")
Back to Journals » OncoTargets and Therapy » Volume 14
Screening of Therapeutic Candidate Genes of Quercetin for Cervical Cancer and Analysis of Their Regulatory Network
Authors Li Y , Kou J, Wu T, Zheng P , Chao X
Received 20 October 2020
Accepted for publication 14 January 2021
Published 5 February 2021 Volume 2021:14 Pages 857—866
DOI https://doi.org/10.2147/OTT.S287633
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Takuya Aoki
Yuanyuan Li,1,2 Jiushe Kou,3 Tao Wu,4 Pengsheng Zheng,1 Xu Chao2
1Department of Reproductive Medicine, The First Affiliated Hospital, College of Medicine, Xi’an Jiaotong University, Xi’an, Shaanxi, 710061, People’s Republic of China; 2Scientific Research Department, The Second Affiliated Hospital, Shaanxi University of Chinese Medicine, Xixian New Area, Shaanxi, 712000, People’s Republic of China; 3Pain Department, The Second Affiliated Hospital, Shaanxi University of Chinese Medicine, Xixian New Area, Shaanxi, 712000, People’s Republic of China; 4College of Acupuncture and Massage, Shaanxi University of Chinese Medicine, Xixian New Area, Shaanxi, 712046, People’s Republic of China
Correspondence: Xu Chao
Scientific Research Department, The Second Affiliated Hospital of Shaanxi University of Chinese Medicine, 831 Longtaiguan Road, Xixian New Area, Shaanxi Province, 712000, People’s Republic of China
Email [email protected]
Pengsheng Zheng
Department of Reproductive Medicine, The First Affiliated Hospital of Xi’an Jiaotong University, 76 West Yanta Road, Xi’an, Shaanxi Province, 710061, People’s Republic of China
Tel +86-29-82657874
Email [email protected]
Purpose: To explore the therapeutic targets and regulatory mechanisms of the antitumor drug quercetin in the treatment of cervical cancer.
Methods: Cervical cancer (HeLa) cells were treated with quercetin and subjected to RNA sequencing using the BGISEQ-500 platform. By combined analysis of GEO database and RNA-seq results, the differentially expressed genes (DEGs) (namely, the genes in the GEO database that were upregulated/downregulated in cervical cancer compared with normal cervix and downregulated/upregulated after quercetin treatment) were identified. Functional enrichment and protein–protein interaction analyses were carried out for the DEGs. The candidate genes were identified using the Gentiscape2.2 and MCODE plug-ins for Cytoscape software, and the upstream miRNAs, lncRNAs, and circRNAs of the candidate genes were predicted using the online tools MirDIP, TarBase, and ENCORI. Finally, the regulatory network was constructed using Cytoscape software.
Results: Quercetin significantly inhibited the proliferation of cervical cancer cells. The combined analyses of the GEO database and RNA-seq results obtained 74 DEGs, and the functional enrichment analysis of the DEGs identified 861 biological processes, 32 cellular components, 50 molecular functions, and 56 KEGG pathways. Five therapeutic candidate genes, including EGFR, JUN, AR, CD44, and MUC1, were selected, and 10 miRNAs, 1 lncRNA, and 71 circRNAs upstream of these genes were identified. Finally, a lncRNA/circRNA-miRNA-mRNA-pathway regulatory network was constructed.
Conclusion: In this study, data mining was used to identify candidate genes and their regulatory network for the treatment of cervical cancer to provide a theoretical basis for targeted therapy of cervical cancer.
Keywords: quercetin, GEO, RNA-seq analysis, DEGs, candidate genes, regulatory network, cervical cancer
Introduction
Cervical cancer (CC) is a malignant tumor that seriously threatens women’s health, and the incidence and mortality of cervical cancer rank fourth among malignant tumors in women worldwide.1 From 2000 to 2015, the incidence and mortality of cervical cancer in China showed a slow-growth trend.2–4 Although traditional surgery, chemoradiotherapy, and adjuvant therapy have made great progress, many serious problems, such as recurrence, metastasis, and drug resistance, still face patients in the later stage of the disease. Vaccine immunization against human papillomavirus (HPV), which is the major risk factor for cervical cancer, has been developed and can prevent up to 70% of HPV-related cervical cancer cases;5 however, vaccines cannot prevent all subtypes of HPV infection, there are age limits and concerns about the safety of the vaccine, and many people have been infected with HPV, so cervical cancer is still a malignant disease harmful to women’s health. With the rapid development of tumor molecular biology, tumor molecular targeted therapy has been applied in clinical practice, and its importance in tumor therapy has been increasingly recognized.
Quercetin (Que) is a well-known plant flavonoid that is reported to have an anticancer effect in vitro and in vivo. The safety and potential utility of quercetin for the prevention and treatment of cancer has been demonstrated in animal experiments and clinical trials.6 Clemente-Soto et al reported that quercetin induced G2 phase cell cycle arrest and apoptosis with the activation of p53 in an E6 expression-independent manner in cervical cancer cells.7 We also found that quercetin significantly inhibited the proliferation of cervical cancer cells (HeLa and SiHa) and had dose-dependent effects. Since the antitumor effects of quercetin were well established, in this study, to explore the therapeutic molecular targets and regulatory mechanisms of cervical cancer, we treated HeLa cells with quercetin and then performed RNA-sequence (RNA-seq) analysis.
As an important component of bioinformatics, gene expression profiling has shown a great potential in cancer research, and widely used to study molecular changes involved in tumor progression, and discover new diagnostic, therapeutic and prognostic targets in various types of cancer. In this study, we downloaded four original human cervical cancer mRNA expression datasets from the NCBI-Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). By combined analysis of GEO and RNA-seq results, we identified differentially expressed genes (DEGs), analyzed functional enrichment and protein–protein interactions, found therapeutic candidate genes and their upstream miRNAs, lncRNAs, and circRNAs, and then constructed a regulatory network. This study might provide a reliable basis for exploring the regulatory mechanism and molecular targets of quercetin in the treatment of cervical cancer.
