")
Back to Journals » Research and Reports in Neonatology » Volume 6
Screening for medium-chain acyl CoA dehydrogenase deficiency: current perspectives
Authors Soler-Alfonso C, Bennett M, Ficicioglu C
Received 3 June 2015
Accepted for publication 16 September 2015
Published 18 January 2016 Volume 2016:6 Pages 1—10
DOI https://doi.org/10.2147/RRN.S60617
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Robert Schelonka
Claudia Soler-Alfonso,1 Michael J Bennett,2 Can Ficicioglu1
1Department of Pediatrics, Section of Metabolic Disease, 2Department of Pathology and Laboratory Medicine, The Children’s Hospital of Philadelphia and Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
Abstract: Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency is the most common disorder associated with fatty acid oxidation. The disorder is characterized by inability to generate sufficient energy from fatty acid metabolism during periods of catabolic stress caused by intercurrent illness or prolonged fasting. The metabolic consequences are severe and include hypoketotic hypoglycemia leading to a Reye-like hepatic encephalopathy syndrome and sudden death. If individuals are detected before a life-threatening episode, the complications of MCAD deficiency are preventable. Newborn metabolic screening enables the early detection of MCAD deficiency in many countries worldwide. The metabolic marker for MCAD deficiency “octanoylcarnitine” (C8) can be detected with a high degree of sensitivity in the newborns by tandem mass spectrometry. The 985A>G (K329E) mutation accounts for the majority of disease alleles, and approximately 47%–80% of MCAD patients are homozygotes for this mutation. Newborns homozygous for the 985A>G mutation have higher octanoylcarnitine levels than those who are heterozygous for 985A>G mutation or possess other genotypes. Time of sampling after birth and prematurity may affect the octanoylcarnitine levels in MCAD-deficient newborns. Tandem mass spectrometry newborn blood spot screening for MCAD deficiency is accurate and effective, and reduces morbidity and mortality in affected children.
Keywords: MCAD, newborn screening, C8, octanoylcarnitine, tandem mass spectrometry, sudden death in childhood
Introduction
Mitochondrial fatty acid β-oxidation (FAO) pathway plays a key role in cellular energy production, especially during physiological response to tissue energy depletion during the periods of fasting, illnesses, and increased muscular activity. FAO provides as much as 80% of energy supply for the functioning of heart and liver at all time. The pathway is complex and includes as many as 20 individual steps (Figure 1). Within the spiral, enzymes with overlapping chain-length specificities catalyze each step.1 In the liver, the oxidation of fatty acids fuels the synthesis of ketone bodies (3-hydroxy-butyrate and acetoacetate), which are utilized as an alternative energy source by extrahepatic organs, particularly the brain.2 Inherited defects of eleven proteins directly involved in this process have been identified in humans. These include defects of the plasma membrane carnitine transporter (MIM 212140); carnitine palmitoyltransferase (CPT) I (MIM 255120) and CPT II (MIM 255110); carnitine/acylcarnitine translocase (MIM 212138); very long-chain, medium-chain, and short-chain acyl-CoA dehydrogenases (VLCAD [MIM 201475], MCAD [MIM 201450], and SCAD [MIM 201470], respectively); 2,4-dienoyl-CoA reductase (MIM 222745); long- and short-chain 3-hydroxyacyl-CoA dehydrogenases (LCHAD [MIM 143450], SCHAD [MIM 601609]); and mitochondrial trifunctional protein (MIM 600890).
