")
Back to Journals » International Journal of General Medicine » Volume 14
Screening of Breast Cancer Methylation Biomarkers Based on the TCGA Database
Authors Wang X, Jia J, Gu X, Zhao W, Chen C, Wu W, Wang J, Xu M
Received 1 June 2021
Accepted for publication 9 November 2021
Published 16 December 2021 Volume 2021:14 Pages 9833—9839
DOI https://doi.org/10.2147/IJGM.S322857
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Xuechun Wang,1,* Jia Jia,2,* Xuehong Gu,3 Wei-wei Zhao,4 Caiping Chen,5 Wanxin Wu,6 Jiayuan Wang,1 Midie Xu7
1Department of Laboratory, Jiaxing First Hospital, Jiaxing, 314000, People’s Republic of China; 2Shanghai Center for Bioinformation Technology, Shanghai, 201202, People’s Republic of China; 3Department of Nursing, Jiaxing First Hospital, Jiaxing, 314000, People’s Republic of China; 4Department of Rehabilitation Medicine, Jiaxing First Hospital, Jiaxing, 314000, People’s Republic of China; 5Department of Breast Surgery, Jiaxing First Hospital, Jiaxing, 314000, People’s Republic of China; 6Department of Pathology, Jiaxing First Hospital, Jiaxing, 314000, People’s Republic of China; 7Department of Pathology, Fudan University Shanghai Cancer Center, Shanghai, 200032, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xuehong Gu
Department of Nursing, Jiaxing First Hospital, No. 1882 of Central South Road, Jiaxing, 314000, People’s Republic of China
Tel +86 13957368443
Fax +86 573-83599079
Email [email protected]
Objective: Breast cancer has become a fatal disease for women world-wide. Its incidence in China has been increasing yearly, and the identification of early-stage biomarkers is urgently required.
Methods: ANOVA was carried out in the case of a primary tumor, adjacent normal tissue, and tumor metastasis of breast cancer, and on pan-cancer samples using the genome-wide methylation data of 31 solid tumor Illumina Methylation 450K chips downloaded from The Cancer Genome Atlas (TCGA) website in September 2018. Methylation sites showing a significant difference (P ≤ 0.05) were screened and compared with the whole-genome methylation data of 31 other solid tumor species in the TCGA database using t-tests in order to screen the methylation sites of breast cancer-specific expression. The expression of the screened methylation sites was confirmed through pyrosequencing in 45 cases of breast cancer, lung cancer, gastric cancer, and colorectal cancer.
Results: A total of 10 specific breast cancer methylation sites (cg13683194, cg07996594, cg21646032, cg07671949, cg21185686, cg03625109, cg16429070, cg23601468, cg24818566, and cg01240931) were analyzed; nine genes (C9orf125, RARB, ESR1, RUNX3, PCDHGB7, DBC1, PDGFRB, TIMP3, and APC) were involved. The overall effect was excellent; a total of 4 methylation sites (2 in the DBC1 gene [cg03625109 and cg24818566], 1 in the C9orf125 gene [cg13683194], and 1 in the PDGFRB gene [cg16429070]) could effectively distinguish breast cancer from 31 other cancer species. The pyrosequencing results revealed that 7 screened methylation sites could significantly distinguish between breast cancer, lung cancer, gastric cancer, and colorectal cancer samples; these sites could also specifically distinguish between luminal A, luminal B, HER2, and Basal-like types of breast cancer.
Conclusion: The 10 breast cancer methylation sites screened in the present study can effectively distinguish breast cancer from 31 other solid tumors, and they are expected to be used as biomarkers for early screening of breast cancer.
Keywords: breast cancer, Basal-like breast cancer, methylation, biomarker, TCGA, ANOVA
Introduction
The breast cancer mortality rate in women around the world is high.1 In recent years, breast cancer has become the most common cancer among Chinese women, accounting for 12.2% of global cases and 9.6% of deaths caused by cancer.2 Early detection of breast cancer is closely related to the disease prognosis. The continuously high number of patients with breast cancer worldwide indicates an urgent need for early detection biomarkers. Epigenetic change, including DNA methylation, is one of the most common molecular changes in human tumor formation; breast cancer is no exception to this.3
DNA methylation is a reversible process of changing gene expression patterns without altering the DNA sequence.4 Hypomethylation and hypermethylation are related to breast cancer;4 when compared with adjacent tissue, hypomethylation in tumors and metastasis tissues is usually found to increase oncogene expression, activate transcription, and then change genome stability.4 A CpG island is located in the promoter region of the tumor suppressor gene, which is usually unmethylated in normal cells.