Materials and Methods
Cell Lines and Cell Culture
Human cervical cancer cell lines (HeLa and SiHa) were purchased from the American Type Culture Collection (ATCC, USA) and cultured in DMEM (Sigma-Aldrich, USA) supplemented with 10% FBS (HyClone, USA) at 37°C in an atmosphere containing 5% CO2.
IC50 and Cell Viability Assays
IC50 and cell viability assays were measured using a 3-(4,5-dimethylthiazole-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich) assay. IC50 assay: Cells in the logarithmic growth stage were trypsinized, neutralized, and then plated at a density of 5×103 cells per well in 96-well plates in 200 µL diluted media.
Quercetin was dissolved in DMSO at a concentration of 100 mM and then diluted to the appropriate dose (0, 6.25, 12.5, 25, 50, 100, or 200 µM; DMSO was used as a control) in the corresponding complete DMEM. Following the standard protocol, the 96-well plates were assessed at 24, 48, 72, and 96 hours by measuring the absorbance at 490 nm. Based on the data, we calculated the inhibition rate, performed curve fitting, and obtained the IC50 value.
Cell viability assay: Cells were plated at a density of 1–2×103 cells per well in 96-well plates in 200 µL diluted quercetin media. The plates were tested on days 1, 3, 5, and 7 by measuring the absorbance at 490 nm. Cell growth curves were generated to measure cell proliferation.
RNA Extraction, Quantification, and Sequencing
HeLa cells were cultured in DMSO (control) or 25 µM quercetin media for 3 days. When they reached 60%–80% confluency, the cells were harvested and washed twice with PBS. Total RNA was extracted and purified from 6 samples (control group: n=3, and quercetin group: n=3) using TRIzol Reagent (Invitrogen) and then quantified and sequenced using the BGISEQ-500 platform by BGI Company (China).
Microarray Datasets
A total of 4 human cervical cancer mRNA expression datasets (GSE7803,8 GSE9750,9 GSE63514,10 GSE52711) were selected and downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). All the datasets contained a comparison between normal cervix and cervical cancer tissue samples. The platform for GSE7803 is the GPL96 [HG-U133A] Affymetrix Human Genome U133A Array, which contains 10 normal cervical samples and 21 cervical cancer samples. The platform for GSE9750 is also GPL96, which contains 24 normal cervical epithelium samples and 33 primary tumor samples. The platform for GSE63514 is the GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array, which contains 24 normal specimens and 28 cancer specimens. The platform for GSE527 is the GPL355 Human 10K cDNA Array, which contains 8 normal cervix samples and 25 cervical cancer samples. The RNA-seq results of HeLa cells treated with quercetin were provided by BGI Company.
Differentially Expressed Gene (DEG) Screening
GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/), an online tool in GEO datasets, was used to analyze the differentially expressed genes between normal cervix and cervical cancer tissue samples. Genes were selected with the following criteria: |logFC| ≥2, P value <0.05, and adj.P. Val <0.05. Then, the significantly upregulated or downregulated genes in the four databases were combined, and duplicate genes were removed.
The criteria of differentially expressed gene screening for the RNA-seq results were as follows: PossionDis: |log2FC| ≥1, FDR ≤0.001, DESeq2: |log2FC| ≥0, and Q value ≤0.05.
Finally, combined with the GEO database and RNA-seq analysis results, we identified the genes in the GEO database that were upregulated/downregulated in cervical cancer compared with the normal cervix and downregulated/upregulated after treatment with quercetin. For convenience, we refer to the final identified differentially expressed genes as “DEGs” in the following text.
Functional Analysis of DEGs
The final screened DEGs were submitted to the Metascape (https://metascape.org/gp/index.html) database for enrichment analysis of Gene Ontology (GO, including biological process, cellular component, molecular function) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The enrichment criteria were as follows: min overlap ≥2, P value cutoff ≤0.05, min enrichment ≥1.5, and default values for the rest.
The Search Tool for the Retrieval of Interacting Genes (STRING, https://string-db.org/cgi/) was used to analyze and visualize interaction relationships between proteins. The final DEGs were uploaded to STRING with the following basic settings: active interaction sources were set as textmining, experiments, databases; minimum required interaction score ≥0.5; display simplification: hide disconnected nodes in the network.
Screening Therapeutic Candidate Genes of Quercetin
The gene sets with an interaction relationship were imported into Cytoscape software to build a visible network. First, the Gentiscape2.2 plug-in was used to calculate the network and topology characteristics of each node. The corresponding genes of nodes with a degree value ≥ mean+SD were hub genes, and those with a betweenness value ≥ mean+SD were bottleneck genes. With the MCODE plug-in, the parameters were set as degree cutoff ≥3, K-core ≥4, and default values for the rest, and then the core genes that constitute the stable structure of the network were identified. Finally, the ‘Calculate and draw custom Venn diagrams’ online tool (http://bioinformatics.psb.ugent.be/webtools/Venn/) was used to select the common genes among the hub genes, bottleneck genes, and core genes as therapeutic candidate genes.
Quantitative Real-Time PCR (qRT-PCR) Analysis
The above total RNA was reverse transcribed into complementary DNA (cDNA) using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific), which was used as a template for qRT-PCR amplification. The PCR mixtures were prepared to a final volume of 20 µL by using PowerUpTM SYBRTM Green Master Mix (Applied Biosystems), and three wells for each sample. PCR assay was performed using the ABI 7500 real-time PCR platform (ABI Life Technologies), and the cycling parameter as follows: 1 cycle of 50°C for 2 minutes, 1 cycle of 95°C for 2 minutes, 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Fold changes were calculated and normalized via the ΔΔCt method using GAPDH as the internal normalization control. Primers used were as follows: (5ʹ-3ʹ): EGFR (F: GCTTGCATTGATAGAAATGG, R: GTCGTCTATGCTGTCCTC); JUN (F: CAGGTGGCACAGCTTAAACA, R: CGCAACTTGTCAAGTTCTCA); AR (F: CCTGGCTTCCGCAACTTACAC; GGACTTGTGCATGCGGTACTC); CD44 (F: CGGACACCATGGACAAGTTT, CCGTCCGAGAGATGCTGTAG); MUC1 (F: GTGCCCCCTAGCAGTACCG, R: GACGTGCCCCTACAAGTTGG); and GAPDH (F: CAATGACCCCTTCATTGACC; R: GACAAGCTTCCCGTTCTCAG).
