")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Screening strategies to identify HSP70 modulators to treat Alzheimer’s disease
Received 3 August 2014
Accepted for publication 23 September 2014
Published 7 January 2015 Volume 2015:9 Pages 321—331
DOI https://doi.org/10.2147/DDDT.S72165
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Jayanthi Repalli, Daniel Meruelo
Department of Pathology, New York University, Langone Medical Center, New York, NY, USA
Abstract: Alzheimer’s disease, the most common type of dementia, is a progressive brain disease that destroys cognitive function and eventually leads to death. In patients with Alzheimer’s disease, beta amyloids and tau proteins form plaques/oligomers and oligomers/tangles that affect the ability of neurons to function properly. Heat shock protein 70 (HSP70) has the ability to prevent aggregation/oligomerization of beta amyloid/tau proteins, making it a potential drug target. To determine this potential, it is essential that we have appropriate in vitro and cell-based assays that help identify specific molecules that affect this aggregation or oligomerization through HSP70. Potential drug candidates could be identified through a series of assays, starting with ATPase assays, followed by aggregation assays with enzymes/proteins and cell-based systems. ATPase assays are effective in identification of ATPase modulators but do not determine the effect of the molecule on beta amyloid and tau proteins. Molecules identified through ATPase assays are validated by thioflavin T aggregation assays in the presence of HSP70. These assays help uncover if a molecule affects beta amyloid and tau through HSP70, but are limited by their in vitro nature. Potential drug candidates are further validated through cell-based assays using mammalian, yeast, or bacterial cultures. However, while these assays are able to determine the effect of a specific molecule on beta amyloid and tau, they fail to determine whether the action is HSP70-dependent. The creation of a novel, direct assay that can demonstrate the antiaggregation effect of a molecule as well as its action through HSP70 would reduce the number of false-positive drug candidates and be more cost-effective and time-effective.
Keywords: heat shock protein 70, beta amyloid, tau, adenosine triphosphatase activity, oligomers, aggregation
Introduction
Alzheimer’s disease (AD) affects approximately 11% of the aging population (older than 65 years) and disables daily functions in affected patients.1 In patients with AD, the brain produces excess amounts of plaques or oligomers composed of beta amyloid, as well as tangles made of tau proteins. This leads to the inability of neurons to function properly, and as a result, patients experience cognitive impairment.2–5There is a positive correlation between dementia and plaque/neurofibrillary tangles in AD patients. The distribution of beta amyloid and tau in AD patients can be observed directly by histopathological and biochemical analysis from postmortem samples.3,6–12
The key to successfully treating AD is elimination of abnormal beta amyloid oligomers/plaques and tau oligomers/neurofibrillary tangles, which might be accomplished, among various alternatives, by targeting a promising protein, heat shock protein 70 (HSP70). This article discusses the importance of eliminating beta amyloid and tau, and reviews state of the art screening methodologies that are suited to this task, discussing their advantages and limitations.
HSP70 in Alzheimer’s disease
A multitude of target proteins that play a role in beta amyloid/tau accumulation are known, including alpha/beta secretases and kinases. However, attempts to target some of these have so far been unsuccessful, while others are currently in various developmental stages.13 We believe that one of the most promising proteins that should be targeted is HSP70, because of its ability to inhibit oligomerization and aggregation of both beta amyloid and tau proteins.
HSP70 maintains homeostasis by protecting against stresses such as heat, oxidative stress, and neurodegenerative disease. HSP70 manifests a neuroprotective effect by binding tightly to misfolded/aggregated proteins to produce properly folded, solubilized proteins or directing them to degradation pathways, thereby stabilizing or degrading cellular proteins to avoid aggregation.14–21
Several neurodegenerative and aggregative disorders, such as AD and Parkinson’s disease,show increased/altered levels of HSP70. In addition, polyglutamine (polyQ) diseases showed polyQ protein colocalization with HSP70.22–25 Furthermore, increased levels of HSP70 protein were found in postmortem cortical tissues of AD brains compared with normal brains, again suggesting that HSP70 associates with the diseased areas of the brain, presumably in order to solubilize aggregates in an attempt to prevent further damage.22
Relationship between HSP70, beta amyloid, and tau
In 2011, Hoshino et al reported improvement in both perception and behavior with a decreased burden of amyloid plaques in an AD mouse model expressing high levels of HSP70 when compared with a control strain.26 Also, the presence of HSP70 increased degradation of beta amyloid peptides in microglial cells.27 Studies by Evans et al using monomeric beta amyloid species, suggested that HSP70 decreases the number of oligomers through refolding of misfolded proteins.17 These studies provide evidence of the interaction between HSP70 and beta amyloid.
Other studies have shown that overexpression of HSP70 is associated with a reduction in tau protein levels in various cultured cell lines as well as in mouse models.28 HSP70 binds to tau and prevents its aggregation by directing it to the ubiquitin degradation pathway through interaction with an E3 ubiquitin ligase.28 Also, HSP70 inhibition may hinder the proper folding of the molecule, thus directing it to other cellular degradation pathways.18
Potential role of HSP70
Together with HSP40, a chaperone protein that forms a complex with HSP70 and regulates its binding to polypeptides by affecting its catalytic activity, HSP70 protects neurons from stress, making it an attractive target in AD drug development.14,15 Catalytic (ATPase) activity involves changing the HSP70 conformation from the adenosine triphosphate (ATP)-bound form, which has less affinity for misfolded, oligomerized, or aggregated proteins, to the adenosine diphosphate (ADP)-bound form, which has high affinity for misfolded, oligomerized, and aggregated proteins. HSP70 breaks the bonds that cause aggregation and helps establish new bonds that are required for the properly folded protein, or directs proteins to degradation if restoration is not possible.14–15,17 Inhibition of HSP70 ATPase activity stabilizes the HSP70 in an ADP-bound form, leading to tight binding of the misfolded protein without releasing it in the correctly folded form. Thus, inhibition reduces refolding activity, leading to activation of degradation pathways, such as the ubiquitin proteasomal pathway, to eliminate excessive aggregated proteins.18 These observations indicate that modulating ATPase activity by activation or inhibition might be beneficial to cells in order to eliminate aggregated proteins.
Therefore, for the purpose of identifying drugs that modulate the ATPase activity of HSP70, several in vitro assays have been developed. However, while identification of ATP catalytic activity helps select lead molecules, it fails to reveal their specific role in neurodegenerative diseases. These lead molecules must then be validated and advanced through in vitro and in vivo assays to estimate their potential in AD. Given the importance of the HSP70-HSP40 complex in cellular physiology, it is imperative that additional HSP70 modulators with unique properties be discovered. Identification of HSP70 modulators is of great importance for uncovering potential novel treatments; hence knowledge of suitable analytical techniques available for identification is essential.
