care Medicine, Zhongda Hospital, School of Medicine, Southeast University, Nanjing, 210009, People’s Republic of China
Correspondence: Yi Yang; Ling Liu
Jiangsu Provincial Key Laboratory of Critical Care Medicine, Department of Critical Care Medicine, Zhongda Hospital, School of Medicine, Southeast University, Nanjing, 210009, People’s Republic of China
Email [email protected]; [email protected]
Purpose: Activated alveolar macrophages (AMs) secrete extracellular vesicles and particles to mediate the inflammatory response in the acute respiratory distress syndrome (ARDS) although the underlying mechanisms are poorly understood. This study investigated whether secretory autophagosomes (SAPs) from AMs contribute to the inflammation-mediated lung injury of ARDS.
Methods: We first isolated SAPs from cell culture supernatants of RAW264.7 cells and AMs and quantified Interleukin (IL)-1β levels in SAPs. Next, we employed a lipopolysaccharide (LPS)-induced ARDS model to investigate whether SAP-derived IL-1β could exacerbate lung injury. Finally, we used siRNA to knockdown Rab8a, both in vitro and in vivo, to investigate the effect of Rab8a on SAP secretion and lung injury in ARDS.
Results: We found that AMs play an important role in ARDS by releasing a novel type of proinflammatory vesicles called SAPs that could exacerbate lung injury. SAPs are characterized as double-membrane vesicles (diameter ∼ 200 nm) with the expression of light chain 3 (LC3). IL-1β in SAPs is the key factor that contributes to the inflammation and lung injury in ARDS. We found that Rab8a is necessary for AMs to release SAPs with IL-1β, and Rab8a knockdown alleviated lung injury in ARDS.
Conclusion: This study showed the novel finding that SAPs released from AMs play a vital role in ARDS by promoting an inflammatory response and the underlying mechanism was associated with IL-1β secretion.
Keywords: secretory autophagosomes, alveolar macrophages, interlukin-1β, Rab8a, acute respiratory distress syndrome
Introduction
Acute respiratory distress syndrome (ARDS) occurs frequently in critically ill patients treated in the ICU with morbidity and mortality rates of ARDS 10.4% and more than 40%, respectively.1 The pathogenesis of ARDS is mediated by the excessive inflammatory response of immune cells to various pulmonary or extrapulmonary factors.2 Despite the high mortality rate, no effective treatment for the underlying pathogenesis of ARDS has been developed. Alveolar macrophages (AMs) constitute approximately 50% of the immune cells in the lungs and, through the synthesis and release of various inflammatory mediators, they play a pivotal role in the pathogenesis of ARDS.3–5 Emerging evidence indicates that AMs autophagy could impact the pathogenesis or progression of ARDS. However, the role of macrophagic autophagy in ARDS appears to be complex, as it can be either cytoprotective or deleterious.6,7
Autophagy is an evolutionarily conserved ubiquitous process for degrading and clearing subcellular components.8,9 In contrast to degradative autophagy, secretory autophagy can bypass lysosome fusion and export various cytoplasmic substrates.10–12 Recently, Bel et al reported that lysozyme-containing autophagosomes from Paneth cells secrete chemokines into the intestinal lumen to participate in the pathogenesis of Crohn’s disease.13 The autophagosomes released from cells are called secretory autophagosomes (SAPs) and are characterized as double-membrane vesicles (diameter ~200 nm) with the expression of light chain 3 (LC3). More recently, yeast, tumor cells, and endothelial cells were shown to release SAPs to amplify pathological signals and to transmit intercellular signals.14–16 Thus, SAPs may participate in the regulation of the inflammatory response. Nonetheless, it is unknown whether AMs can release SAPs to modulate the inflammatory response in ARDS.
Interleukin (IL)-1β is a well-known proinflammatory cytokine that is secreted by activated macrophages. In addition to conventional protein secretion by the endoplasmic reticulum (ER) and the Golgi apparatus, IL-1β can be secreted through several unconventional pathways. In recent years, several studies have shown that IL-1β secretion can be regulated via autophagy.17,18 However, there is no evidence that IL-1β can be transported directly via intact SAPs.
This study aimed to provide insight into the inflammatory regulation in ARDS and to identify potential drug targets for the treatment of ARDS. Therefore, we evaluated the role of AM-released SAPs in promoting the inflammatory response in ARDS and investigated whether the mechanism underlying this effect is associated with IL-1β secretion.
Materials and Methods
Isolation and Identification of Mouse AMs
Male wild-type (WT) C57BL/6 mice aged 8–10 weeks were used in all experiments. AMs were harvested from the bronchoalveolar lavage fluid (BALF) of mice. Briefly, the lungs of the mice were perfused with 1 mL phosphate-buffered saline (PBS; 10 times, 0.1 mL/perfusion/min) and a 20-gauge endotracheal catheter was used to collect BALF. Each BALF sample was centrifuged at 300 g for 5 min, and the supernatant was discarded. The pellet was re-suspended in the RPMI 1640 medium (RPMI 1640; Wisent Biotechnology, Nanjing, China) to obtain a cell concentration of 2×106 cells/mL. After 2 h of incubation at 37°C and with 5% CO2, unattached cells were removed, and the adherent cells were cultured in RPMI 1640 containing 10% fetal bovine serum (FBS; Coring, Australia), 100 IU/mL penicillin, and 100 µg/mL streptomycin. Flow cytometry for F4/80, CD11c, and Siglec F was used to identify the phenotype of AMs.
Cell Culture
The murine-derived macrophage cell line RAW264.7 was purchased from the Cells Resource Center of Shanghai Institutes for Biological Sciences, The Chinese Academy of Science. The cells were cultured at 37°C in Dulbecco’s modified Eagle’s medium (DMEM; Wisent Biotechnology, China) containing 10% FBS, 100 IU/mL penicillin, and 100 µg/mL streptomycin in a humidified atmosphere with 5% CO2. When the cells reached 80% confluence, the adherent cells were trypsinized using 0.25% trypsin-EDTA (Invitrogen, American) and underwent passage in new flasks for further expansion. The cells were untreated or treated with 100 ng/mL lipopolysaccharide (LPS) for 12, 24, 48, or 72 h to determine the effect of LPS-stimulated RAW264.7 activation. Mouse AMs were cultured at 37°C in RPMI 1640 containing 10% FBS, 100 IU/mL penicillin, and 100 µg/mL streptomycin in a humidified atmosphere with 5% CO2.
Cell Viability Assay
The cytotoxic effect of LPS (MCE, USA) on RAW264.7 cells was evaluated via trypan blue exclusion assay. The cells were seeded in 24-well plates (5 × 104/well), incubated at 37°C overnight, and then treated with 0, 10, 100, or 1000 ng/mL LPS. Cell viability was assessed after 24 h with trypan blue exclusion to determine the viable cell percentage.
Animal Procedures
Briefly, male C57BL/6J mice (n = 5–7 per group) were randomly administered intratracheal injections of LPS (5 mg/kg; MCE, USA), SAPs from LPS-stimulated RAW264.7 cells (109 particles/mouse), or recombinant IL-1β (250 U/mouse; BD Pharmingen, USA) with or without pretreatment of IL-1RA (25 mg/kg, 24 h before injection of other drugs; MCE, USA). PBS (Biological Industries, Israel) was used as control and as the vehicle of LPS, SAPs, recombinant IL-1β, and IL-1RA. After 24 h, lung and BALF samples were collected. The left lung of each mouse was processed for hematoxylin and eosin staining. The levels of IL-6, IL-10, IL-12, and tumor necrosis factor-alpha (TNF-α) in lung tissue homogenates were quantified using an ELISA kit (Ela Science, China).
Isolation and Purification of SAPs
We isolated and purified SAPs according to the previously reported methods for isolating tumor cell-released autophagosomes (TRAPs).19 The culture supernatants and BALF samples were centrifuged at 400 g for 5 min to remove whole cells and debris. The supernatants were centrifuged at 12,000 g for 30 min. Subsequently, the SAPs-containing pellets were collected, washed thrice with PBS, and isolated with magnetic beads (Miltenyi Biotec) combined with LC3B antibody (Cell Signaling Technology) for SAPs. Finally, the pellets were resuspended in PBS and stored at −80°C or used for nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM), or Western blotting (WB) analysis.