Statistical Analysis
Statistical analyses were performed using GraphPad Prism V8.3.0 software (La Jolla). The significance of the differences between control (DMSO) and quercetin group was determined by the Student’s t-test. All data were shown as the mean±SEM, and a value of p <0.05 was defined as statistically significant.
Upstream miRNAs, lncRNAs, and circRNAs Analysis of Candidate Genes
We submitted the candidate genes to the MirDIP database (http://ophid.utoronto.ca/mirDIP/index_confirm.jsp) to predict their upstream interacting miRNAs with the following parameters: score class, very high; integrated score ≥0.6. We then verified the authenticity online with the TarBase v.8 database (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=tarbasev8); then, the miRNAs were submitted to the ENCORI (http://starbase.sysu.edu.cn/index.php) online tool for analysis of upstream lncRNAs and circRNAs with the following parameters: Clip-data ≥5, Degradome-data ≥3, and default values for the rest.
Construction of the lncRNA/circRNA-miRNA-mRNA-Pathway Regulatory Network
The main pathways related to the candidate genes and the corresponding miRNAs, lncRNAs, and circRNAs that met the screening criteria were summarized. The file was established and imported into Cytoscape software to construct a visual lncRNA/circRNA-miRNA-mRNA-pathway regulatory network.
Results
Quercetin Inhibited the Proliferation of Cervical Cancer Cells
To confirm the antitumor effect of quercetin, HeLa and SiHa cells were treated with different concentrations of quercetin, and MTT assays were used to detect cell proliferation. The results showed that quercetin significantly inhibited the proliferation of cervical cancer cells (P <0.001) and had a dose-dependent effect (Figure 1A and B).
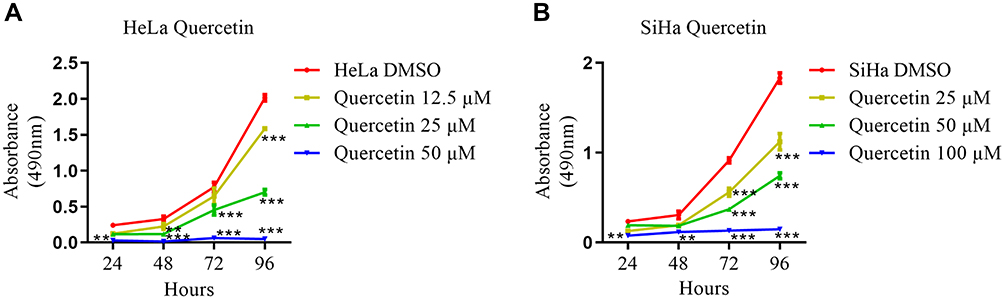 |
Figure 1 Quercetin inhibited the proliferation of cervical carcinoma cells. (A) HeLa cells were treated with DMSO (vehicle control) or 12.5, 25, or 50 µM quercetin, and an MTT assay was used to assess cell proliferation. (B) SiHa cells were treated with DMSO (vehicle control) or 25, 50, or 100 µM quercetin, and an MTT assay was used to assess cell proliferation. Values are shown as the mean±SEM (two-way ANOVA, **p <0.01, ***p <0.001 vs the corresponding control). |
Identification of DEGs in the Treatment of Cervical Cancer with Quercetin
Four cervical cancer raw microarray datasets (GSE7803, GSE9750, GSE63514, and GSE527) were analyzed in GEO online, and the results showed that there were 1887 differentially expressed genes between normal cervix and cervical cancer tissue samples, including 698 upregulated genes and 1189 downregulated genes.
Because quercetin significantly inhibited the proliferation of cervical cancer cells, we treated HeLa cells with quercetin and then performed RNA-seq analysis. A total of 1122 differentially expressed genes were identified, including 688 upregulated genes and 434 downregulated genes.
We combined the results of the GEO database and RNA-seq analyses and identified the upregulated/downregulated genes in the GEO database in cervical cancer compared with normal cervix; we also identified the genes that showed the opposite changes in expression after quercetin treatment as downregulated/upregulated genes. Finally, as shown in Table 1, we identified 74 differentially expressed genes (DEGs), including 21 that were upregulated in GEO but downregulated after treatment with quercetin and 53 that were downregulated in GEO but upregulated after treatment with quercetin.