Developing an assay that could determine early on the interaction of drug candidates with specific key players in AD development could ultimately save time, effort, and money by allowing researchers to focus on fewer drug candidates with greater potential for successful treatment of disease.
Detection technologies to identify HSP70 modulators
Dephosphorylation of ATP is a fundamental function of HSP70.15 Different principles have been applied to probe the rate of HSP70 ATPase activity; for example, by evaluating either the product formation or the substrate binding levels by tagging the substrate or measuring the refolding of proteins (Figure 1).
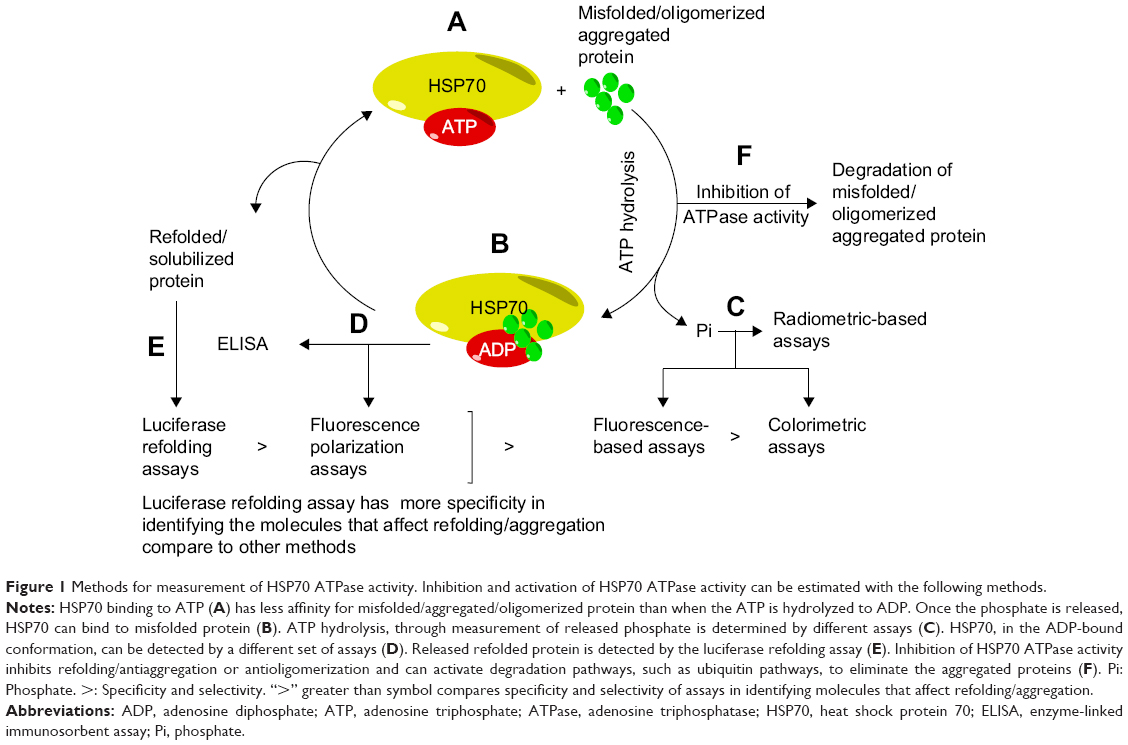 | Figure 1 Methods for measurement of HSP70 ATPase activity. Inhibition and activation of HSP70 ATPase activity can be estimated with the following methods. |
There are currently several types of assays that are valuable in these studies, including radiometric,29–32 absorbance-based,18,33–35 fluorescence-based,35–38 luciferase refolding,39–44 enzyme-linked immunosorbent assay,35,45 and tryptophan fluorescence,35,46 assays (Table 1). These assays possess both advantages and limitations. Overall, these assays are very useful in identifying HSP70 modulators, but they fail to detect any effects of these modulators on aggregation of beta amyloid and tau proteins, which is crucial if one is to have an impact in understanding the role of HSP70 in the development of AD. Among these assays, the luciferase assay is the most specific and sensitive because it measures the refolding of proteins. Assays based on thioflavin T (ThT)/thioflavin S (ThS) dyes can be used to identify HSP70 modulators that inhibit aggregation through HSP70. However, to examine the effects of HSP70 on beta amyloid/tau oligomerization, dot-blots with specific antioligomer antibodies must be used, which are not easily accessible for high throughput screening.