Nanoparticle Tracking Analysis (NTA)
We measured the size and concentration of the collected vesicles via NTA by using ZetaView PMX 110 (Particle Metrix, Meerbusch, Germany) and the corresponding software ZetaView 8.04.02. Isolated vesicle samples were appropriately diluted using 1 × PBS to measure the particle size and concentration. NTA measurements were recorded and analyzed at 11 positions. The ZetaView system was calibrated using 110 nm polystyrene particles. The temperature was maintained between 23°C and 30°C.
Transmission Electron Microscopy (TEM)
An isolated SAPs sample (5 µL) was deposited on a formvar-carbon grid (01753-F, PELCO) and incubated at room temperature for 5 min. The excess liquid was removed using a small piece of Whatman filter paper (WHA10010155, Sigma-Aldrich). Then, 5 µL of 1% uranyl acetate was applied to the sample for 5–10 s. Subsequently, three drops of water were placed on parafilm of the sample to wash the grid three times. After the grid was dry, the sample was observed using a transmission electron microscope at 80 kV (1230, JEOL).
Western Blot Analysis
The total proteins from macrophages and SAPs were extracted using the RIPA lysis buffer. The protein concentrations of the cell lysates were measured using the BCA protein assay (Beyotime, China). Samples containing equal amounts of proteins were resolved via 10% or 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane (Bio-Rad, American). Subsequently, the membrane was blocked with 5% BSA and incubated overnight at 4°C with specific primary antibodies. We used antibodies against LC3B, Beclin-1, Rab8a, Apaf-1, CD40L, and β-actin (1:1000 for all; #43566, #3495, #6975, #8723, #15094, and #4970, respectively, Cell Signaling Technology), and CD63 (1:1000, 25682–1-AP, Proteintech). Following incubation with the primary antibody, the membrane was washed and incubated with a horseradish peroxidase-conjugated secondary anti-rabbit antibody (Beyotime, China, 1:5000) for 1 h at 4°C and visualized using electrochemiluminescence (ECL) detection kits (Beyotime, China). Each experiment was repeated at least three times, and the gray value of each band was measured by ImageJ (version 1.8.0, ImageJ Software, USA) for quantification and further analysis.
ELISA
IL-1β, IL-6, IL-10, IL-12, and TNF-α were quantified via ELISA in accordance with the manufacturer’s instructions (ELISA kits for mouse IL-1β, E-EL-M0037c; mouse IL-6, E-EL-M0044c; mouse IL-10, E-EL-M0046c; mouse IL-12, E-EL-M0726c; mouse TNF-α, E-EL-M0049c; all from Ela Science, China). The OD450 values of the samples were obtained using a microplate reader. Each sample was analyzed in duplicate. The intra- and inter-assay coefficients of variation were both <20%.
Flow Cytometry
The AMs collected from the BALF samples and the isolated EVs were characterized via flow cytometry. The Fc receptor-blocking agent (Miltenyi Biotech, Germany) was used to block the Fc receptors for 5 min at 4°C. Next, AMs and SAPs were incubated with the antibodies in the dark at 4°C for 30 min and then washed with PBS. The antibodies used were anti-F4/80 phycoerythrin (PE), anti-CD11c allophycocyanin (APC), anti-Siglec F fluorescein isothiocyanate (FITC), and anti-LC3 allophycocyanin (APC) (all from BD Biosciences, USA). All data were collected using a flow cytometer and Novo Express (ACEA NovoCyte, China) and then analyzed using FlowJo, version X (Tree Star, USA).
In-vitro and in-vivo RNAi of Rab8a
The siRNAs used for in vitro and in vivo RNAi of Rab8a were purchased from RiboBio Co., Ltd. (Guangzhou, People’s Republic of China). RAW264.7 cells were transfected with the siRNA using the riboFECT CP Transfection Kit (C10511-05) according to the manufacturer’s instructions. The efficiency of transfection was evaluated via WB analysis. We designed three different siRNAs for Rab8a knockdown: RNAi-1(5′-CUGCAGACGUGGAGAAGAUTT-3′), RNAi-2(5′-GAAUAAGUGUGAUGUGAAUTT-3′), and RNAi-3(5′-GUGCAAAGGCCAACAUCAATT-3′). After in vitro transfection with the three siRNAs, we chose RNAi-1, which had the best efficacy, for further in vitro and in vivo experiments. The multiplicity of infection of the transfection was 60. The in vivo Rab8a-siRNA was injected into the caudal vein of mice.
Statistical Analysis
All experiments were repeated using different animals and cells in at least triplicates. Data were analyzed using the GraphPad Prism 8.0 (GraphPad Software, San Diego, USA). The descriptive statistics are presented as the mean and SD for continuous variables. The data shown in the graphs are presented as the mean ± SD. All data were tested for normal distribution using the Kolmogorov–Smirnov test. The Student’s t-test or one-way ANOVA was used to compare the statistical significance of the differences. P < 0.05 was considered indicative of statistical significance.
Results
LPS-Induced Inflammatory Reaction and Autophagy in Macrophages
To determine the effect of LPS on RAW264.7 cells, we measured cell viability 24 h after LPS stimulation. Compared to the control group, 100 ng/mL LPS significantly decreased RAW264.7 cell viability (Figure 1A). To examine the effect of LPS on RAW264.7 cell activation, we stimulated the cells with 100 ng/mL LPS for 12, 24, 48, or 72 h. Compared with the control levels, LPS increased the levels of the proinflammatory cytokine IL-1β in cell culture supernatants, and the concentration of cytokines increased over time (Figure 1B). In addition, LC3 II and beclin-1 were used as markers of autophagy, and results of WB indicated that the autophagy in RAW264.7 cells was enhanced by LPS administration (Figure 1C). We also cultured primary mouse AMs and confirmed via flow cytometry that they expressed F4/80, CD11c, and Siglec F (Figure 1D). The LC3 II and beclin-1 levels in LPS-stimulated AMs increased in tandem with those in LPS-stimulated RAW264.7 cells (Figure 1E). Therefore, 24-h 100 ng/mL LPS stimulation was considered appropriate to induce inflammation and autophagy in macrophages.