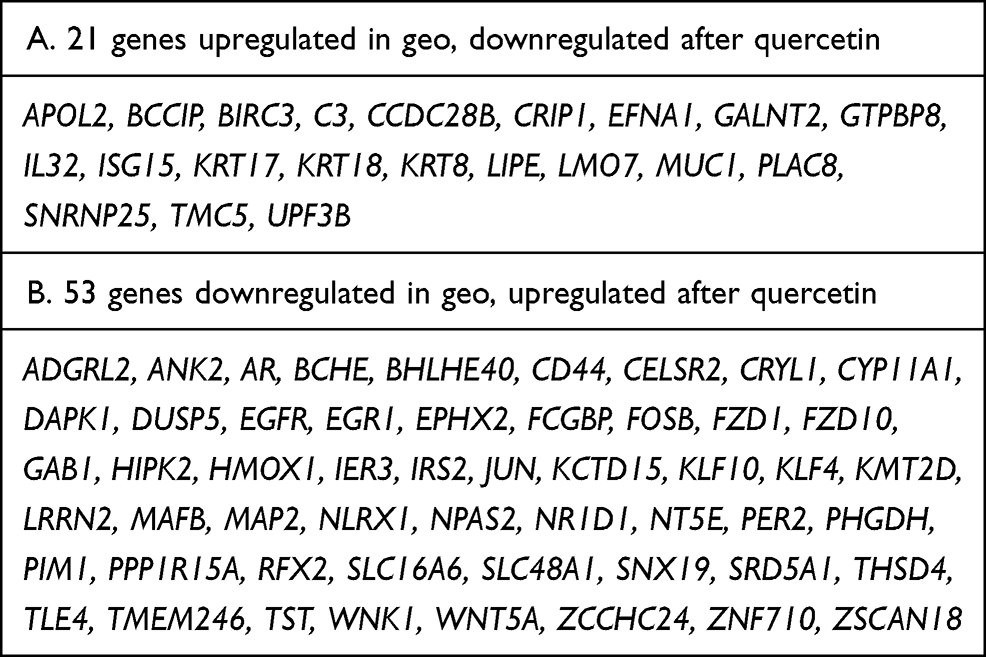 |
Table 1 Identifying 74 Differentially Expressed Genes (DEGs) by Combining the GEO Database and RNA-Seq Analyses |
GO Function and KEGG Pathway Enrichment Analysis of DEGs
The identified DEGs were analyzed in the Metascape database for enrichment analysis of Gene Ontology (GO, including biological process, cellular component, molecular function) and KEGG pathways.
There were 861 enriched biological processes, 32 enriched cellular components, and 50 enriched molecular functions. As shown in Figure 2, the top 20 clusters of biological processes with their representative enriched terms included circadian rhythm, apoptotic signaling pathway, gland development, positive regulation of vasculature development, fat cell differentiation, and regulation of DNA-binding transcription factor activity, etc. (Figure 2A). The top 8 clusters of cellular components included transcription factor complex, costamere, membrane raft, apical plasma membrane, perinuclear region of cytoplasm, and azurophil granule lumen (Figure 2B). The top 15 clusters of molecular functions included transcription factor binding, transcription cofactor binding, DNA-binding transcription activator activity, cofactor binding, Wnt-activated receptor activity, ATPase binding, and phosphatase binding (Figure 2C).
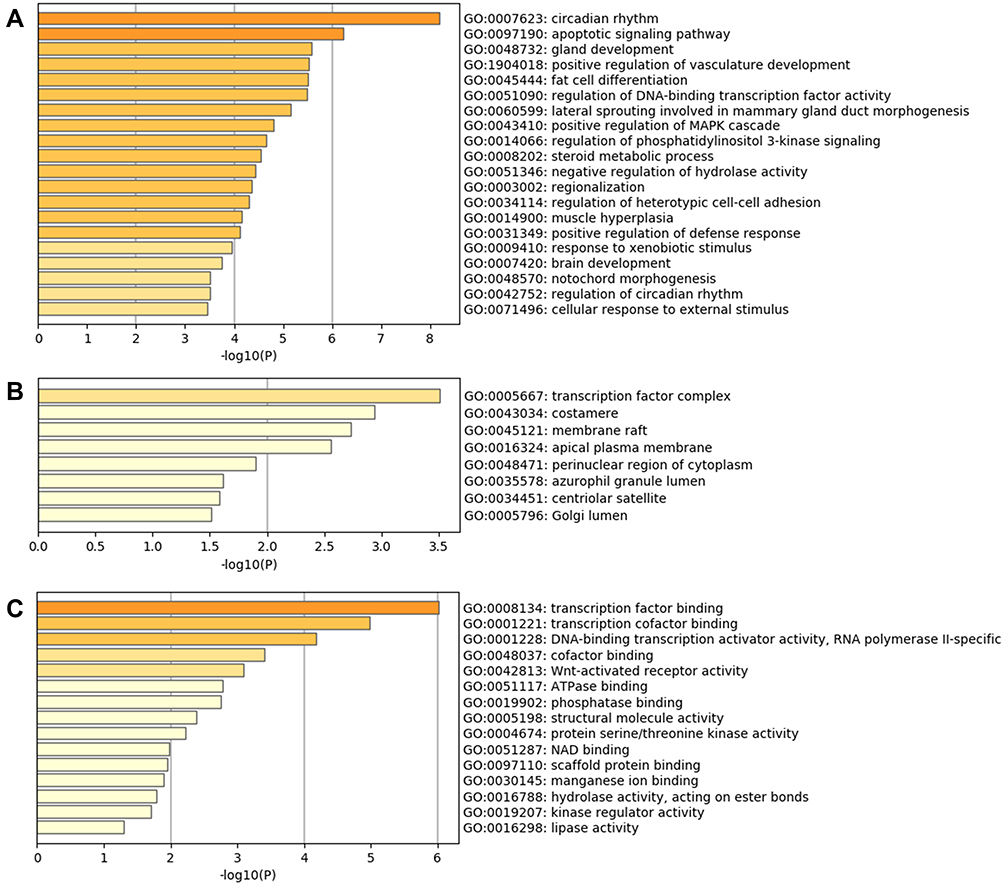 |
Figure 2 Gene Ontology (GO) enrichment analysis results of DEGs. (A) Top 20 clusters of biological processes with their representative enriched terms. (B) Top 8 clusters of cellular components with their representative enriched terms. (C) Top 15 clusters of molecular functions with their representative enriched terms. |
Furthermore, 56 KEGG pathways were enriched, and the top 8 clusters with their representative enriched terms are shown in Figure 3, including circadian rhythm, proteoglycans in cancer, microRNAs in cancer, ErbB signaling pathway, AGE-RAGE signaling pathway in diabetic complications, and regulation of lipolysis in adipocytes.