Mammalian DNA methylation levels over the course of a mammal’s life vary significantly: in the first few divisions of a fertilized egg, demethylases remove almost all methylation markers inherited from the parent DNA molecule.5 When the embryo develops in the uterus, a new kind of methylation occurs within the genome, builds sex that DNA methylation enzyme to establish a new mode of methylation. Once new methylation patterns are built in the cell, they are passed to all cells in the DNA molecule by maintaining the methylation enzyme. This explains why imprinting is neither a mutation nor a permanent change.5
Imprinting is reversible; it only lasts for the lifetime of the individual. In the next generation of individual gamete formation, the old gene imprinting is eliminated, and new gene imprinting occurs.5 Thus, one of the molecular mechanisms of genetic imprinting may be DNA methylation.5 In the study of genetic imprinting and tumors, it has also been found that the tumor suppressor gene P16 is inactivated by methylation; meanwhile, demethylation can restore the original characteristics of the gene.6 Abnormal methylation may be an important cause of tumor formation,6 suggesting that DNA methylation modification has a wide range of functions.6
Methylated DNA can undergo demethylation. DNA demethylation is regulated by segments within genes and the factors that bind to them.7 There are two hypotheses explaining the molecular mechanisms of DNA demethylation.7
The first hypothesis is associated with semi-reserved DNA replication, which is called passive demethylation.7 If the DNA is not methylated after semi-preserved replication, it is in the semi-methylated state.7 If the semi-preserved DNA replication occurs again and the DNA methylation activity is still inhibited, 50% of the cells are in the semi-methylated state.
The second hypothesis involves an active process unrelated to semi-reserved replication.7 DNA demethylation is catalyzed by DNA demethylase. DNA demethylation is a reaction in which methylated bases are removed under the action of DNA glycosidase; this is equivalent to the repair reaction of damaged DNA catalyzed by the cleavage and coupling of glycosidase and nuclease without a base.7 5-methylcytosine glycosylase is an in vivo candidate demethylase. In addition, methylated CpG binding proteins, such as MBD2, also exhibit demethylase activity.7
During cell development, various epigenetic phenomena are not isolated, but are closely related.7 The DNA methylation and histone methylation co-regulation of gene expression was first demonstrated in Neurospora Crassa; further biochemical studies have shown that DNA methylation is regulated by histone methylation. Mammal studies have found that DNA methylation is the basis for establishing and maintaining other epigenetic phenomena, such as the recruitment of inhibitory complexes (eg, histone deacetylases at DNA methylation sites and the removal of histone acetylation markers near the sites). DNA methylation is also believed to be regulated by histone modification;7 it has been reported that histone modification of H3K9me can promote the process of DNA methylation.7
However, in cancer cells, abnormal hypermethylation of these promoter regions is involved in the transcriptional silencing of tumor suppressor genes;4 these epigenetic changes occur in the early stage of cancerization in normal tissue and eventually lead to the development of breast cancer.5 The reported specific methylation genes of breast cancer include ACADL, DAMTSL1, CAV1, NPY, PTGS2, UNX3, BRCA1, ATM, IGF2, CDH1, ESR1, SYK, TIMP3, RARB, APC, RASSF1A, GSTP1, SFN, CST6, DAPK, and AKT1.4,6–9 These gene-specific DNA methylation changes are expected to be used as biomarkers for early breast cancer detection.
In the present study, the whole-genome methylation chip data of patients with breast cancer in the TCGA database, combined with methylation genes related to breast cancer reported in relevant literature, were used.10 A total of 10 methylation sites in nine genes were analyzed and screened before they were used in early breast cancer screening.