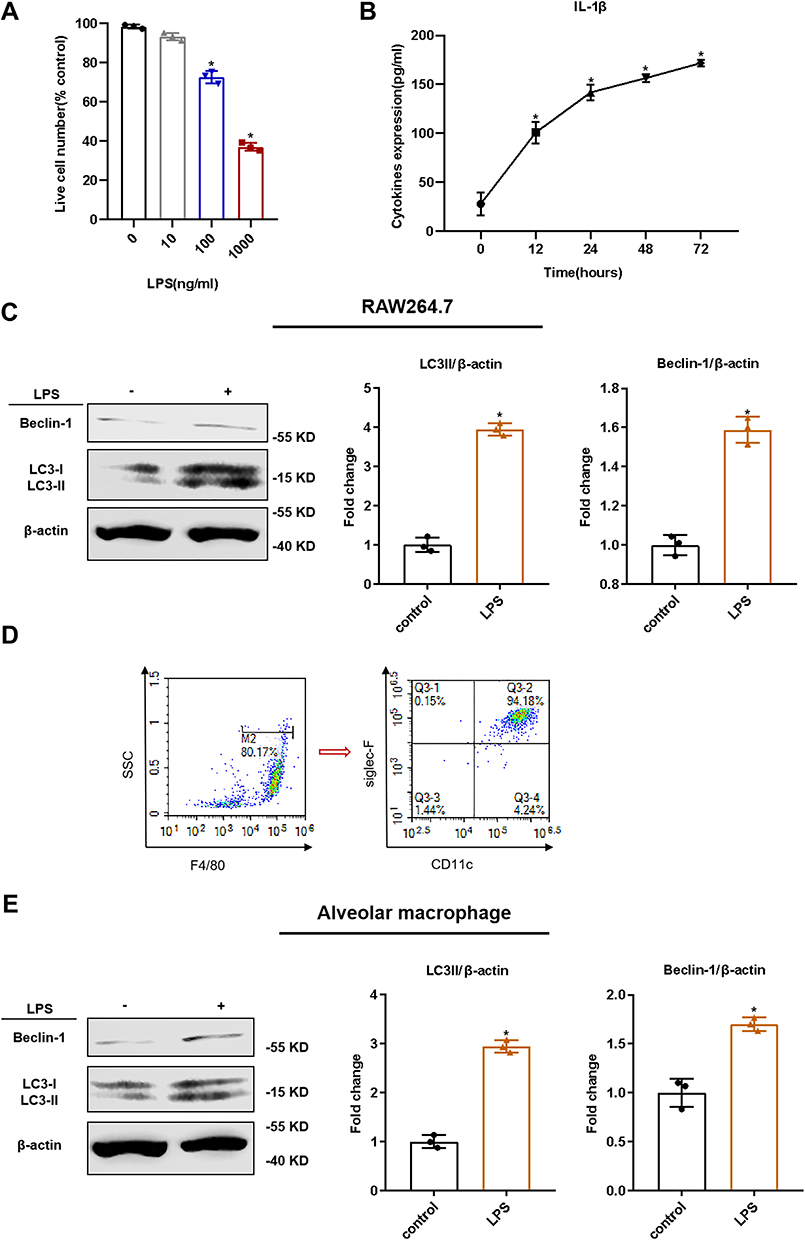 |
Figure 1 LPS-induced inflammatory reaction and autophagy in macrophages. RAW264.7 cells were stimulated with or without LPS for 12, 24, 48, or 72 h. (A) The effects of various concentrations of LPS on the viability of RAW264.7 cells upon 24 h of treatment, as tested via trypan blue exclusion assay; (B) The effect of LPS (100 ng/mL) on the level of interleukin (IL)-1β in cell supernatants, as tested via ELISA; (C) Identification of mouse alveolar macrophages via flow cytometry. Positive for F4/80, CD11c, and Siglec F; (D) The effect of LPS on the autophagy in RAW264.7 cells was tested via Western blotting (WB); (E) The effect of LPS on the autophagy in alveolar macrophages was tested via WB. The experiments were repeated at least three times. Each value represents the mean SD of three independent experiments (n = 3). *p < 0.05 vs the control, analyzed via the t-test. |
SAPs Released by LPS-Stimulated Macrophages
SAPs were isolated from the culture supernatant of RAW264.7 cells. NTA showed that the size of the vesicles was consistent with that of autophagosomes (~200 nm; Figure 2A). We validated the double-membrane configuration of the isolated vesicles with TEM (Figure 2B). The autophagosome membrane marker LC3-II was detected by WB (Figure 2C) and flow cytometric assays (Figure S1). Moreover, compared with apoptotic bodies, microvesicles, and exosomes from RAW264.7 cells, no expression of Apaf-1 (a marker of apoptotic bodies), CD40L (a marker of microvesicles), or CD63 (a marker of exosomes) was detected in SAP vesicles (Figure S2). After LPS stimulation, the number of vesicles in the culture supernatants of both RAW264.7 cells and AMs increased significantly, as shown by NTA (Figure 2D). WB showed that LC3-II was more abundant in the cell supernatant of LPS-stimulated RAW264.7 cells and AMs, because equivalent amounts of proteins were present between the lanes (Figures 2E and S3). Therefore, the secretion of SAPs from macrophages could be induced by LPS administration.
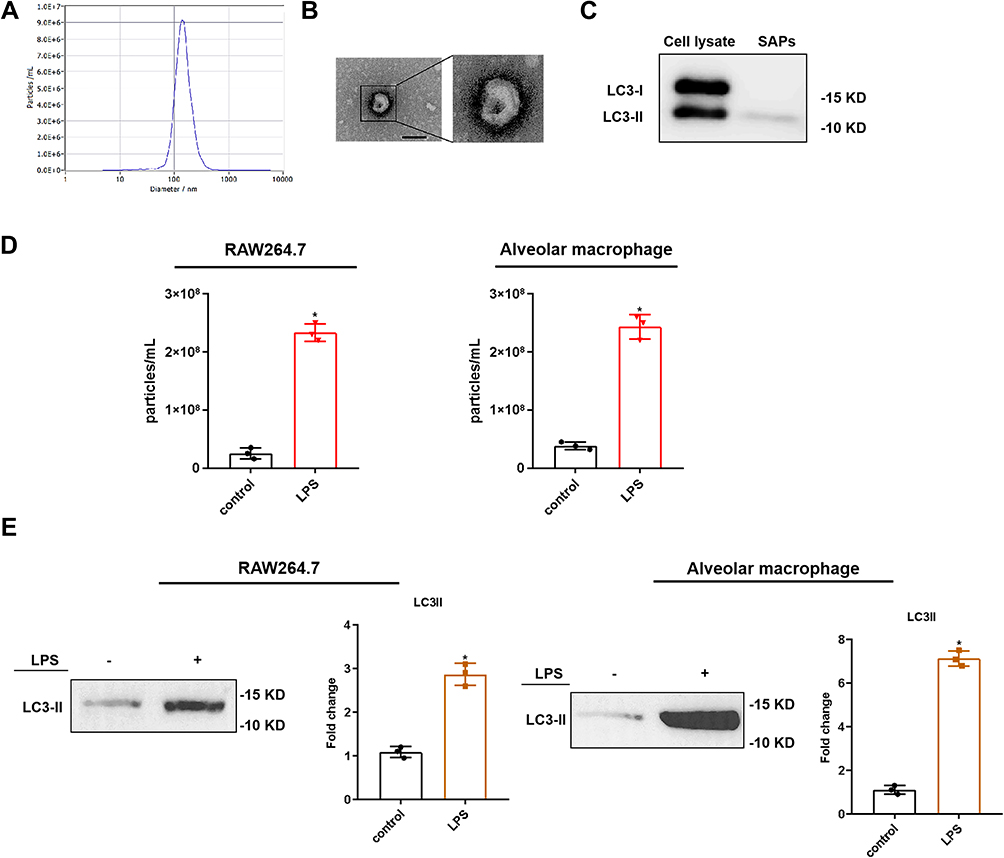 |
Figure 2 SAPs released by LPS-stimulated macrophages. SAPs were isolated from the culture supernatant of RAW264.7 cells via centrifugation. (A) The size and concentration of the vesicles from RAW264.7 cells were assessed via NTA; (B) The vesicles from RAW264.7 cells were identified via transmission electron microscopy (Scale bar = 200 nm); (C) Western blot analysis revealed that the vesicles from the RAW264.7 cells expressed LC3-II; (D) Results from nanoparticle tracking analysis demonstrated that there were significant increases in the number of vesicles secreted by RAW264.7 cells and alveolar macrophages upon LPS stimulation, compared with the number in the control; (E) Western blot analysis revealed that there were significant increases in the levels of LC3-II in vesicles from RAW264.7 cells and alveolar macrophages upon LPS stimulation, compared with the number in the control. The experiments were repeated at least three times. Each value represents the mean±SD of three independent experiments (n = 3).*p < 0.05 vs the control, analyzed via the t-test. |
SAPs from LPS-Stimulated Macrophages Exacerbated Lung Injury in ARDS Mice
Mice were administered SAPs from LPS-induced RAW264.7 cells through intratracheal injection. Compared to those in the control group, the application of SAPs clearly exacerbated the pathological changes in the lung tissues and the number of infiltrated inflammatory cells increased markedly (Figure 3A and B). SAPs administration could induce an ARDS lung injury akin to that obtained by LPS stimulation, and the administration of both SAPs and LPS could induce more severe lung injury compared to LPS alone. ELISA results demonstrated that the levels of proinflammatory cytokines (IL-1β, IL-6, and TNF-α) increased significantly after SAPs administration, whereas those of anti-inflammatory cytokines (IL-10 and IL-12) decreased markedly (Figure 3C). Furthermore, we observed a significant decrease in the survival rate of mice after the injection of both SAPs and LPS compared with LPS injection alone (Figure 3D). We thus concluded that the injection of SAPs from LPS-stimulated macrophages exacerbated LPS-induced ARDS in mice.