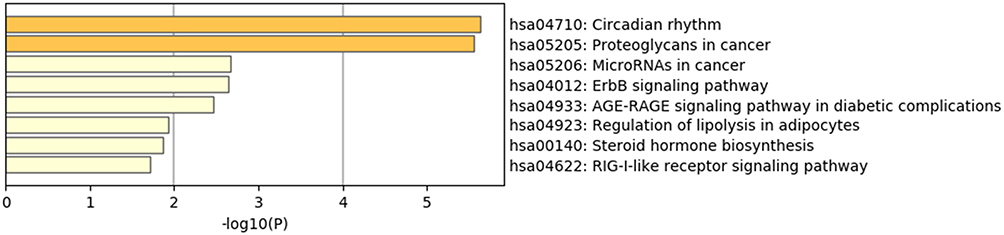 |
Figure 3 KEGG pathway enrichment analysis results of DEGs. |
the STRING online tool was used to analyze and visualize the protein–protein interaction relationships of the DEGs. According to the set parameter criteria, 32 proteins without interaction were hidden, and a protein interaction network with 65 edges was constructed from the remaining 42 node proteins (Figure 4).
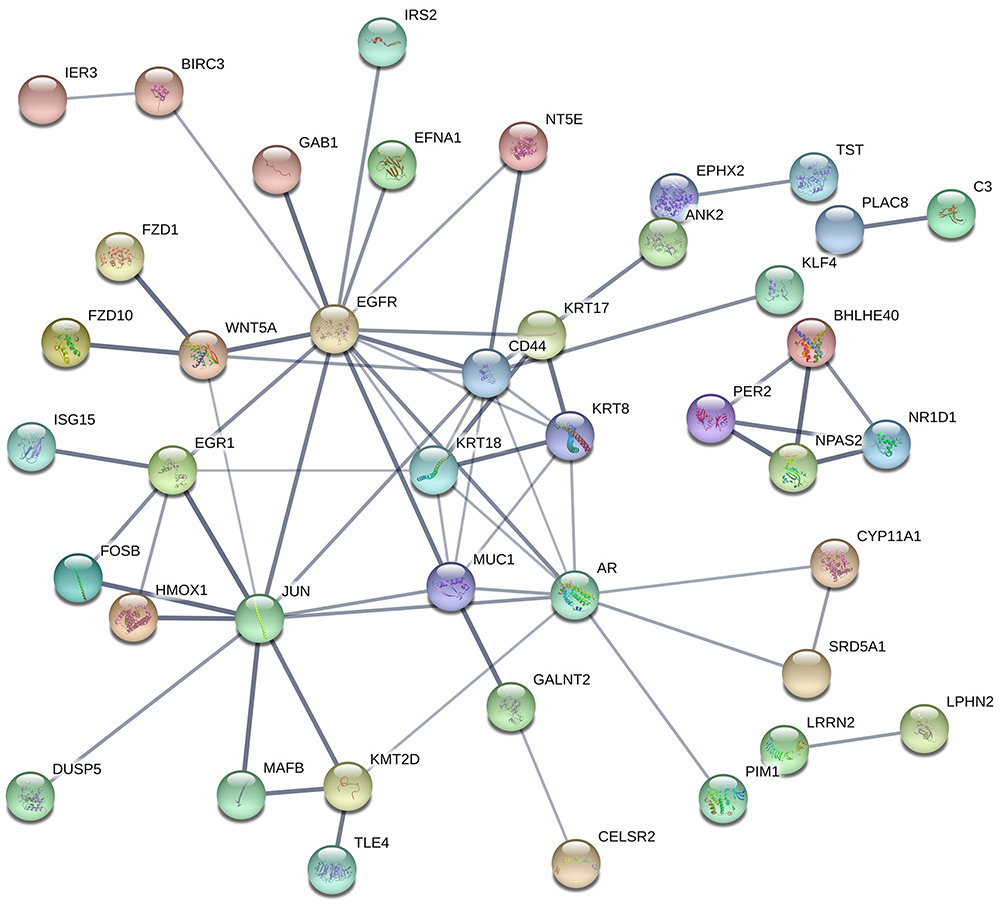 |
Figure 4 Protein–protein interaction analysis results of DEGs. |
Identification of Therapeutic Candidate Genes and Analysis of Upstream miRNAs, lncRNAs, and circRNAs
The above protein–protein interaction network was imported into Cytoscape software, and with the MCODE plug-in, 7 core genes that constitute the stable structure of the network were identified (Figure 5A, Table 2). The Gentiscape2.2 plug-in was used and identified 6 hub genes and 5 bottleneck genes (Table 2). The common core genes, hub genes, and bottleneck genes, which are critical to the network, were considered candidate genes and included epidermal growth factor receptor (EGFR), Jun proto-oncogene (JUN), androgen receptor (AR), cluster of differentiation 44 (CD44), and mucin 1 (MUC1) (Figure 5B, Table 2). We also used a qRT-PCR assay to detect the RNA expression level of candidate genes in HeLa control and quercetin group. The results showed that the expression of four genes (EGFR, AR, CD44, MUC1) matched the RNA-seq analysis, and the expression of JUN had no significant difference between the two groups (Figure 5C). About the targeting of quercetin to JUN gene, the literature reported that quercetin increased the expression of c-Jun and p-c-Jun in a dose-dependent manner in prostatic cell lines.12
 |
Table 2 Screening Results of Therapeutic Candidate Genes |
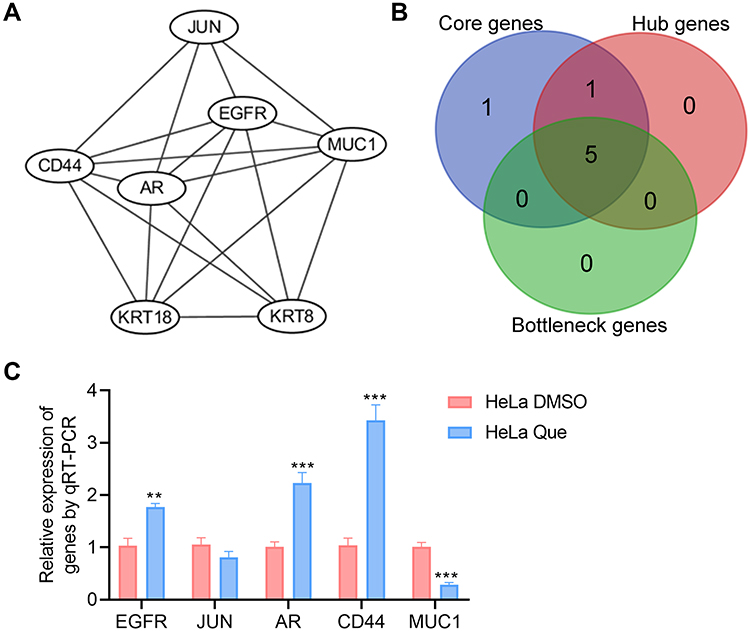 |
Figure 5 Screening of therapeutic candidate genes. (A) Interaction diagram of the core genes; (B) Venn analysis of the core genes, hub genes, and bottleneck genes; (C) Treated HeLa cells with DMSO (vehicle control) and 25 µM quercetin, the RNA expression of genes were detected using qRT-PCR assay, and the statistical analysis was shown. Student's t-test was used for the statistical analysis, **p<0.01, ***p<0.001. |
After the 5 candidate genes were submitted to the MirDIP database to predict their upstream interacting miRNAs and verified for authenticity in TarBase v.8 online, 10 miRNAs were identified (Table 3). Then, the above miRNAs were submitted to the ENCORI online tool for analysis of their upstream lncRNAs and circRNAs, and 2 miRNA–lncRNA relationship pairs and 174 miRNA–circRNA relationship pairs were obtained. After the deletion of duplicate genes, a total of 1 lncRNA and 71 circRNAs were identified (Table 3).