Information and Methods
Acquisition and Analysis of TCGA Solid Tumor Methylation Data
In the present study, the data of 450k Illumina human whole-genome methylation chips and phenotypes from 15 solid tumors were comprehensively analyzed; the data were downloaded from the TCGA database in September 2018 (https://portal.gdc.cancer.gov/). The methylation level of all probes on the chip was expressed as the β value (range = 0–1), which represented unmethylation and complete methylation, respectively. The R packages TCGA biolinks, dplyr, DT, and Summarized Experience were used for data download and analysis. ANOVA was used for the site annotation and differential analysis.
Tumor Samples
A total of 45 surgical tumor samples were clinically selected from our hospital. Among these samples, 15 were of breast cancer (luminal A, luminal B, HER2, and Basal-like breast cancer), 10 were of lung cancer, 10 were of gastric cancer, and 10 were of colorectal cancer.
Candidate Breast Cancer-Specific Expression Methylation Sites
The breast cancer samples were divided into three categories according to their phenotype information: in situ tumor, metastatic tumor, and normal tissue. These were then analyzed using the one-way ANOVA procedure, and a stepwise polynomial regression analysis procedure was used for all the variables.
The assessment criteria of statistical significance were determined in accordance with the Benjamini–Hochberg (BH) False Discovery Rates (FDR), and probe identifiers (cg numbers) (probe cg ID) with a BH FDR P-value of ≤0.05 were selected and used to annotate the candidate breast cancer methylation gene. The R aov function was used for analysis.
The probe cg ID on the corresponding gene was selected and ordered from small to large according to the P-value. The first 100 probe cg IDs were selected as the candidate methylation sites of breast cancer. Next, the methylation data of 100 probe cg IDs corresponding with 31 other solid tumor species were further evaluated using one-way ANOVA and BH FDR correction step in order to screen out specific methylation sites of breast cancer, thus distinguishing breast cancer from other solid tumors.
A layer of screening was performed to select discriminating breast cancer biomarkers. First, the selected methylation site was required to be capable of significantly differentiating breast tumors from at least 6 tumors with a P-value of ≤0.005. A total of 48 probe cg IDs were selected during this step, and indifferently expressed methylation sites existing in >3 tumors with a P-value of ≥0.05 were filtered during the second step. Only 14 probe cg IDs were left after the filtering. Finally, only 10 methylated probe sites were successfully designed, tested, and used for subsequent experiments. The Database for Annotation, Visualization and Integrated Discovery 6.8 (https://david.ncifcrf.gov/) was used for gene ontology (GO) and the pathway analysis of genes involved in methylation sites.
Pyrosequencing
Specific polymerase chain reaction (PCR) primers were designed to target the screened methylation sites, and the genomic DNA of tumor FFPE samples was extracted using the QIAGEN Qiaamp DNA FFPE Tissue Kit (QIAGEN, 56404). The DNA was then methylated using the QiagenEpitect Bisulfite Kit (Qiagen, 59104). The methylation site primers were designed using PyroMark Assay Design 2.0. The DNA was amplified using PCR and then detected via pyrosequencing.
Statistical Analysis
A self-programmed R script (R version 3.5.3) was used for the statistical analysis.
Results
Specific Breast Cancer Methylation Site
All genome methylation data on the primary tumor, adjacent normal tissue, and tumor metastasis of breast cancer were analyzed. The methylation sites with an ANOVA P-value of ≤0.05 were selected. The methylation probes on the corresponding gene were selected after site annotation and ordered from small to large according to the P-value. The first 100 methylation probes were selected as the differential expression of breast cancer methylation sites. The 100 methylation probes involved 25 genes (Supplementary Table 1). The main GO types and signaling pathways (P ≤ 0.05) of genes involved in the differential expression of breast cancer methylation sites are shown in Supplementary Table 2.