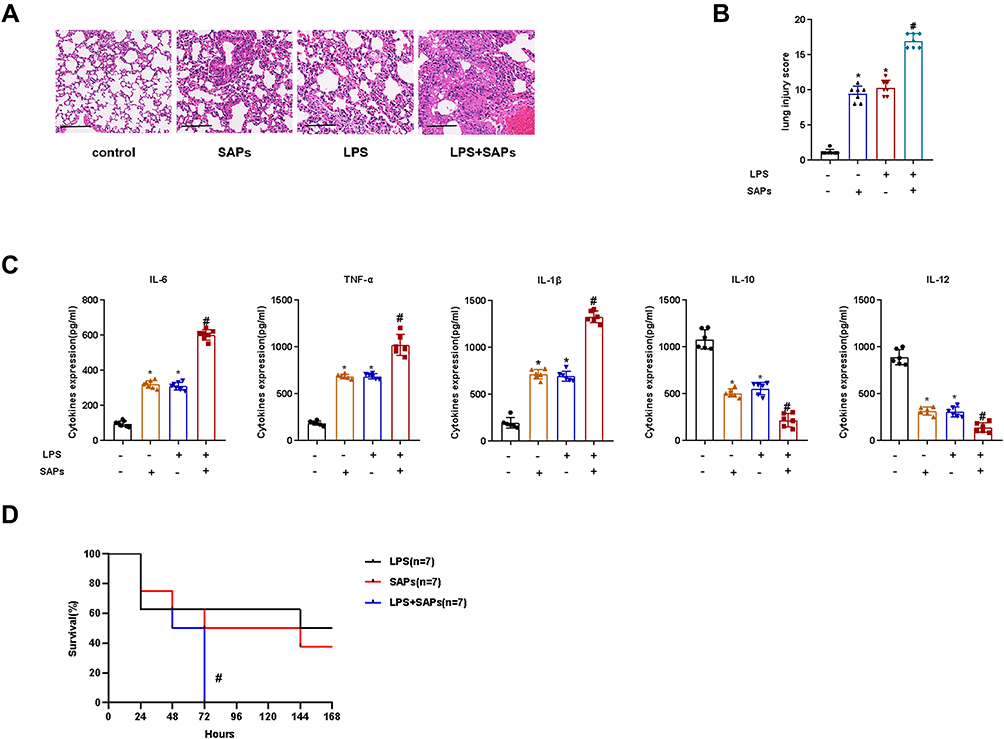 |
Figure 3 SAPs from LPS-stimulated macrophages exacerbated lung injury in ARDS mice. SAPs from RAW264.7 cells were administered to mice through intratracheal injection. (A) Histopathological images of the lung samples (haematoxylin-and-eosin staining, Scale bar = 200 µm); (B) The injury scores of the lung samples (n = 7 per group); (C) The effects of SAPs from RAW264.7 cells on the inflammatory cytokine levels in the lungs were assessed using ELISA (n = 6 per group); (D) The effect of SAPs from RAW264.7 cells on the survival rate of mice (n = 7 per group). The experiments were repeated at least three times. Each value represents the mean±SD of three independent experiments.*p < 0.01 vs the control; #p < 0.05 vs the LPS group, analyzed via the t-test. |
IL-1β from SAPs Secreted by LPS-Stimulated Macrophages Caused Lung Injury in Mice
To investigate the mechanism of lung injury caused by SAPs, we used ELISA to test the levels of proinflammatory cytokines (IL-1β) in SAPs after the SAP membrane was broken by Triton X-100. The results revealed considerably more IL-1β in SAPs than in the RAW264.7 cell supernatant (Figure 4A). Additionally, we found that the levels of IL-1β in the cell supernatant increased evidently after the SAPs membrane was broken (Figure 4B). To validate that IL-1β could be secreted by SAPs, we isolated SAPs from cell supernatants at 0, 1, 2, 3, 5, 7, and 14 days after cell supernatant collection and then measured the IL-1β levels in SAPs and supernatants. The results indicated that the levels of IL-1β in SAPs decreased, while those in the cell supernatant increased over time (Figure 4C). As we had already demonstrated that SAPs from macrophages could cause lung injury in mice, we used interleukin 1 receptor antagonist (IL-1RA) to treat mice before SAPs injection to verify that IL-1β in SAPs was the key factor in inducing lung injury. We found that IL-1RA pretreatment significantly ameliorated the pathological changes in the lung tissues (Figure 4D and E), and the survival rate of mice increased markedly with IL-1RA administration (Figure 4F). Therefore, IL-1β secreted by SAPs from macrophages was likely the key molecule that caused lung injury in mice.
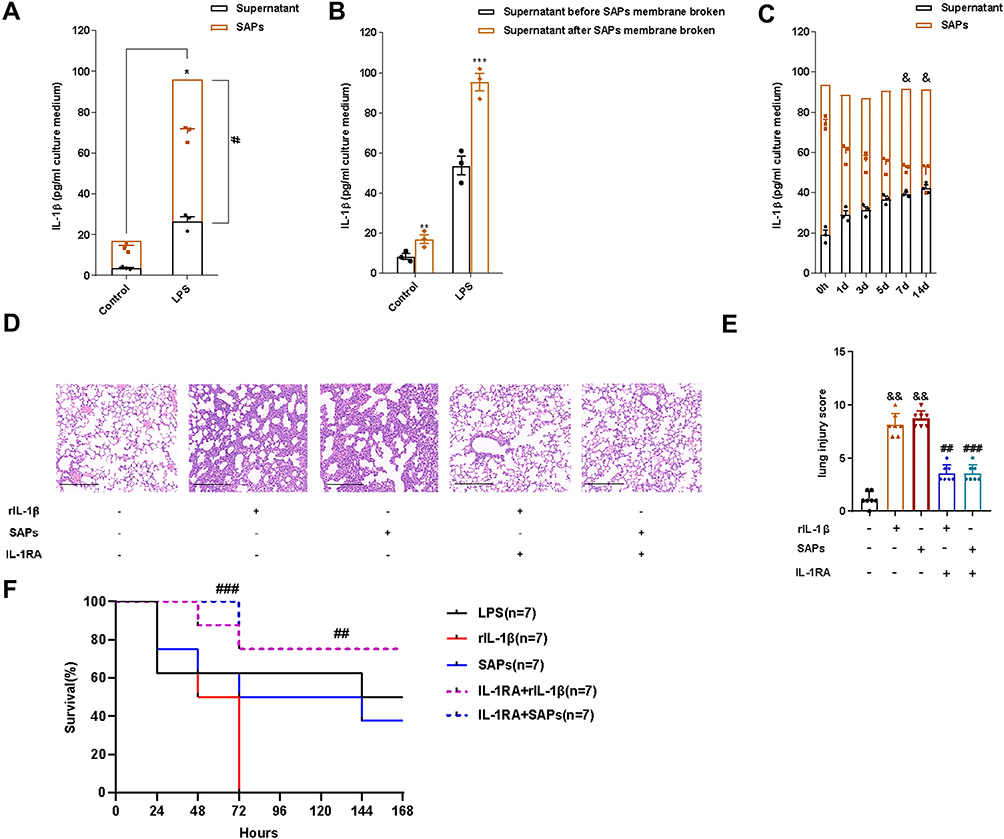 |
Figure 4 IL-1β from SAPs secreted by LPS-stimulated macrophages caused lung injury in mice. (A) The levels of IL-1β in SAPs and cell-culture supernatant were assessed via ELISA after the SAPs were lysed using Triton X-100 (n = 3 per group); (B) The level of IL-1β in the culture supernatant of RAW264.7 cells was assessed via ELISA before and after the SAPs were lysed using Triton X-100 (n = 3 per group); (C) SAPs were isolated from the cell-culture supernatant 1, 2, 3, 5, 7, or 14 d after collecting the supernatant. The levels of IL-1β in SAPs and supernatant were assessed via ELISA after the SAPs were lysed using Triton X-100 (n = 3 per group); (D) Histopathological images of the lungs from mice administered with SAPs or rIL-1β with or without pre-treatment with IL-1RA (haematoxylin-and-eosin staining, Scale bar = 200 µm); (E) The injury scores of the lung samples (n = 7 per group); (F) The effects of IL-1RA and SAPs from RAW264.7 cells on the survival rate of mice (n = 7 per group). The experiments were repeated at least three times. Each value represents the mean±SD of three independent experiments. *p < 0.05 vs the SAPs + supernant in the control group; #p < 0.05 vs the supernatant in the LPS group; ***p < 0.05 vs the supernatant in the LPS group before SAPs membrane broken; &p < 0.05 vs the SAPs at day 0; ##p < 0.05 vs the rIL-1β group; ###p < 0.05 vs the SAPs group; &&p < 0.05 vs the control group, analyzed via the t-test. |
Rab8a Expression in SAPs Secreted by LPS-Stimulated Macrophages
As SAPs could induce lung injury, we sought to inhibit their secretion from activated macrophages. Rab8a is essential for Paneth cells to secrete SAPs;13 therefore, we used a WB assay to ascertain the presence of Rab8a on macrophage-derived SAPs, and detected Rab8a expression on SAPs from both LPS-stimulated RAW264.7 cells and AMs (Figure 5A and B). Therefore, Rab8a may be crucial for macrophages to secrete SAPs.