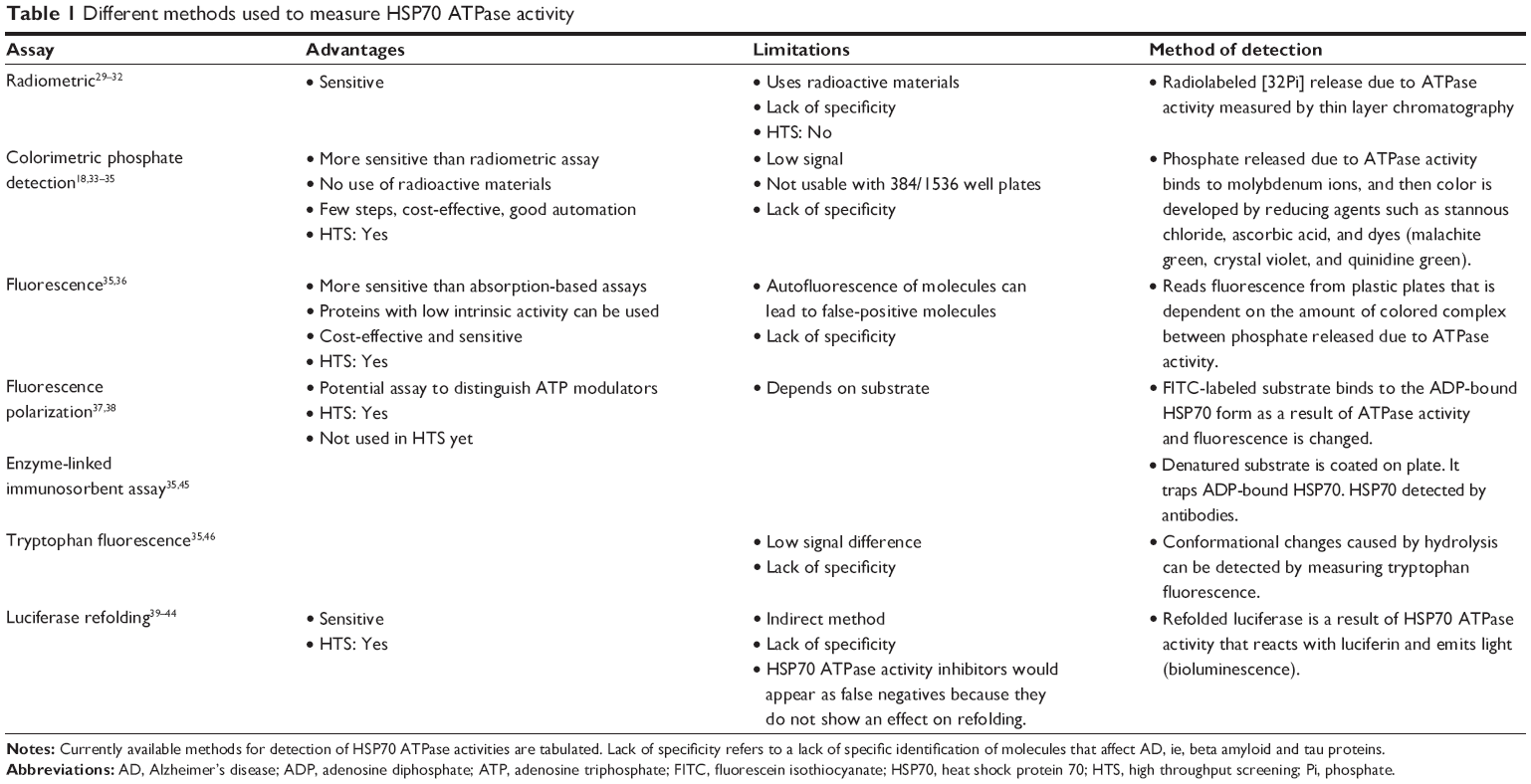 | Table 1 Different methods used to measure HSP70 ATPase activity |
Approaches to determine the effect of modulators on aggregation
Thioflavin T aggregation assay
Beta amyloid in its solubilized form exists in a random coil form, but when it aggregates, it forms a beta sheet structure. Beta sheet structures formed by beta amyloid can be identified and quantified by a specific ThT assay, which is performed using synthetic beta amyloid peptide. The assay measures enhanced fluorescence in the presence of aggregated structures (Table 2)17,47–54 HSP70 functionality in the interruption of aggregation can be detected and quantified by using ThT, an aggregate-specific dye.50
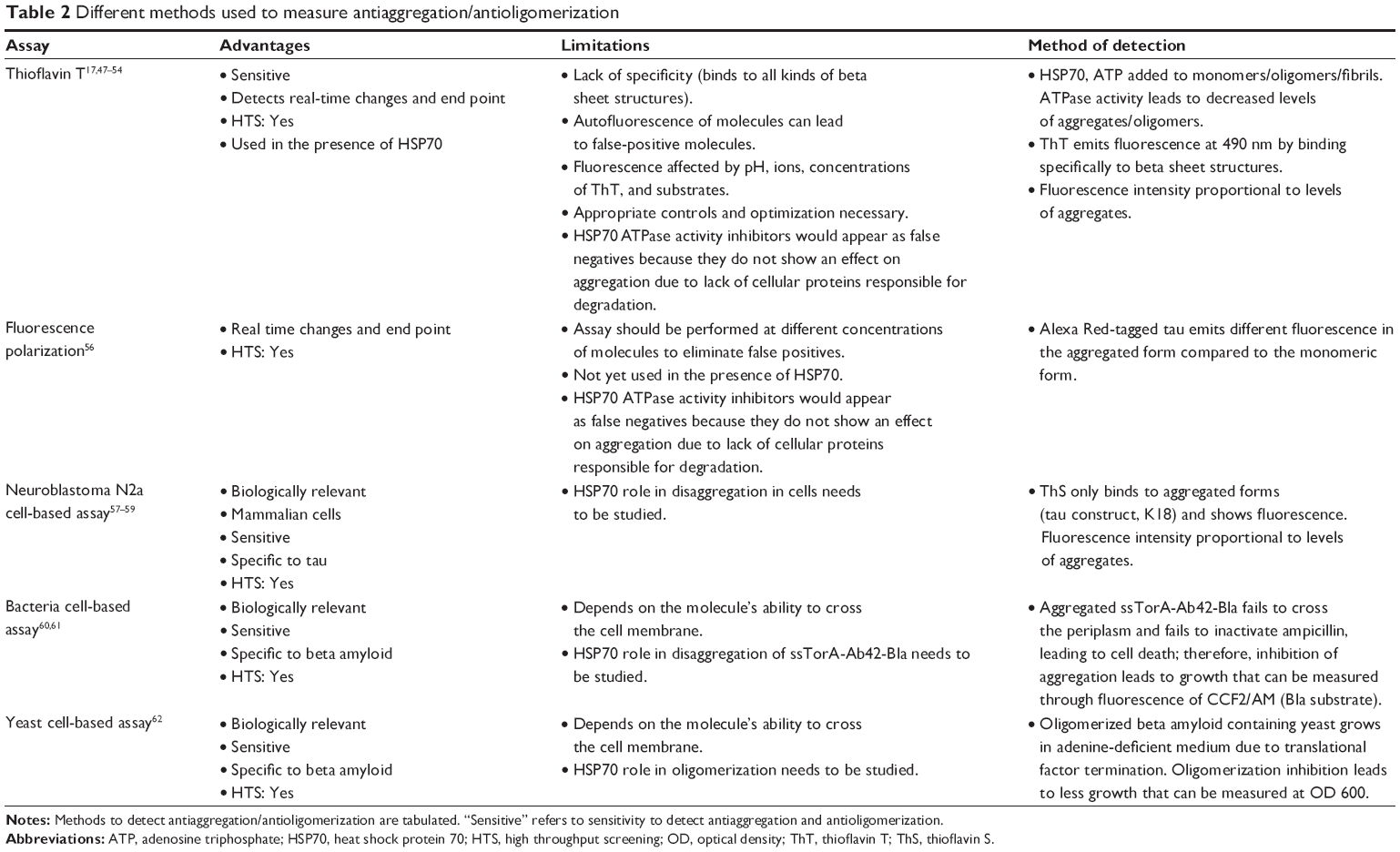 | Table 2 Different methods used to measure antiaggregation/antioligomerization |
Advantages
The results of a ThT assay not only provide information about the effects of HSP70 on aggregation but also reveal evidence of any modulating factors that regulate the antiaggregation effects,50 so the effects of a drug candidate on aggregation can be established.