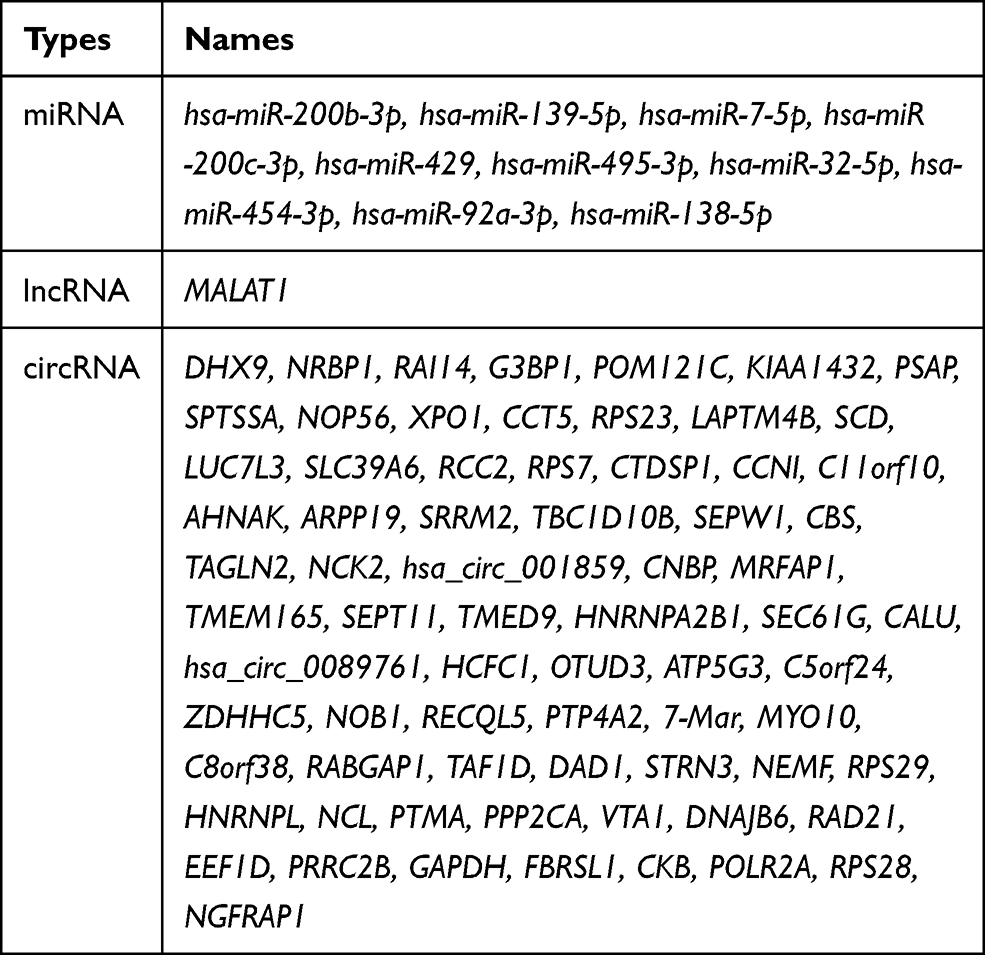 |
Table 3 Analysis of the Upstream Interacting miRNAs, lncRNA, and circRNAs of Candidate Genes |
lncRNA/circRNA-miRNA-mRNA-Pathway Regulation Network
The candidate genes and their related KEGG pathways, as well as all the interaction relationships involved in the upstream miRNAs, lncRNA, and circRNAs, were imported into Cytoscape software to construct a visualization of the lncRNA/circRNA-miRNA-mRNA-pathway regulatory network, which included a total of 90 nodes (4 candidate genes, 4 KEGG pathways, 10 miRNAs, 1 lncRNA, and 71 circRNAs) and 116 pathways (Figure 6).