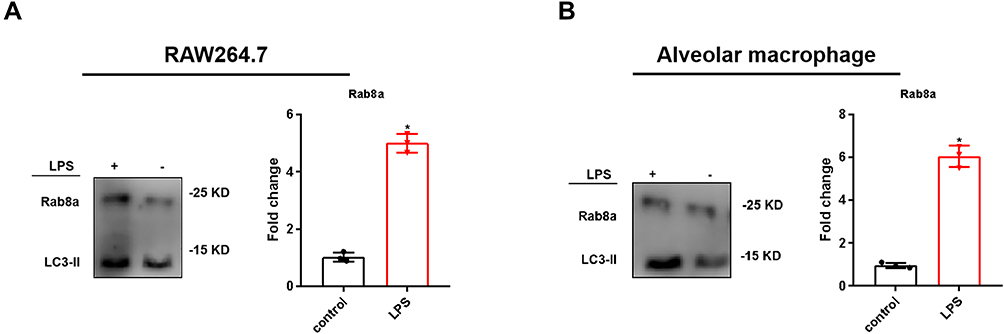 |
Figure 5 Expression of Rab8a in SAPs secreted by LPS-stimulated macrophages. (A and B) Western blot analysis revealed that there were significant increases in the levels of Rab8a in the vesicles from RAW264.7 cells and alveolar macrophages in the LPS-stimulated groups, compared with those in the controls (n = 3 per group). The experiments were repeated at least three times. Each value represents the mean±SD of three independent experiments.*p < 0.05 vs the control, analyzed via the t-test. |
Rab8a-Modulated SAP Secretion in LPS-Induced Macrophages
To evaluate the effect of Rab8a on SAP secretion by macrophages, we knocked down Rab8a in RAW264.7 cells and verified the efficiency of Rab8a knockdown using WB analysis (Figure 6A and B), which showed a notable decrease in the number of vesicles in the cell supernatant measured by NTA (Figure 6C). The WB results demonstrated that LC3-II was less abundant in the culture supernatant of Rab8a-knockdown RAW264.7 cells (Figure 6D). Additionally, we found that the IL-1β levels in SAPs clearly decreased after Rab8a knockdown (Figure 6E). Therefore, the secretion of IL-1β by SAPs from macrophages could be inhibited by Rab8a knockdown, and Rab8a was essential for macrophages to secrete SAPs.
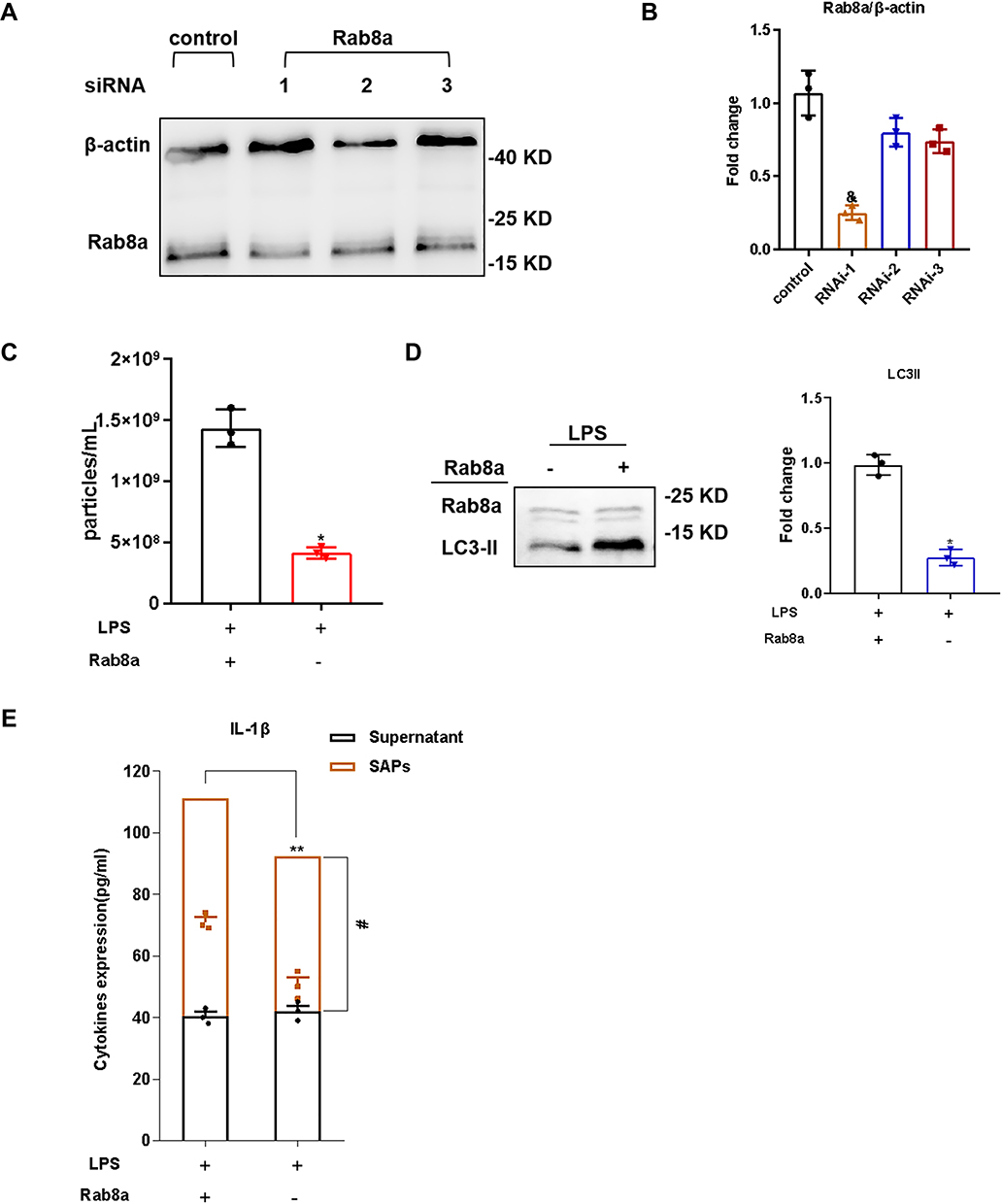 |
Figure 6 Rab8a modulated SAPs secretion in LPS-induced macrophages. (A and B) Western blot analysis illustrated efficient siRNA-mediated knockdown of Rab8a in RAW264.7 cells in comparison with the control; (C) Western blot analysis illustrated efficient siRNA-mediated knockdown of Rab8a in RAW264.7 cells in comparison with the control; (D) Western blot analysis demonstrated that there were significant decreases in the levels of LC3-II in vesicles from LPS-stimulated RAW264.7 cells in the Rab8a knockdown groups compared with those in the control groups; (E) The levels of IL-1β in SAPs and cell-culture supernatant were assessed via ELISA after the SAPs were lysed using Triton X-100. The experiments were repeated at least three times. Each value represents the mean±SD of three independent experiments (n = 3). &p < 0.05 vs the control group; *p < 0.05 vs the LPS+Rab8a+ group; **p < 0.05 vs supernatant+SAPs in the LPS+Rab8a+ group; #p < 0.05 vs supernatant in the LPS+Rab8a− group, analyzed via the t-test. |
Rab8a Knockdown Reduced SAP Secretion and Ameliorated Lung Injury in ARDS Mice
To validate the effect of Rab8a knockdown on SAP secretion in LPS-stimulated ARDS in mice, we knocked down Rab8a in mice with ARDS induced by intratracheal injection of LPS and verified the efficacy of Rab8a silencing by WB (Figure 7A), and then isolated vesicles from their BALF. NTA results indicated that the number of vesicles decreased prominently in BALF from ARDS mice with Rab8a knockdown compared with the ARDS group (Figure 7B). The WB results demonstrated that LC3-II was less abundant in the BALF of Rab8a-knockdown ARDS mice (Figure 7C). Therefore, Rab8a knockdown significantly reduced SAPs secretion in ARDS mice. To verify the effect of Rab8a knockdown on lung injury in LPS-stimulated ARDS