The expression values of 100 screened methylation sites were further retrieved from the genome-wide methylation data on solid tumors. A t-test was used to screen data with a P-value of ≤0.05 and in which ≤3 cancer species could not be significantly distinguished. The 10 eventually screened methylation sites of specific expression in breast cancer (cg13683194, cg07996594, cg21646032, cg07671949, cg21185686, cg03625109, cg16429070, cg23601468, cg24818566, and cg01240931) are listed in Supplementary Table 1. Nine genes (C9orf125, RARB, ESR1, RUNX3, PCDHGB7, DBC1, PDGFRB, TIMP3, and APC) were involved. A total of 4 methylation sites (2 in the DBC1 gene [cg03625109 and cg24818566], 1 in the C9orf125 gene [cg13683194], and 1 in the PDGFRB gene [cg16429070]) could effectively distinguish breast cancer from the other cancer species. Furthermore, 6 methylation sites in the other six genes could be used to distinguish cancer species.
Among the other solid tumor species, cholangiocarcinoma, colon cancer, diffuse large B cell lymphoma, kidney chromophobe cell carcinoma, low-grade gliomas, lung squamous cell carcinoma, ovarian serous cystadenocarcinoma, pheochromocytoma and paraganglioma, and rectal adenocarcinoma could be distinguished from breast cancer by 9 methylation sites; skin melanoma and uterine sarcoma could be distinguished from breast cancer by 8 methylation sites; and the remaining 20 cancer species could be completely differentiated from breast cancer by 10 methylation sites. The classification results were excellent (Supplementary Tables 3 and 4).
The data of the table corresponding to Figure 1 are shown in Supplementary data 1. The expression of 10 methylation markers was obtained through the use of the methylation chip data of 443 Colon cancer samples, 907 Lung cancer samples, 888 breast cancer samples, and 398 gastric cancer samples in the TCGA database. After analysis, 10 screened markers capable of significantly distinguishing breast cancer from other tumors were found in each tumor.
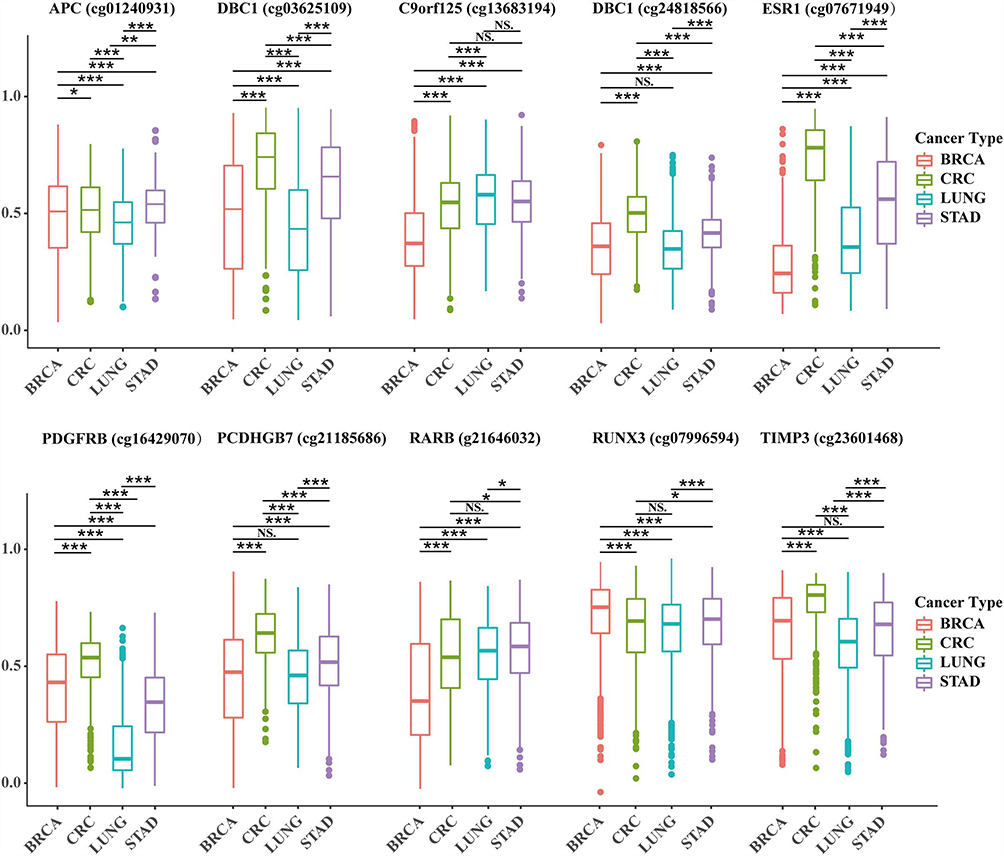 |
Figure 1 Difference analysis of 10 specific methylation sites in breast cancer, lung cancer, gastric cancer and colorectal cancer. Abbreviations: BRCA, breast cancer; CRC, colon cancer; LUNG, lung cancer; STAD, gastric cancer; NS, the difference was not statistically significant. Notes: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. |
Next, pyrosequencing was carried out for 10 methylation markers in 15 breast cancer tissues to detect methylation levels. The specific detection results are shown in Figure 2, and the detection values are shown in Supplementary data 2.