Evans et al showed that HSP70-treated beta amyloid monomers have less fluorescence compared with untreated monomers when subjected to aggregating conditions, indicating both the ability of HSP70 to decrease aggregate formation and the usability of the ThT assay. Using this format, the authors determined the effect of the HSP70 regulator, SW02. Samples treated with SW02, which is an activator of HSP70 ATPase activity, showed decreased fluorescence, indicating that activation of HSP70 by SW02 could alter the conformation of misfolded beta amyloid peptides into a less aggregated state. Thus, these studies confirm the applicability of the ThT assay to identify HSP70-mediated beta amyloid aggregation inhibitors.17
There are few assays available to measure the effects of molecules affecting the ATPase activity of HSP70. ThS, like ThT, specifically binds to beta sheet (aggregate) structures and has been used to measure the antiaggregation effects of HSP70 on tau. When HSP70 was incubated with tau in aggregate-inducing conditions, HSP70-treated tau showed less fluorescence compared to untreated samples.19 Thus, this assay can be used to identify HSP70-mediated tau aggregation inhibitors. In a study from 2007, Crowe et al screened 51,000 potential drug candidates that inhibit tau aggregation in 384 wells by using only tau protein without the presence of HSP70. This initial screening was followed by confirmatory tests and additional sedimentation tests to check the antiaggregation effect of each molecule. These experiments ultimately resulted in identification of two molecules of a newly identified class of fibril inhibitors, 2,3-di(furan-2-yl)-quinoxalines, 113F08 and 330B06.55
Fluorescence polarization assay
In the fluorescence polarization assay, tau is tagged with Alexa Red and will emit fluorescence based on its aggregation stage. By using this methodology, 292,000 molecules at six different concentrations were screened and resulted in identification of several sets of previously recognized molecules and new aminothienopyridazines.56 This assay may be done in the presence of HSP70 and molecules together with appropriate controls. For example, HSP70 in combination with other molecules that inhibit aggregation would show different fluorescence readouts compared with aggregated tau (Table 2).
HSP70 binding assay
In this binding assay, tau is tagged with a His-tag that specifically binds to metal affinity resin. By using this methodology, the association of HSP70 with tau can be measured by adding different concentrations of HSP70 to tau in the presence/absence of aggregating agents such as arachidonic acid. The mixture can be separated through spinning, and pelleted/soluble fractions can be run on sodium dodecyl sulfate polyacrylamide gel electrophoresis and probed with HSP70 and His-tag tau antibodies. In the presence of arachidonic acid, association of HSP70 with aggregated species is observed, while there is no association in the absence of arachidonic acid, indicating that HSP70 binds to tau and prevents its aggregation.57 This does not clearly distinguish between the refolding or degradation of tau proteins and is difficult to adapt for high throughput screening. However, this assay may be useful to delineate the interactions between HSP70 and tau together with additional molecules. HSP70 in combination with aggregation-inhibitory molecules would show association of HSP70 with aggregated species, whereas HSP70 without molecules would not show association.
Effects of modulators in cell culture
Molecules obtained through the use of the above approaches can be validated through cell-based assays. Neuroblastoma N2a cells are transfected with an aggregation-prone tau construct, and molecules that inhibit tau aggregation can be detected by using ThS dye. ThS specifically binds to beta sheet structures and shows enhanced fluorescence upon binding. Any molecules with antiaggregation properties will result in less aggregation of tau and thereby less availability of beta sheet structures, ultimately leading to less fluorescence. Cell death caused by tau aggregates can be measured by the lactate dehydrogenase assay (Table 2).58–60 For testing the effect of specific molecules on beta amyloid, cell-based assays in bacterial and yeast systems are available.61–63 In bacterial cells expressing aggregation-prone beta amyloid, ssTorA-Ab42-Bla, molecules that inhibit aggregation will result in increased growth of these cells, which can be measured through the fluorescence of CCF2/AM (Bla substrate). By using this model, four antiaggregation molecules were obtained from a triazine derivatives library (Table 2).61,62 In yeast cell-based systems expressing the oligomerization-prone beta amyloid construct, Aβ-MRF (Aβ42 fused to yeast translational termination factor, Sup35p), which allows growth of yeast in adenine-deficient medium, inhibition of oligomerization by target molecules reduces the growth of the yeast cells. Using this model, two antioligomeric molecules were identified from 12,800 molecules.63 These assays are very useful in elucidating the effects of specific molecules on beta amyloid and tau proteins in live cells and are also helpful tools to validate and identify lead molecules. Performance of the molecules depends on those molecules reaching their target(s) in vivo (eg, permeability), so molecules should be validated through different systems (Table 2).61–63 The three assays have common benefits, such as low cost, reliability, speed, and high throughput capability. Mammalian cell-based assays, especially those expressing tau protein in neuroblastoma N2a cells, are more analogous to humans than other assays.58,59 Therefore, the findings would likely have greater relevance to human neuronal cells.
Oligomers in yeast systems are similar to those found in the human brain. The results may therefore be more reflective of the human situation than bacterial assays. Yeast-based assays can facilitate the identification of molecules that can inhibit Aβ42 oligomerization (early stages of aggregation).63 Bacterial systems also allow identification of molecules that inhibit Aβ42 aggregation. However, these systems have not been used to determine which stage of the aggregation cascade is inhibited, and this information is important for eliminating the production of toxic intermediates of Aβ42 such as dimers and trimers that form earlier in the aggregate formation.61–63 Thus, yeast assays present advantages over bacterial systems in terms of possessing oligomers similar to those in the human brain and allowing for determination of molecules intervening at specific stages of aggregation. However, these methods are somewhat limited due to their inability to show the interaction between HSP70 and the target protein in preventing aggregation of beta amyloid and tau.
Studying the effects of HSP70 using these approaches might help to more accurately and specifically identify new molecules that interact with HSP70 to remove oligomers and fibrils. This will be helpful in differentiating HSP70-dependent and HSP70-independent molecules.