mice, we examined the lung tissue of ARDS mice with Rab8a knockdown. The results demonstrated that Rab8a knockdown notably ameliorated pathological changes in the lung tissues (Figure 7D and E), and the survival rate of mice increased with Rab8a knockdown (Figure 7F). Additionally, ELISA results revealed that the levels of proinflammatory cytokines (IL-6, TNF-α, and IL-1β) decreased after Rab8a knockdown, whereas the levels of anti-inflammatory cytokines (IL-10 and IL-12) increased notably, which indicated that the inflammation of lung tissue from ARDS mice was markedly attenuated by Rab8a knockdown (Figure 7G). In addition, we found that the levels of IL-1β in BALF noticeably decreased after Rab8a knockdown, especially for SAPs in BALF (Figure 7H).
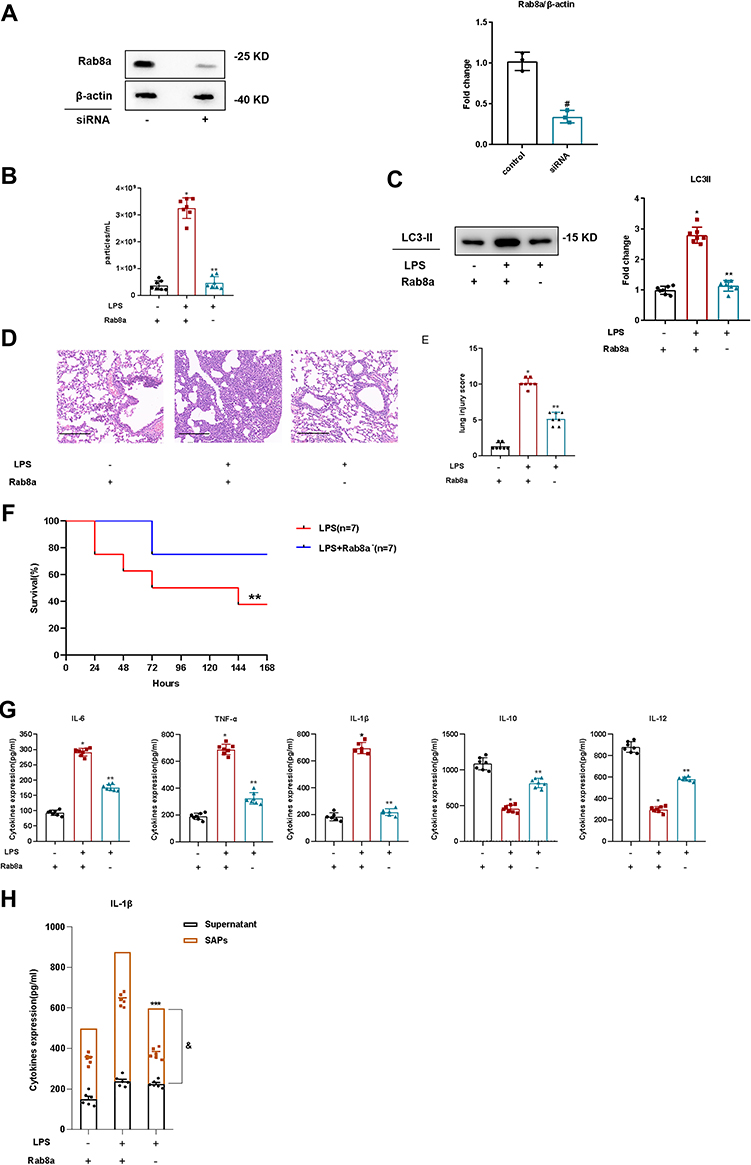 |
Figure 7 Rab8a knockdown reduced SAPs secretion and ameliorated lung injury in ARDS mice. (A) Western blot analysis illustrated efficient in vivo siRNA-mediated knockdown of Rab8a in mice in comparison with the control; (B) Nanoparticle tracking analysis revealed that there were significant decreases in the levels of vesicles in the BALF of the mice with knocked down Rab8a, compared with those in the control groups (n = 7 per group); (C) Western blot analysis demonstrated that there were significant decreases in the levels of LC3-II in the vesicles from the BALF of the mice with knocked down Rab8a, compared with those in the control groups (n = 7 per group); (D) Histopathological images of the lung samples from the mice with or without Rab8a knockdown (haematoxylin-and-eosin staining, Scale bar = 200 µm); (E) The injury scores of the lung samples (n = 7 per group); (F). The effect of Rab8a knockdown on the survival rate of lipopolysaccharide (LPS)-induced ARDS mice (n = 7 per group); (G) The effects of Rab8a knockdown on the inflammatory cytokine levels in the lungs were assessed using ELISA (n = 7 per group); (H) The levels of IL-1β in SAPs and supernatant of BALF were assessed via ELISA after the SAPs were lysed using Triton X-100 (n = 6 per group). The experiments were repeated at least three times. Each value represents the mean±SD of three independent experiments. *p < 0.05 vs the LPS−Rab8a+ group; **p < 0.05 vs the LPS+Rab8a+ group; ***p < 0.05 vs supernatant+SAPs in the LPS+Rab8a+ group; &p < 0.05 vs supernatant in the LPS+Rab8a. #p < 0.05 vs supernatant in the LPS+Rab8a − group, analyzed via the t-test. |
Discussion
This study is the first to illustrate that SAPs from AMs play a crucial role in ARDS by enhancing the inflammatory response and exacerbating lung injury and this finding may indicate a novel therapeutic approach for reducing SAPs secretion to ameliorate the inflammatory response in ARDS. Moreover, this study found that IL-1β is a critical factor that contributes to the AMs-derived SAPs-induced inflammation and lung injury and that IL-1RA diminished the stimulatory effects of SAPs on lung inflammation and injury. Furthermore, our findings indicate that Rab8a is essential for AMs to release SAPs with IL-1β and that Rab8a knockdown significantly alleviates lung injury in ARDS.