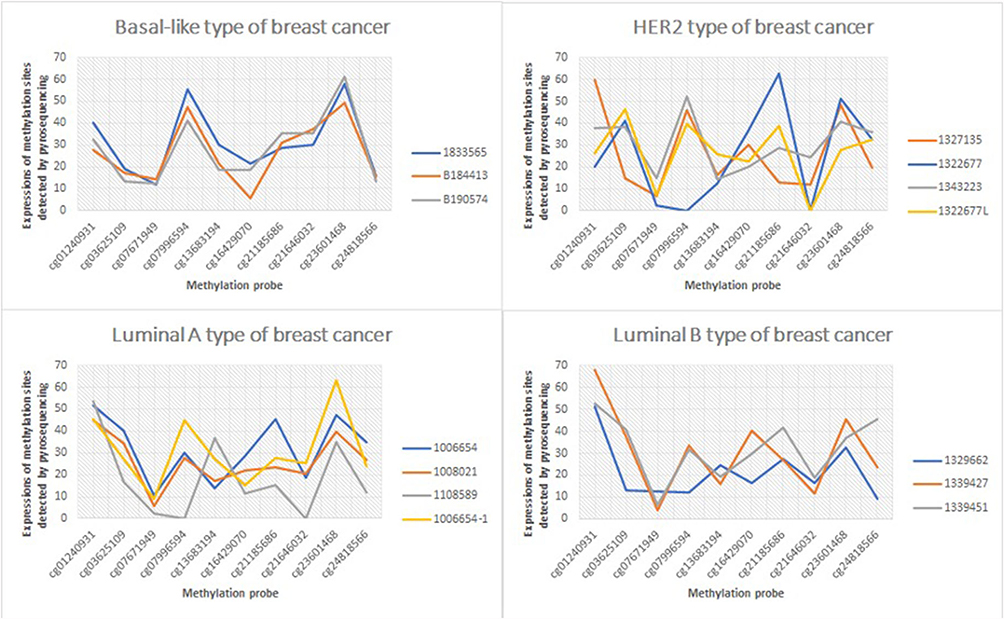 |
Figure 2 The expression of Luminal A, Luminal B, HER2 and Basal-like in different breast cancer subtypes. |
Cases that were not analyzed: 10 cases of lung cancer, 10 cases of stomach cancer, and 10 cases of colorectal cancer. The analysis should have been planned before the study was conducted; however, it was not carried out later due to limited samples (only 15 tissue samples of breast cancer were collected).
Expressions of Specific Methylation Sites in Different Tumors
A total of 45 samples of breast cancer (luminal A, luminal B, HER2, and Basal-like breast cancer), lung cancer, gastric cancer, and colorectal cancer in our hospital were selected for the present study. Lung cancer, gastric cancer, and colorectal cancer all had 10 samples. A total of 10 specific methylation sites were detected using pyrosequencing. Among these, 3 methylation sites (DBC1 [cg24818566], PCDHGB7 [cg21185686], and TIMP3 [cg23601468]) could not significantly distinguish breast cancer from lung cancer or gastric cancer. Meanwhile, other sites could distinguish breast cancer from other cancer species, with significant differences (P < 0.05, Figure 1).