Testing of lead molecules in mouse models
Lead molecules can be tested in mouse models of AD, such as Tg2576, APP23, and 5X FAD,64 for the clearance of amyloid-β/tau levels and improvement of cognition symptoms. Levels of Aβ/tau can be measured with immunohistochemistry studies using ThS. Cognition symptoms can be measured and monitored using spatial, contextual, and working memory tests with the Morris water maze, radial arm maze, fear conditioning, Y-maze, and object recognition tests. These tests and mouse models of AD are not within the scope of this review but are explained by Kathryn et al65 and Hall and Roberson.64 Overexpression of HSP70 or geranylgeranylacetone treatment (increases HSP70 expression) in AD APP23 mice that manifest Aβ plaques and memory deficits showed decreased abnormal Aβ levels and improved cognition symptoms.26,66
Currently known HSP70 targeting molecules
The examples described below further reinforce the interest in pursuing molecules that target HSP70. As noted previously, HSP70 in the ADP-bound conformation has high affinity for misfolded tau and degrades it through a ubiquitin-mediated proteasomal pathway.67,68 Several molecules, including methylene blue (MB),18,68–72 MKT-077 67,73–76 YM-01,67–73,77 and YM-08,67 were discovered based on this mechanism whereby the molecules bind to the ADP-bound conformation of HSP70 and block ATP binding or ATPase activity.
MB is the first generation of these molecules, and inhibits HSP70 ATPase activity by oxidizing Cys306.69 MB decreased abnormal tau/Aβ levels in cellular and mouse AD models and improved cognitive function in AD patients in Phase IIb trials.18,69–71,78 As MB inhibits acetylcholinesterase, butyrylcholinesterase, and monoamine oxidase and facilitates transport of electrons in the respiratory chain,72 it is not likely to be HSP70-selective.
Three other notable molecules, ie, MKT-077, YM-01, and YM-08, reduced abnormal tau levels in cell-based models in which tau is overexpressed. This is achieved by molecules binding to the nucleotide-binding domain of HSC70, a cytoplasmic member of the HSP70 family.67,73 YM-01 decreased long-term potentiation in brain slices of a tau transgenic mouse model, rTg4510.73 Additionally, it inhibited eye degeneration and corrected failed eclosion in a Spinobulbar Muscular Atrophy model of Drosophila melanogaster flies, through inhibition of aggregation/oligomerization of polyQ AR.77 However, use of both these molecules (MKT-077 and YM-01) in AD is limited by their inability to cross the blood–brain barrier and by their nephrotoxicity.67,76
YM-08, a neutral analog of MKT-077, is synthesized by replacing the cationic pyridinium ring of MKT-077 with a neutral pyridinium ring to make it blood–brain barrier penetrable. It has a more suitable pharmacokinetic profile in the central nervous system, showing an ~0.25 brain/plasma value for at least 18 hours in CD1 mice (greater than 0.3 is considered stronger central nervous system candidate). Additionally, it showed rapid clearance from the kidney, with retention of 55.2 ng/g at 1 hour compared with YM-01 at 63,231 ng/g, indicating a potential for less nephrotoxicity. YM-08 presents itself as a template that inhibits HSP70 and reduces tau, with the potential to delay progression of AD.67
Summary
Even though there are many kinase assays available to measure ATPase activity, only a few have been validated specifically for screening of HSP70 modulators. While these assays are able to identify molecules with catalytic activity, they fail to establish the specificity of the molecules for beta amyloid or tau, which are involved in AD. The luciferase assay39 is more specific than other assays and does allow for identification of modulators that affect HSP70-mediated refolding of proteins. Since refolding is one of the mechanisms that prevent aggregation, this assay can be used to identify the antiaggregation efficacy of a molecule; however, it is limited by the inability to detect specific effects on beta amyloid and tau. This is due to an inability to simulate in vivo conditions in the assay, suggesting a need for a cell-based system that can relate endpoints to HSP70-mediated actions.
While the ThT/ThS assay is effective in measuring aggregate levels as well as elucidating the relationship between HSP70-mediated effects and aggregation, it has been used in relation to beta amyloid/tau. Another approach may be adaptation of the fluorescence polarization assay, where tau is tagged with Alexa Red, enhancing the fluorescence in aggregate forms. Introducing HSP70 and measuring the polarization might be beneficial, but this assay needs to be validated. However, the effects of HSP70 inhibitors that reduce aggregation through proteasomal degradation are difficult to validate due to the absence of proteins responsible for degradation, so proteins that actually work as inhibitors may appear as false negatives. In combination with other proteins involved in HSP70-mediated degradation, such as the carboxyl terminus of HSC70-interacting protein and ubiquitin, these assays could potentially be adapted to identify molecules that inhibit aggregation through degradation.21 ThT assays are not usable to detect oligomeric forms of beta amyloid, and both ThT and fluorescence polarization assays presently use molecules in the absence of protein to eliminate false positives, eg, molecules that have antiaggregative effects independent of HSP70. The development of other negative controls by using HSP70-independent aggregates might be useful. Studies by Marcinowski et al identified the HTFAAVL peptide, which has no binding affinity to HSP70.79 Similar peptides that do not interact with HSP70, but can form misfolded protein and aggregates, would be ideal molecules to use as negative controls, since any effects seen on these molecules would be independent of direct interaction with HSP70. Thus, the development of peptides that can aggregate but do not interact with HSP70 can be used as negative controls and be helpful in eliminating false positives.
Assays using mammalian, bacterial, and yeast cells are excellent systems to measure the overall effect of molecules on aggregation, but they do not explain the involvement of HSP70 in this process. The lack of cell-based systems to specifically identify HSP70-mediated, aggregation-inhibitory and oligomerization-inhibitory molecules makes it difficult to delineate the mechanism of a specific molecule. Overexpression of HSP70 in cell-based systems would be a useful tool to assess the effect of HSP70 on aggregation levels. For example, overexpression of HSP70 might inhibit aggregation completely, while partially overexpressing cells would show less inhibition. Furthermore, an HSP70 knockdown model80 would show no effect on aggregation. In that type of system, when additional molecules are included, it would be possible to differentiate HSP70-independent versus HSP70-dependent effects. Even though the role of HSP70 in AD has been documented,17,26 there are no cell-based assays that can currently be used for high throughput screens to identify HSP70-mediated antiaggregation/antioligomerization molecules.