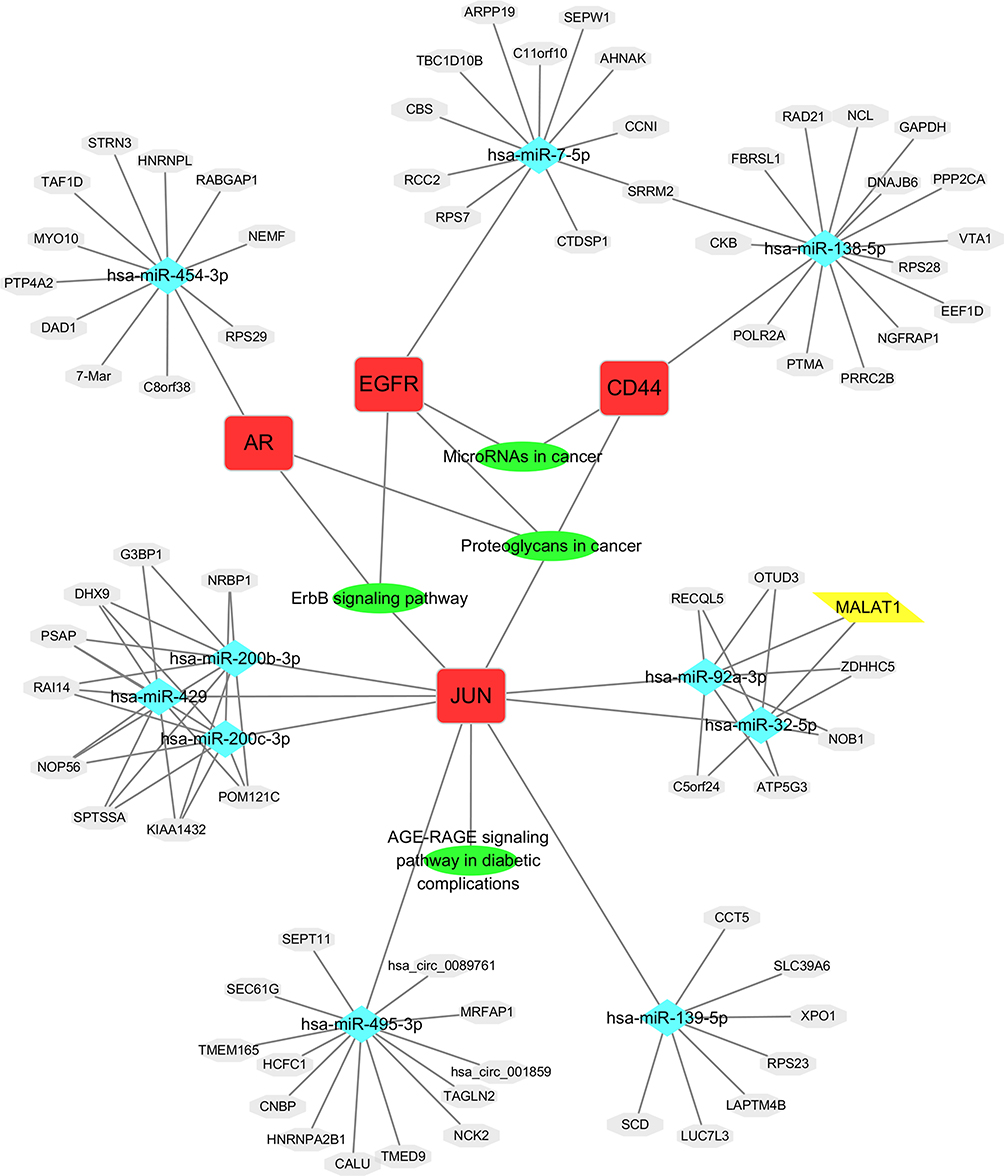 |
Figure 6 The lncRNA/circRNA-miRNA-mRNA-pathway regulatory network. |
Discussion
At present, biomedicine has entered the era of big data. With the help of massive datasets from multi-omics at multiple levels of biomedicine, it is possible to study the functional mechanisms of organisms in unprecedented breadth and depth. Because biomedical data are often scattered and complex, bioinformatics, which is based on massive data mining and analysis, has been widely used in the process of seeking diagnostic and therapeutic targets of various diseases (including tumors) in applications such as identifying the etiology, clinical diagnostic markers, and targets of disease as well as discovering and predicting the functions of key molecules and regulatory mechanisms.
GEO and The Cancer Genome Atlas (TCGA) database are the most well-known public biomedical databases. In TCGA, there were 296 cervical cancer samples and 3 normal samples, which was too few, so we chose the GEO database for analysis. The GEO database includes GSE7803, GSE9750, GSE63514, GSE527, and GSE46857.13 There were 25 cervical cancer samples and 4 normal samples in the GSE46857 dataset; thus, the number of the normal group was less than 5 in this dataset. Additionally, analysis of a few of the differentially expressed genes, such as ARMC8, H6PD, and TTN, revealed contradictory results in which the expression in the control group was higher or lower than that in the cervical cancer group, so the GSE46857 dataset was excluded.
With the Cytoscape software Gentiscape2.2 and MCODE plug-ins, we identified 5 candidate genes (EGFR, JUN, AR, CD44, MUC1) that were differentially expressed after quercetin treatment in cervical cancer cells and predicted the upstream miRNAs of the candidate genes using the MirDIP and TarBase online tools. According to the screening criteria and verification results, we did not find an upstream miRNA of MUC1 (Figure 6), but we cannot rule out the possibility that MUC1 is targeted in cervical cancer. Many works in the literature have reported that the candidate genes identified in this study play important roles in the treatment of tumors. MUC1 induced cancer stem cell (CSC) enrichment in paclitaxel-resistant cells via activation of EGFR, which enhanced IL-6 transcription, and this effect was eliminated by erlotinib, suggesting a novel strategy to treat paclitaxel-resistant cervical cancer.14 EGFR is highly expressed in cervical cancer tissues,15 and EGFR activation is associated with poor prognosis in cervical cancer patients.16 EGFR tyrosine kinase inhibitors (TKIs) are widely used for the treatment of cancers, such as lung cancer,17 non-small cell lung cancer,18 and colorectal cancer,19 especially in patients with EGFR mutations. Detection of AR splice variant 7 is associated with worse outcomes for patients with castration-resistant prostate cancer who are treated with AR signaling (ARS) inhibitors.20 Quercetin dramatically induced the protein expression of c-Jun which in turn inhibited the AR function.12 Our results showed that the RNA expression of JUN was increased in SiHa cells after treated with DMSO and 50 µM quercetin (data not shown). Therefore, the targeting of quercetin to JUN gene may be affected by cell type and state. The presence of stem cell proteins (such as SOX2) and the loss of CD44 independently predict reduced locoregional control in locally advanced cervical cancer.21 We also identified upstream lncRNAs and circRNAs of miRNAs using the ENCORI online tool, imported all relationships into Cytoscape software, and constructed a visual lncRNA/circRNA-miRNA-mRNA-pathway regulatory network.