The expressions of methylation sites in different breast cancer types were further analyzed by the Pearson correlation and Student’s t-test. The results revealed that according to the Pearson correlation analysis, the expressions of methylation sites in luminal A and luminal B breast cancer samples were highly similar (r = 0.9, P = 0.00041). A high degree of correlation was also found between Basal-like and luminal A samples (r = 0.71, P = 0.021), luminal A and HER2 samples (r = 0.8, P = 0.0058), and luminal B and HER2 samples (r = 0.69, P = 0.026); however, this was not the case in HER2 and Basal-like type samples (r = 0.63, P = 0.053). A total of 2 methylation sites (cg21646032 and cg23601468) could significantly distinguish Basal-like types from other types of breast cancer (P < 0.05) (Figures 2 and 3).
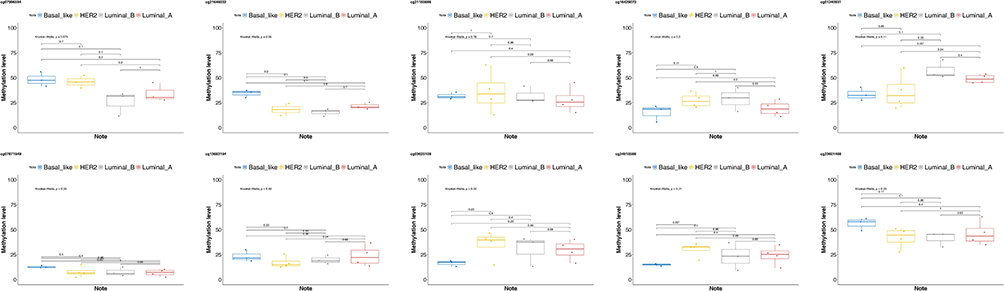 |
Figure 3 The expressions of specific methylation sites in different breast cancer types. |
Specific Breast Cancer Methylation Genes
According to the relevant literature on breast cancer methylation reported in PubMed, dozens of genes related to differential breast cancer methylation have been summarized (Supplementary Table 5). A relation between these genes and differential breast cancer methylation has been reported; furthermore, the use of certain genes as biomarkers for breast cancer screening, prognosis, and subtype classification has been reported. The discovery of NPY, RUNX3, CST6, GSTP1, SOX17, BRCA1, ESR1, TIMP3, RARB, APC, RASSF1A, CST6, DAPK, and P16 in this study is consistent with the results of other research. Hence, the differential expression methylation sites in breast cancer found in this study are common markers.
Discussion
In the present study, specific breast cancer methylation genes were examined through TCGA methylation data mining and based on literature. A total of 10 breast cancer methylation sites were screened. The differentiation was considered based on as many cancer species as possible. A P-value of 0.05 indicated a statistically significant difference among methylation sites (Supplementary Table 1). The differentiation effect was also good. Therefore, the 10 screened breast cancer methylation sites are feasible and effective in the early screening of breast cancer.
Of the 4 methylation sites that could completely distinguish breast cancer from the other 31 solid tumors species, 2 were in the DBC1 gene, 1 was in the C9orf125 gene, and 1 was in the PDGFRB gene. Previous literature has reported that these two gene panels, which include 15 genes, could distinguish ER-positive and AR-positive subtypes in breast cancer cases in China.11 It has been reported that the C9orf125 gene is hypermethylated in triple-negative breast cancer; this is significantly correlated with poor prognoses.12 The methylation of the RUNX3 gene plays an important role in breast cancer tumor formation; its hypermethylation is significantly associated with the risk of ductal carcinoma in situ and infiltrative ductal carcinoma. Thus, it can be used as a biomarker for early screening of breast cancer.6,13 A study reported that, as part of a 6-gene methylated panel, the PCDHGB7 gene could effectively distinguish among breast cancer, breast disease, and health control.14 The ESR1, TIMP3, RARB, and APC genes have been found in several review articles on breast cancer methylation markers;4,8 the credibility and significance of the articles indicate that these genes can be used as biomarkers for early screening of breast cancer.