Despite progress in the diagnosis and treatment of ARDS, current treatments for ARDS are mostly based on supportive methods for mechanical lung ventilation.16 The lack of specific therapies for the underlying pathogenesis of the uncontrolled inflammation results in the high mortality and poor prognosis of ARDS.20 Extracellular vesicles (EVs) and particles secreted from AMs play important roles in the progression of lung inflammation and injury in ARDS and that AMs are the main sources of EVs in BALF from mice with early-stage LPS-induced ARDS.21–23 Currently, most studies focus on the roles of EVs, including MVs or exosomes. However, the vesicles we identified, which are derived from AMs, did not express markers of EVs. To the best of our knowledge, our study demonstrated for the first time that, in addition to EVs, AMs release SAPs following LPS stimulation. Recent studies have suggested that EVs and particles from macrophages contain various proinflammatory biological mediators, and even lipid fractions of EV membranes possess proinflammatory activity.24–27 Thus, it was logical to hypothesize that SAPs from AMs may mediate inflammation and lung injury. Therefore, our study focused on the role of SAPs in the pathogenesis of the inflammation in ARDS. We found that the administration of SAPs from LPS-stimulated macrophages could exacerbate ARDS in mice, which was consistent with the effect of other EVs and particles from macrophages reported from previous studies.28,29 Accordingly, SAPs from macrophages could be considered novel vesicles that contribute to the overwhelming inflammation in ARDS.
Our findings demonstrated that the proinflammatory effect of AMs-derived SAPs is elicited, at least partially, via an IL-1β–dependent mechanism. Our results indicate that, in LPS-stimulated ARDS, SAPs from AMs are loaded with IL-1β, and thus, anti–IL-1β treatment was exceedingly effective in mitigating the SAPs-induced inflammation and lung injury. Several recent studies have shown that IL-1β, as a member of the IL-1 family, can be released without transiting through the conventional secretory pathway. Macrophages can secrete IL-1β in an autophagy-dependent manner called secretory autophagy.30–32 Dupont et al demonstrated that autophagy contributes to IL-1β biogenesis and secretion of via an export pathway that depends on Atg5 and inflammasomes.17 Previous studies showed that IL-1β is confined to the intermembrane space between the inner and outer membranes of the double-membrane autophagosomes during secretory autophagy and then released into the extracellular space when the outer membrane fuses with the plasma membrane.33–37 However, this finding differs from the results presented herein, which suggests that IL-1β can be packaged in SAPs – a type of double-membrane vesicle – and can then be released from macrophages. To our knowledge, this is the first time that the cytokine content of SAPs has been explored. Our results clearly identify IL-1β as a bioactive molecular cargo of AMs-derived SAPs, and this observation has not been described previously. Additionally, we found that the biological activity of the IL-1β to lung injury was the same regardless of whether we provided SAP-encapsulated IL-1β, and this finding was consistent with the effect of other cytokines of EVs reported from a previous study.38 However, the mechanism of release of the inflammatory mediator IL-1β from double-membraned SAPs are not fully understood. Neves et al demonstrated that the liberation of cytokines from cell-free eosinophil granules could be based on a stimulus-dependent mechanism.39 Other studies on EVs-encapsulated cytokines, including IL-6 and IL-10, suggested that entrapped cytokines could be released when EVs interact with the cell surface, as lipid vesicles could become leaky on contact with the plasma membrane.40 Moreover, we unexpectedly observed that SAPs from AMs could remain intact in vitro for 14 days, which suggests that IL-1β may be more stable and thus retain its proinflammatory effect in ARDS longer when packaged in SAPs than as soluble IL-1. Therefore, the key point of our study was that IL-1β, as the inflammatory cargo transported by AMs-derived SAPs, was able to mediate proinflammatory effects in ARDS.
As LC3 conjugation is a hallmark of autophagosome formation, the manipulation of autophagy may be a potential mechanism to regulate SAPs release to mediate inflammation. Surprisingly, in our study, inhibition of autophagy did not alleviate lung injury in mice with ARDS (Figure S4). These results are in agreement with those of recent studies that indicate that the role of autophagy in ARDS is controversial and varies with the disease background and the stages of lung inflammation.41 Thus, instead of regulating SAPs secretion by manipulating autophagy, our study focused on the mechanisms underlying the intracellular transport of vesicles and their secretion in macrophages. Rab8a is a small GTPase that plays a key role in the intracellular transport of proteins and vesicles from the ER to the Golgi apparatus and the plasma membrane.37 Several studies have shown that Rab8a contributes to the autophagy-based unconventional secretory pathway.42–45 Our study demonstrates, for the first time, that Rab8a is the pivotal factor which mediates SAP secretion via macrophages. These results reflect those of Bel et al, who found that SAPs derived from Paneth cells contain Rab8a, which is essential for Paneth cells to secrete SAPs.13 Furthermore, we proved that knocking down Rab8a can decrease SAPs secretion in vivo and improve inflammation and lung injury in mice with ARDS; thus, Rab8a may be a novel therapeutic target for the treatment of ARDS.
There are several limitations to this study. First, we investigated SAPs derived from macrophages only in vitro and in mice. Additional studies are needed to explore the role of SAPs in the BALF of ARDS patients. Second, we used only one ARDS model, which was induced via intratracheal LPS injection. However, ARDS can be direct (pulmonary) or indirect (extrapulmonary). Given this difference in the pathogenesis of ARDS, our results should be interpreted cautiously. Third, although we purified SAPs with magnetic beads combined with LC3b antibody, the possibility of other kinds of vesicles being present in the SAPs we harvested exists because LC3 can be present in several extracellular vesicles besides SAPs. However, we examined the expressions of markers of different extracellular vesicles with SAPs to rule out other types of vesicles and found that the majority of the vesicles we collected were SAPs. Fourth, as we did not detect inflammatory cytokines other than IL-1β in SAPs, it is possible that the lung injury could be induced by other proinflammatory cytokines in SAPs or even by SAPs themselves. Although we used an IL-1RA to verify that IL-1β played a vital role in lung injury stimulated by AMs-derived SAPs, it is necessary to investigate other components of SAPs that could cause lung injury and inflammatory response. Last, we only examined the levels of SAPs in BALF after ARDS modeling; therefore, it could be meaningful to determine the dynamic changes of SAPs levels at different stages of ARDS in future studies.
Conclusion
This is the first study to show that IL-1β in AMs-derived SAPs participate in the pathogenesis of ARDS by mediating the inflammatory response and lung injury via IL-1β secretion. Both cellular and animal experiments demonstrated that the mechanism underlying SAPs secretion by macrophages is associated with Rab8a. Accordingly, SAPs may serve as a novel biomarker, and Rab8a may be a potential therapeutic target in ARDS.
Data Sharing Statement
All the data are illustrated in the figures and in the supplementary data.
Ethics Approval
The study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Committee of Animal Care and Use of Southeast University (Protocol number 20180106007).
Acknowledgments
We would also like to acknowledge the professors and colleagues from Southeast University including: Dr. Fei Peng, Yuying Tang and Cuilin Chu for their technical support guidance on the manuscript of this article.