Funding
This Work was supported by the following grants: (1) Natural Science Basic Research Program of Shaanxi Province (No.2019JQ-871); (2) National Natural Science Foundation of China (No.81904310); (3) Horizontal Subject of Postdoctoral Joint Training (No.202101139); and (4) Subject Innovation Team of the Second Affiliated Hospital of Shaanxi University of Chinese Medicine (2020XKTD-A03).
Disclosure
All authors declare that they have no conflicts of interest in the article.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Chen W, Zheng R, Zhang S, et al. Cancer incidence and mortality in China, 2013. Cancer Lett. 2017;401:63–71. doi:10.1016/j.canlet.2017.04.024
3. Chen W, Sun K, Zheng R, et al. Cancer incidence and mortality in China, 2014. Chinese J Cancer Res. 2018;30(1):1–12. doi:10.21147/j.issn.1000-9604.2018.01.01
4. Zheng RT, Sun KT, Zhang ST, et al. Report of cancer epidemiology in China, 2015. Chin J Oncol. 2019;41(1):19–28. doi:10.3760/cma.j.issn.0253-3766.2019.01.005
5. Zhang S, Batur P. Human papillomavirus in 2019: an update on cervical cancer prevention and screening guidelines. Cleve Clin J Med. 2019;86(3):173–178. doi:10.3949/ccjm.86a.18018
6. Jana N, Bretislav G, Pavel S, Pavla U. Potential of the flavonoid quercetin to prevent and treat cancer - current status of research. Klin Onkol. 2018;31(3):184–190. doi:10.14735/amko2018184
7. Clemente-Soto AF, Salas-Vidal E, Milan-Pacheco C, Sanchez-Carranza JN, Peralta-Zaragoza O, Gonzalez-Maya L. Quercetin induces G2 phase arrest and apoptosis with the activation of p53 in an E6 expression-independent manner in HPV-positive human cervical cancer-derived cells. Mol Med Rep. 2019;19(3):2097–2106. doi:10.3892/mmr.2019.9850
8. Zhai Y, Kuick R, Nan B, et al. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007;67(21):10163–10172. doi:10.1158/0008-5472.CAN-07-2056
9. Scotto L, Narayan G, Nandula SV, et al. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosomes Cancer. 2008;47(9):755–765. doi:10.1002/gcc.20577
10. den Boon JA, Pyeon D, Wang SS, et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: role of stromal estrogen receptor signaling. Proc Natl Acad Sci U S A. 2015;112(25):E3255–3264. doi:10.1073/pnas.1509322112
11. Wong YF, Selvanayagam ZE, Wei N, et al. Expression genomics of cervical cancer: molecular classification and prediction of radiotherapy response by DNA microarray. Clin Cancer Res. 2003;9(15):5486–5492.
12. Yuan H, Pan Y, Young CY. Overexpression of c-Jun induced by quercetin and resverol inhibits the expression and function of the androgen receptor in human prostate cancer cells. Cancer Lett. 2004;213(2):155–163. doi:10.1016/j.canlet.2004.04.003
13. Thomas A, Mahantshetty U, Kannan S, et al. Expression profiling of cervical cancers in Indian women at different stages to identify gene signatures during progression of the disease. Cancer Med. 2013;2(6):836–848. doi:10.1002/cam4.152
14. Lv Y, Cang W, Li Q, et al. Erlotinib overcomes paclitaxel-resistant cancer stem cells by blocking the EGFR-CREB/GRbeta-IL-6 axis in MUC1-positive cervical cancer. Oncogenesis. 2019;8(12):70. doi:10.1038/s41389-019-0179-2
15. Yan J, Zhang Y, Ren C, Shi W, Chen L. Involvement of nuclear protein C23 in activation of EGFR signaling in cervical cancer. Tumour Biol. 2016;37(1):905–910. doi:10.1007/s13277-015-3889-x
16. Peng Z, Wang Q, Zhang Y, He J, Zheng J. EBP50 interacts with EGFR and regulates EGFR signaling to affect the prognosis of cervical cancer patients. Int J Oncol. 2016;49(4):1737–1745. doi:10.3892/ijo.2016.3655
17. Offin M, Rizvi H, Tenet M, et al. Tumor mutation burden and efficacy of EGFR-tyrosine kinase inhibitors in patients with EGFR-mutant lung cancers. Clin Cancer Res. 2019;25(3):1063–1069. doi:10.1158/1078-0432.CCR-18-1102
18. Masood A, Kancha RK, Subramanian J. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in non-small cell lung cancer harboring uncommon EGFR mutations: focus on afatinib. Semin Oncol. 2019;46(3):271–283. doi:10.1053/j.seminoncol.2019.08.004
19. Chan DLH, Segelov E, Wong RS, et al. Epidermal growth factor receptor (EGFR) inhibitors for metastatic colorectal cancer. Cochrane Database Syst Rev. 2017;6:CD007047. doi:10.1002/14651858.CD007047.pub2
20. Bastos DA, Antonarakis ES. CTC-derived AR-V7 detection as a prognostic and predictive biomarker in advanced prostate cancer. Expert Rev Mol Diagn. 2018;18(2):155–163. doi:10.1080/14737159.2018.1427068
21. Chopra S, Deodhar K, Pai V, et al. Cancer stem cells, CD44, and outcomes following chemoradiation in locally advanced cervical cancer: results from a prospective study. Int J Radiat Oncol Biol Phys. 2019;103(1):161–168. doi:10.1016/j.ijrobp.2018.09.003
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.