The data from the 10 sites screened in the present study were obtained from the TCGA database; these data mainly included information regarding people in Europe and the USA. A larger sample group is required before the data can be used as biomarkers for early screening of breast cancer in the Chinese population.
Conclusion
The 10 methylation sites of breast cancer screened in the present study can effectively distinguish breast cancer from solid tumors, and they are expected to be used as biomarkers for early screening of breast cancer. In the future, screened methylation markers will be verified on an expanded sample. The authors of this study hope to provide a more scientific basis for the sites’ accuracy and applicability as early breast cancer screening markers.
Ethics Approval and Consent to Participate
This study was conducted with approval from the Ethics Committee of Jiaxing First Hospital. This study was conducted in accordance with the declaration of Helsinki. Written informed consent was obtained from all participants.
Acknowledgments
We would like to acknowledge the hard and dedicated work of all the staff that implemented the intervention and evaluation components of the study. The author also would like to acknowledge the data preprocess provided by Dr. Yongtian Zhao and Dr. Haiming Li of Fulgent Technologies Inc.
Funding
Research on molecular markers for early diagnosis and prognosis of breast cancer based on tumor ctDNA detection technology(2017AY33023).
Disclosure
The authors declare that they have no competing interests.
References
1. Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941–1953. doi:10.1002/ijc.31937
2. Fan L, Strasser-Weippl K, Jun-Jie L, et al. Breast cancer in China. Lancet Oncol. 2014;15(7):e279–89. doi:10.1016/S1470-2045(13)70567-9
3. Muller HM, Widschwendter A, Fiegl H, et al. DNA methylation in serum of breast cancer patients: an independent prognostic marker. Cancer Res. 2003;63:7641–7645.
4. Cheuk IW, Shin VY, Kwong A. Detection of methylated circulating DNA as noninvasive biomarkers for breast cancer diagnosis. J Breast Cancer. 2017;20(1):12–19. doi:10.4048/jbc.2017.20.1.12
5. Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284–299. doi:10.1038/nrg.2016.13
6. Lu DG, Ma YM, Zhu AJ, Han YW. An early biomarker and potential therapeutic target of RUNX 3 hypermethylation in breast cancer, a system review and meta-analysis. Oncotarget. 2016;8(13):22166–22174. doi:10.18632/oncotarget.13125
7. Heng J, Guo X, Wenhan W, et al. Integrated analysis of promoter mutation, methylation and expression of AKT1 gene in Chinese breast cancer patients. PLoS One. 2017;12(3):e0174022. doi:10.1371/journal.pone.0174022
8. Tang Q, Cheng J, Cao X, Surowy H, Burwinkel B. Blood-based DNA methylation as biomarker for breast cancer: a systematic review. Clin Epigenetics. 2016;8:115. doi:10.1186/s13148-016-0282-6
9. Yadav P, Masroor M, Nandi K, Kaza RCM, Jain SK, Khurana N, Saxena A. Promoter methylation of BRCA1, DAPK1 and RASSF1A is associated with increased mortality among Indian women with breast cancer. APJCP. 2017;19(2):443–448.
10. Hill VK, Ricketts C, Bieche I, et al. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res. 2011;71(8):2988–2999. doi:10.1158/0008-5472.CAN-10-4026
11. Li Z, Guo X, Wu Y, et al. Methylation profiling of 48 candidate genes in tumor and matched normal tissues from breast cancer patients. Breast Cancer Res Treat. 2015;149(3):767–779. doi:10.1007/s10549-015-3276-8
12. Stirzaker C, Zotenko E, Song JZ, et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat Commun. 2015;6:5899. doi:10.1038/ncomms6899
13. Lau QC, Raja E, Salto-Tellez M, et al. RUNX3 is frequently inactivated by dual mechanisms of protein mislocalization and promoter hypermethylation in breast cancer. Cancer Res. 2006;66(13):6512–6520. doi:10.1158/0008-5472.CAN-06-0369
14. Shan M, Yin HZ, Li J, et al. Detection of aberrant methylation of a six-gene panel in serum DNA for diagnosis of breast cancer. Oncotarget. 2016;7(14):18485–18494. doi:10.18632/oncotarget.7608
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.