Author Contributions
XY-X and XL participated in the collection and assembly of data, data analysis and interpretation, and manuscript writing. XC-D participated in acquisition, analysis, and interpretation of data. LL, YY and HB-Q participated in the design and supervision of research. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was funded by National Natural Science Foundation of China [Grant numbers 81870066, 81670074, 81930058, 81971888], Six Talent Peaks Project in Jiangsu Province [Grant numbers TD-SWYY-003], Social Development Specifc Projects of Jiangsu Province [Grant number BE2018743], and Natural Science Foundation of Jiangsu Province [Grant number BK20181271].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315:788–800. doi:10.1001/jama.2016.0291
2. Calfee CS. ARDS in 2015: new clinical directions, new biological insights. Lancet Respir Med. 2015;3:912–913. doi:10.1016/S2213-2600(15)00425-7
3. Deprez M, Zaragosi LE, Truchi M, et al. A single-cell atlas of the human healthy airways. Am J Respir Crit Care Med. 2020;202:1636–1645. doi:10.1164/rccm.201911-2199OC
4. Hashimoto D, Chow A, Noizat C, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi:10.1016/j.immuni.2013.04.004
5. Jardine L, Haniffa M. Human lung macrophages: roll up for the MISTRG tour. Immunity. 2021;54:194–196. doi:10.1016/j.immuni.2021.01.006
6. Aggarwal S, Mannam P, Zhang J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol. 2016;311:L433–L452. doi:10.1152/ajplung.00128.2016
7. Mizumura K, Cloonan S, Choi ME, et al. Autophagy: friend or foe in lung disease? Ann Am Thorac Soc. 2016;13:40–47. doi:10.1513/AnnalsATS.201508-529OC
8. Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664–673. doi:10.1016/j.bbamcr.2008.07.014
9. Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi:10.1152/physrev.00030.2009
10. Jiang S, Dupont N, Castillo EF, et al. Secretory versus degradative autophagy: unconventional secretion of inflammatory mediators. J Innate Immun. 2013;5:471–479. doi:10.1159/000346707
11. Ponpuak M, Mandell MA, Kimura T, et al. Secretory autophagy. Curr Opin Cell Biol. 2015;35:106–116. doi:10.1016/j.ceb.2015.04.016
12. Minakaki G, Menges S, Kittel A, et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosomelike phenotype. Autophagy. 2018;14:98–119. doi:10.1080/15548627.2017.1395992
13. Bel S, Pendse M, Wang Y, et al. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science. 2017;357:1047–1052. doi:10.1126/science.aal4677
14. Wang J, Davis S, Zhu M, et al. Autophagosome formation: where the secretory and autophagy pathways meet. Autophagy. 2017;13:973–974. doi:10.1080/15548627.2017.1287657
15. Gonzalez CD, Resnik R, Vaccaro MI. Secretory autophagy and its relevance in metabolic and degenerative disease. Front Endocrinol (Lausanne). 2020;11:266. doi:10.3389/fendo.2020.00266
16. Davis S, Wang J, Ferro-Novick S. Crosstalk between the secretory and autophagy pathways regulates autophagosome formation. Dev Cell. 2017;41:23–32. doi:10.1016/j.devcel.2017.03.015
17. Dupont N, Jiang S, Pilli M, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011;30:4701–4711. doi:10.1038/emboj.2011.398
18. Kimura T, Jia J, Kumar S, et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017;36:42–60. doi:10.15252/embj.201695081
19. Zhou M, Wen ZF, Cheng F, et al. Tumor-released autophagosomes induce IL-10-producing B cells with suppressive activity on T lymphocytes via TLR2-MyD88-NF-kappa B signal pathway. Oncoimmunology. 2016;5:7. doi:10.1080/2162402X.2016.1180485
20. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med. 2017;377:562–572. doi:10.1056/NEJMra1608077
21. Soni S, Wilson MR, O’Dea KP, et al. Alveolar macrophage-derived microvesicles mediate acute lung injury. Thorax. 2016;71:1020–1029. doi:10.1136/thoraxjnl-2015-208032
22. Hu Q, Lyon CJ, Fletcher JK, et al. Extracellular vesicle activities regulating macrophage-and tissue-mediated injury and repair responses. Acta Pharm Sin B. 2021;11:1493–1512. doi:10.1016/j.apsb.2020.12.014
23. O’Dea KP, Tan YY, Shah S, et al. Monocytes mediate homing of circulating microvesicles to the pulmonary vasculature during low-grade systemic inflammation. J Extracell Vesicles. 2020;9:1706–1708. doi:10.1080/20013078.2019.1706708
24. Wang JG, Williams JC, Davis BK, et al. Monocytic microparticles activate endothelial cells in an IL-1β–dependent manner. Blood. 2011;118:2366–2374. doi:10.1182/blood-2011-01-330878
25. Lee TH, D’Asti E, Magnus N, et al. Microvesicles as mediators of intercellular communication in cancer-the emerging science of cellular “debris”. Semin Immunopathol. 2011;33:455–467. doi:10.1007/s00281-011-0250-3
26. Islam MN, Das SR, Emin MT, et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012;18:759–765. doi:10.1038/nm.2736
27. Thomas LM, Salter RD. Activation of macrophages by P2X7-induced microvesicles from myeloid cells is mediated by phospholipids and is partially dependent on TLR4. J Immunol. 2010;185:3740–3749. doi:10.4049/jimmunol.1001231
28. Zhu Z, Zhang D, Lee H, et al. Macrophage-derived apoptotic bodies promote the proliferation of the recipient cells via shuttling microRNA-221/222. J Leukoc Biol. 2017;101:1349–1359. doi:10.1189/jlb.3A1116-483R
29. Neri T, Armani C, Pegoli A, et al. Role of NF-kappaB and PPAR-gamma in lung inflammation induced by monocyte-derived microparticles. Eur Respir J. 2011;37:1494–1502. doi:10.1183/09031936.00023310
30. Duran JM, Anjard C, Stefan C, et al. Unconventional secretion of Acb1 is mediated by autophagosomes.J Cell Biol. 2010;188(4):527–536. doi:10.1083/jcb.200911154
31. Manjithaya R, Anjard C, Loomis WF, et al. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol. 2010;188:537–546. doi:10.1083/jcb.200911149
32. Gee HY, Noh SH, Tang BL, et al. Rescue of DeltaF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell. 2011;146:746–760. doi:10.1016/j.cell.2011.07.021
33. Munding C, Keller M, Niklaus G, et al. The estrogen-responsive B box protein: a novel enhancer of interleukin-1beta secretion. Cell Death Differ. 2006;13:1938–1949. doi:10.1038/sj.cdd.4401896
34. Mandell MA, Jain A, Arko-Mensah J, et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell. 2014;30:394–409. doi:10.1016/j.devcel.2014.06.013
35. Chauhan S, Kumar S, Jain A, et al. TRIMs and galectins globally cooperate and TRIM16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016;39:13–27. doi:10.1016/j.devcel.2016.08.003
36. Cebrian I, Visentin G, Blanchard N, et al. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell. 2011;147:1355–1368. doi:10.1016/j.cell.2011.11.021
37. Petkovic M, Jemaiel A, Daste F, et al. The SNARE Sec22b has a non-fusogenic function in plasma membrane expansion. Nat Cell Biol. 2014;16:433–444. doi:10.1038/ncb2937
38. Fitzgerald W, Freeman ML, Lederman MM, et al. A system of cytokines encapsulated in extracellular vesicles. Sci Rep. 2020;10:18935. doi:10.1038/s41598-020-75735-w
39. Neves JS, Radke AL, Weller PF. Cysteinyl leukotrienes acting via granule membrane-expressed receptors elicit secretion from within cell-free human eosinophil granules. J Allergy Clin Immunol. 2010;125:477–482. doi:10.1016/j.jaci.2009.11.029
40. Aiello A, Giannessi F, Percario ZA, et al. An emerging interplay between extracellular vesicles and cytokines. Cytokine Growth Factor Rev. 2020;51:49–60. doi:10.1016/j.cytogfr.2019.12.003
41. Hoyer FF, Naxerova K, Schloss MJ, et al. Tissue-specific macrophage responses to remote injury impact the outcome of subsequent local immune challenge. Immunity. 2019;51:899–914. doi:10.1016/j.immuni.2019.10.010
42. Wall AA, Luo L, Hung Y, et al. Small GTPase Rab8a-recruited Phosphatidylinositol 3-Kinase γ regulates signaling and cytokine outputs from endosomal toll-like receptors. J Biol Chem. 2017;292:4411–4422. doi:10.1074/jbc.M116.766337
43. Wall AA, Condon ND, Luo L, et al. Rab8a localisation and activation by Toll-like receptors on macrophage macropinosomes. Philos Trans R Soc Lond B Biol Sci. 2018;374:1765. doi:10.1098/rstb.2018.0151
44. Luo L, Wall AA, Tong SJ, et al. TLR Crosstalk activates LRP1 to Recruit Rab8a and PI3Kγ for suppression of inflammatory responses. Cell Rep. 2018;24:3033–3044. doi:10.1016/j.celrep.2018.08.028
45. Condon ND, Heddleston JM, Chew TL, et al. Macropinosome formation by tent pole ruffling in macrophages. J Cell Biol. 2018;217:3873–3885. doi:10.1083/jcb.201804137
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.