The establishment of an assay that can measure the interactions between HSP70 and beta amyloid and tau is important in order to understand the exact mechanism of inhibition. Molecules that target HSP70 can be mechanistically validated through HSP70 ATPase assays, which will help determine whether they are working through modulation of the ATPase activity of HSP70. Subsequently, target molecules can be used in aggregation assays with polyQ and huntingtin protein along with beta amyloid to check the specificity of these molecules given that HSP70 has the ability to interact with multiple proteins (Figure 2). In addition, simply using appropriate negative controls with HSP70-independent aggregates helps to eliminate molecules that are working independently of HSP70. Furthermore, a specific HSP70-mediated mechanism can be delineated through luciferase assays, protease sensitivity assessment, and nondenaturing gels. These systems can help to determine if molecules work through HSP70-mediated corrected folding. Protease assays involve the labeling of proteins and monitoring of degradation sensitivity. Proteins in their folded form are more stable compared with the unfolded form in the presence of unspecific proteases. Substrate stability can then be measured by sodium dodecyl sulfate polyacrylamide gel electrophoresis. Nondenaturing gels are helpful for separation of folded and unfolded protein as they migrate differently.81 Cells expressing tau/beta amyloid protein, when treated with lead molecules in the presence/absence of proteasomal inhibitors, could determine if a molecule is working through HSP70-mediated degradation. Proteasomal inhibitors that prevent HSP70-mediated degradation will show more protein compared with untreated samples. Another method proposed by Chang82 is to label tau with green fluorescent protein and measure the differences in signal. If protein is degraded, it will show less fluorescence, and more fluorescence will be observed when protein is not degraded.
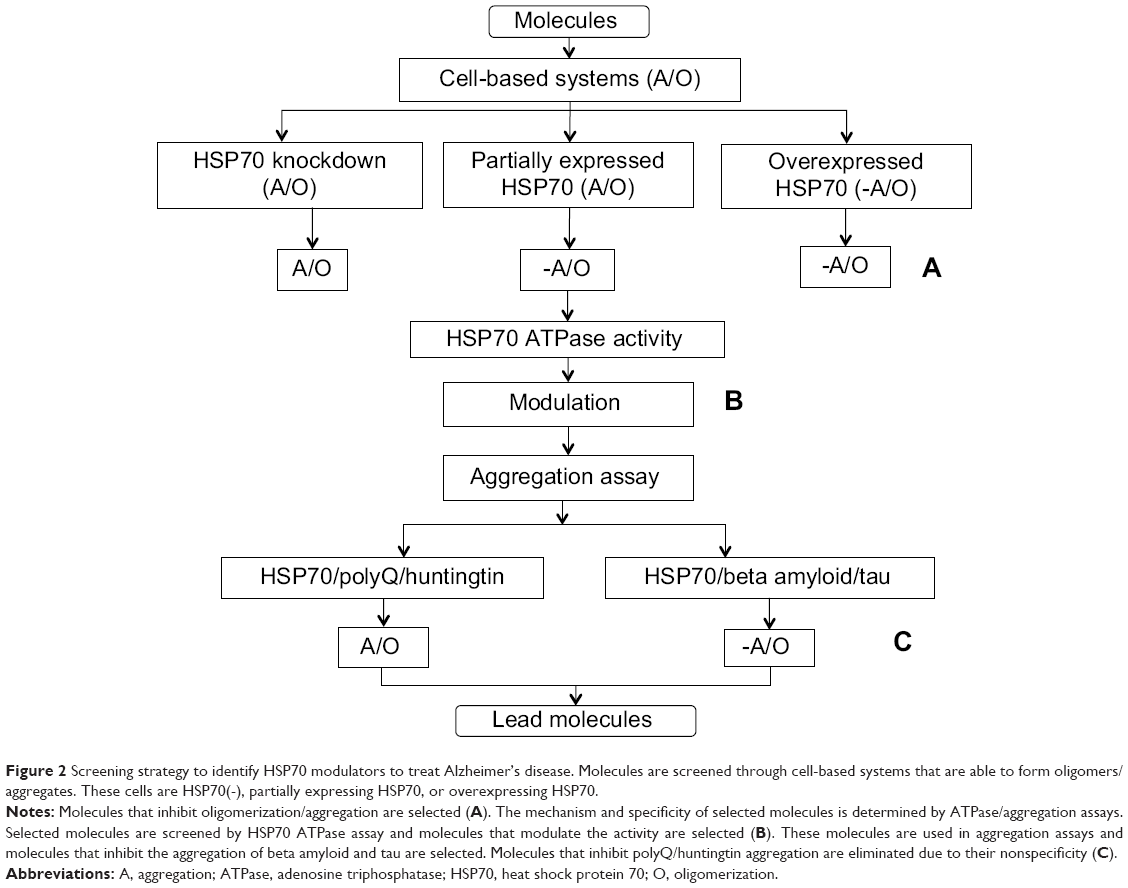 | Figure 2 Screening strategy to identify HSP70 modulators to treat Alzheimer’s disease. Molecules are screened through cell-based systems that are able to form oligomers/aggregates. These cells are HSP70(-), partially expressing HSP70, or overexpressing HSP70. |
Mechanistically validated molecules increase the potential for success, decrease the number of false positives and/or false negatives, and establish the credibility of using molecules in mouse models to determine toxicity and efficacy. It is expensive to study molecules in mice, and having a reliable assay that can identify potential candidates would be useful and cost-effective. Thus, an appropriate system would be both cost-effective and time-effective and would ultimately lead to more efficient identification of potential drug candidates for AD.
Disclosure
The authors report no conflicts of interests in this work.
References
Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimers Dement. 2014;10(2):e47–e92. | ||
Kumar S, Walter J. Phosphorylation of amyloid beta (Abeta) peptides – a trigger for formation of toxic aggregates in Alzheimer’s disease. Aging (Albany NY). 2011;3(8):803–812. | ||
Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430(7000):631–639. | ||
Sakono M, Zako T. Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J. 2010;277(6):1348–1358. | ||
Kim HJ, Chae SC, Lee DK, et al. Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. FASEB J. 2003;17(1):118–120. | ||
Takashima A. Amyloid-beta, tau, and dementia. J Alzheimers Dis. 2009;17(4):729–736. | ||
Dolan D, Troncoso J, Resnick SM, Crain BJ, Zonderman AB, O’Brien RJ. Age, Alzheimer’s disease and dementia in the Baltimore Longitudinal Study of Ageing. Brain. 2010;133 Pt 8:2225–2231. | ||
Dolan H, Crain B, Troncoso J, Resnick SM, Zonderman AB, O’Brien RJ. Atherosclerosis, dementia, and Alzheimer disease in the Baltimore Longitudinal Study of Aging cohort. Ann Neurol. 2010;68(2):231–240. | ||
O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185–204. | ||
Medina M, Avila J. The role of extracellular Tau in the spreading of neurofibrillary pathology. Front Cell Neurosci. 2014;8:113. | ||
Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004;11(3):213–228. | ||
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189. | ||
Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148(6):1204–1222. | ||
Turturici G, Sconzo G, Geraci F. HSP70 and its molecular role in nervous system diseases. Biochem Res Int. 2011;2011:618127. | ||
Mayer MP, Bukau B. HSP70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62(6):670–684. | ||
Liberek K, Lewandowska A, Zietkiewicz S. Chaperones in control of protein disaggregation. EMBO J. 2008;27(2):328–335. | ||
Evans CG, Wisen S, Gestwicki JE. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1–42) aggregation in vitro. J Biol Chem. 2006;281(44):33182–33191. | ||
Jinwal UK, Miyata Y, Koren J, 3rd, et al. Chemical manipulation of HSP70 ATPase activity regulates tau stability. J Neurosci. 2009;29(39): 12079–12088. | ||
Voss K, Combs B, Patterson KR, Binder LI, Gamblin TC. HSP70 alters tau function and aggregation in an isoform specific manner. Biochemistry. 2012;51(4):888–898. | ||
Cyr DM, Hohfeld J, Patterson C. Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem Sci. 2002; 27(7):368–375. | ||
Jiang J, Ballinger CA, Wu Y, et al. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of HSC70 as a target for ubiquitylation. J Biol Chem. 2001;276(46):42938–42944. | ||
Perez N, Sugar J, Charya S, et al. Increased synthesis and accumulation of heat shock 70 proteins in Alzheimer’s disease. Brain Res Mol Brain Res. 1991;11(3–4):249–254. | ||
Labrador-Garrido A, Bertoncini CW, Roodveldt C. The HSP70 chaperone system in Parkinson’s disease. In: Rana AQ, editor. Etiology and Pathophysiology of Parkinson’s disease. Available from: http://www.intechopen.com/books/etiology-and-pathophysiology-of-parkinson-s-disease/the-hsp70-chaperone-system-in-parkinson-s-disease. | ||
Mayer RJ, Arnold J, Laszlo L, Landon M, Lowe J. Ubiquitin in health and disease. Biochim Biophys Acta. 1991;1089(2):141–157. | ||
Fernandez-Estevez MA, Casarejos MJ, Lopez Sendon J, et al. Trehalose reverses cell malfunction in fibroblasts from normal and Huntington’s disease patients caused by proteosome inhibition. PLoS One. 2014;9(2): e90202. | ||
Hoshino T, Murao N, Namba T, et al. Suppression of Alzheimer’s disease-related phenotypes by expression of heat shock protein 70 in mice. J Neurosci. 2011;31(14):5225–5234. | ||
Kakimura J, Kitamura Y, Takata K, et al. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. 2002;16(6):601–603. | ||
Petrucelli L, Dickson D, Kehoe K, et al. CHIP and HSP70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13(7): 703–714. | ||
Fewell SW, Smith CM, Lyon MA, et al. Small molecule modulators of endogenous and co-chaperone-stimulated HSP70 ATPase activity. J Biol Chem. 2004;279(49):51131–51140. | ||
McCarty JS, Buchberger A, Reinstein J, Bukau B. The role of ATP in the functional cycle of the DnaK chaperone system. J Mol Biol. 1995;249(1): 126–137. | ||
Nadler SG, Eversole AC, Tepper MA, Cleaveland JS. Elucidating the mechanism of action of the immunosuppressant 15-deoxyspergualin. Ther Drug Monit. 1995;17(6):700–703. | ||
Brodsky JL. Selectivity of the molecular chaperone-specific immunosuppressive agent 15-deoxyspergualin: modulation of HSC70 ATPase activity without compromising DnaJ chaperone interactions. Biochem Pharmacol. 1999;57(8):877–880. | ||
Cogan EB, Birrell GB, Griffith OH. A robotics-based automated assay for inorganic and organic phosphates. Anal Biochem. 1999;271(1):29–35. | ||
Chang L, Bertelsen EB, Wisen S, Larsen EM, Zuiderweg ER, Gestwicki JE. High-throughput screen for small molecules that modulate the ATPase activity of the molecular chaperone DnaK. Anal Biochem. 2008;372(2):167–176. | ||
Miyata Y, Chang L, Bainor A, et al. High-throughput screen for Escherichia coli heat shock protein 70 (HSP70/DnaK): ATPase assay in low volume by exploiting energy transfer. J Biomol Screen. 2010;15(10):1211–1219. | ||
Zuck P, O’Donnell GT, Cassaday J, et al. Miniaturization of absorbance assays using the fluorescent properties of white microplates. Anal Biochem. 2005;342(2):254–259. | ||
Kang Y, Taldone T, Clement CC, et al. Design of a fluorescence polarization assay platform for the study of human HSP70. Bioorg Med Chem Lett. 2008;18(13):3749–3751. | ||
Ricci L, Williams KP. Development of fluorescence polarization assays for the molecular chaperone HSP70 family members: HSP72 and DnaK. Curr Chem Genomics. 2008;2:90–95. | ||
Wisen S, Gestwicki JE. Identification of small molecules that modify the protein folding activity of heat shock protein 70. Anal Biochem. 2008; 374(2):371–377. | ||
Thulasiraman V, Matts RL. Luciferase renaturation assays of chaperones and chaperone antagonists. Methods Mol Biol. 1998;102:129–141. | ||
Frydman J, Hartl FU. Principles of chaperone-assisted protein folding: differences between in vitro and in vivo mechanisms. Science. 1996; 272(5267):1497–1502. | ||
Frydman J, Nimmesgern E, Ohtsuka K, Hartl FU. Folding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones. Nature. 1994;370(6485):111–117. | ||
Schumacher RJ, Hurst R, Sullivan WP, McMahon NJ, Toft DO, Matts RL. ATP-dependent chaperoning activity of reticulocyte lysate. J Biol Chem. 1994;269(13):9493–9499. | ||
Thulasiraman V, Matts RL. Effect of geldanamycin on the kinetics of chaperone-mediated renaturation of firefly luciferase in rabbit reticulocyte lysate. Biochemistry. 1996;35(41):13443–13450. | ||
Wawrzynow A, Zylicz M. Divergent effects of ATP on the binding of the DnaK and DnaJ chaperones to each other, or to their various native and denatured protein substrates. J Biol Chem. 1995;270(33): 19300–19306. | ||
Ha JH, McKay DB. Kinetics of nucleotide-induced changes in the tryptophan fluorescence of the molecular chaperone HSC70 and its subfragments suggest the ATP-induced conformational change follows initial ATP binding. Biochemistry. 1995;34(36):11635–11644. | ||
Hawe A, Sutter M, Jiskoot W. Extrinsic fluorescent dyes as tools for protein characterization. Pharm Res. 2008;25(7):1487–1499. | ||
Bolder SG, Sagis LM, Venema P, van der Linden E. Thioflavin T and birefringence assays to determine the conversion of proteins into fibrils. Langmuir. 2007;23(8):4144–4147. | ||
Hudson SA, Ecroyd H, Kee TW, Carver JA. The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J. 2009;276(20):5960–5972. | ||
Zhang H Xu LQ, Perrett S. Studying the effects of chaperones on amyloid fibril formation. Methods. 2011;53(3):285–294. | ||
LeVine H 3rd. Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2(3):404–410. | ||
Nilsson MR. Techniques to study amyloid fibril formation in vitro. Methods. 2004;34(1):151–160. | ||
Eisert R, Felau L, Brown LR. Methods for enhancing the accuracy and reproducibility of Congo red and thioflavin T assays. Anal Biochem. 2006; 353(1):144–146. | ||
Krebs MR, Bromley EH, Donald AM. The binding of thioflavin-T to amyloid fibrils: localisation and implications. J Struct Biol. 2005; 149(1):30–37. | ||
Crowe A, Ballatore C, Hyde E, Trojanowski JQ, Lee VM. High throughput screening for small molecule inhibitors of heparin-induced tau fibril formation. Biochem Biophys Res Commun. 2007;358(1):1–6. | ||
Crowe A, Huang W, Ballatore C, et al. Identification of aminothienopyridazine inhibitors of tau assembly by quantitative high-throughput screening. Biochemistry. 2009;48(32):7732–7745. | ||
Patterson KR, Ward SM, Combs B, et al. Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry. 2011;50(47): 10300–10310. | ||
Khlistunova I, Pickhardt M, Biernat J, Wang Y, Mandelkow EM, Mandelkow E. Inhibition of tau aggregation in cell models of tauopathy. Curr Alzheimer Res. 2007;4(5):544–546. | ||
Khlistunova I, Biernat J, Wang Y, et al. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281(2):1205–1214. | ||
Santa-Maria I, Perez M, Hernandez F, Avila J, Moreno FJ. Characteristics of the binding of thioflavin S to tau paired helical filaments. J Alzheimers Dis. 2006;9(3):279–285. | ||
Lee LL, Ha H, Chang YT, DeLisa MP. Discovery of amyloid-beta aggregation inhibitors using an engineered assay for intracellular protein folding and solubility. Protein Sci. 2009;18(2):277–286. | ||
Fisher AC, Kim W, DeLisa MP. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 2006;15(3):449–458. | ||
Park SK, Pegan SD, Mesecar AD, Jungbauer LM, LaDu MJ, Liebman SW. Development and validation of a yeast high-throughput screen for inhibitors of Abeta(4)(2) oligomerization. Dis Model Mech. 2011;4(6):822–831. | ||
Hall AM, Roberson ED. Mouse models of Alzheimer’s disease. Brain Res Bull. 2012;88(1):3–12. | ||
Bryan KJ, Lee H, Perry G, Smith MA, Casadesus G. Transgenic mouse models of Alzheimer’s disease: behavioral testing and considerations. In: Buccafusco JJ, editor. Methods of Behavior Analysis in Neuroscience. Boca Raton, FL, USA: CRC Press; 2009. | ||
Hoshino T, Suzuki K, Matsushima T, Yamakawa N, Suzuki T, Mizushima T. Suppression of Alzheimer’s disease-related phenotypes by geranylgeranylacetone in mice. PLoS One. 2013;8(10):e76306. | ||
Miyata Y, Li X, Lee HF, et al. Synthesis and initial evaluation of YM-08, a blood-brain barrier permeable derivative of the heat shock protein 70 (HSP70) inhibitor MKT-077, which reduces tau levels. ACS Chem Neurosci. 2013;4(6):930–939. | ||
Miyata Y, Koren J, Kiray J, Dickey CA, Gestwicki JE. Molecular chaperones and regulation of tau quality control: strategies for drug discovery in tauopathies. Future Med Chem. 2011;3(12):1523–1537. | ||
Miyata Y, Rauch JN, Jinwal UK, et al. Cysteine reactivity distinguishes redox sensing by the heat-inducible and constitutive forms of heat shock protein 70. Chem Biol. 2012;19(11):1391–1399. | ||
Medina DX, Caccamo A, Oddo S. Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011;21(2):140–149. | ||
Wischik C BP, Wischik D, Seng K. Tau aggregation inhibitor (TAI) therapy with Rember™ arrests disease progression in mild and moderate Alzheimer’s disease over 50 weeks. Alzheimers Dement. 2008:T167. | ||
Oz M, Lorke DE, Hasan M, Petroianu GA. Cellular and molecular actions of methylene blue in the nervous system. Med Res Rev. 2011; 31(1):93–117. | ||
Abisambra J, Jinwal UK, Miyata Y, et al. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol Psychiatry. 2013;74(5):367–374. | ||
Wadhwa R, Sugihara T, Yoshida A, et al. Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the HSP70 family protein mot-2 and reactivation of p53 function. Cancer Res. 2000;60(24):6818–6821. | ||
Koya K, Li Y, Wang H, et al. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996;56(3): 538–543. | ||
Propper DJ, Braybrooke JP, Taylor DJ, et al. Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann Oncol. 1999;10(8):923–927. | ||
Wang AM, Miyata Y, Klinedinst S, et al. Activation of HSP70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol. 2013;9(2):112–118. | ||
O’Leary JC, 3rd, Li Q, Marinec P, et al. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. | ||
Marcinowski M, Rosam M, Seitz C, et al. Conformational selection in substrate recognition by HSP70 chaperones. J Mol Biol. 2013;425(3): 466–474. | ||
Shiber A, Breuer W, Brandeis M, Ravid T. Ubiquitin conjugation triggers misfolded protein sequestration into quality control foci when HSP70 chaperone levels are limiting. Mol Biol Cell. 2013;24(13): 2076–2087. | ||
Vanitha Thulasiraman RGF, Frydman J. Folding assays. In: Schneider C, editor. Methods in Molecular Biology. Chaperonin Protocols. Volume 140. Totowa, NJ, USA: Humana Press Inc; 2000. | ||
Chang CY. High Throughput Screens Against Heat Shock Protein 70 (HSP70), University of Michigan; 2011. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.