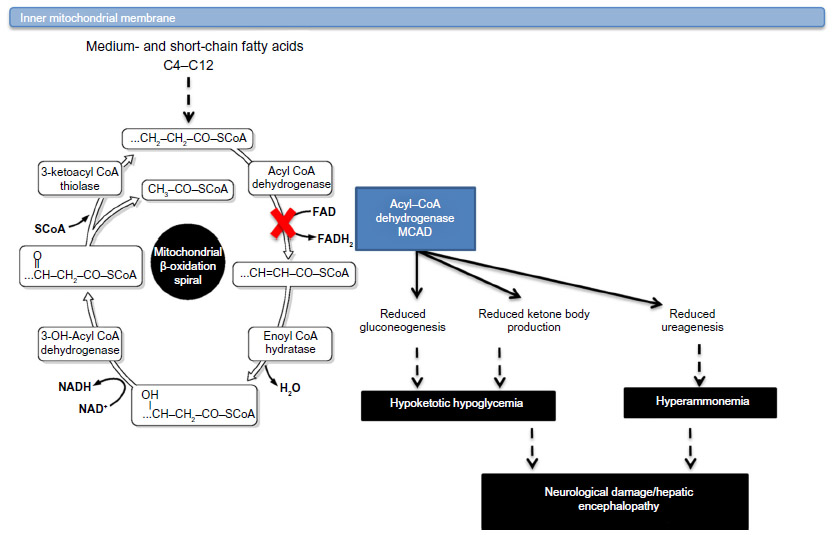 | Figure 1 MCAD deficiency: biochemistry and pathological consequences. |
MCAD deficiency is the most common inborn error associated with FAO. The enzyme is responsible for catalyzing the initial step in the β-oxidation of medium-chain fatty acids (C6–C12) in the mitochondrial matrix. During prolonged fasting or illness, individuals are initially heavily dependent on energy generation from glucose oxidation until glycogen reserves are depleted. Following this depletion of glycogen reserves, glucose is generated through gluconeogenesis and then energy maintenance becomes dependent on the utilization and oxidation of fatty acids. FAO is defective in MCAD deficiency and may rapidly lead to hypoglycemia and hypoketosis when body needs FAO to produce energy. The accumulating medium-chain fatty acids such as C8 (octanoate) and other medium-chain acyl-CoAs may have toxic effects, which disrupt urea cycle and may cause hyperammonemia. Since the first patients with MCAD deficiency were described,3,4 this has become recognized as one of the most common inherited disorders of FAO seen commonly in Caucasians of northern European origin. The natural history of the disease changed since the introduction of newborn screening (NBS) technique in the early 1990s. Here, we review different aspects of this condition contrasting the evolution of natural history before and after NBS was instituted, and we describe two MCAD deficiency cases from our practice, a major metabolic reference center in the USA. These two cases exemplify the major features of this disease and the current trends of research and present-day issues related to NBS. The Children’s Hospital of Philadelphia, Committee for the Protection of Human Subjects Policies and Procedures did not require ethical approval or consent to present these cases.
Case reports
Case 1
The patient was a male infant, born at 41 weeks of gestation, and was large for gestational age (4,100 g). Prenatal history was contributory for gestational diabetes and adolescent mother. The parents were unrelated and had Caucasian origin. He was discharged on the second day of life. Next day, the infant was seen by a primary care physician and found to have poor weight gain and poor feeding since birth (13% weight loss since birth) and lethargy. The infant was referred to a small local emergency department, where he was found to be nonresponsive. Initial laboratory examination showed severe hypoglycemia and metabolic acidosis (blood glucose was 12 mg/dL, HCO3 3.5 mmol/L, pH 7.0, base excess –27 mEq). Intravenous access was very difficult to obtain. Ketones in urine were not checked. An intraosseus line was placed as an emergency and D10w was given as a bolus, which resulted in improved blood glucose level to 40 mg/dL. Subsequently, the infant was transferred to the closest neonatal intensive care unit (NICU) in their geographical area. While being transported, he developed respiratory failure and was intubated for support. The NBS laboratory called our center and reported this patient’s abnormal NBS. He had elevated C8 (octanoylcarnitine) level concerning for MCAD deficiency. NBS report was issued on the day of life three when he had the metabolic crisis. Due to snow storm in the area, the transport was not possible to our center. The infant received multiple bicarbonate boluses throughout the night, and struggled to control glycemic values that were as high as 300 through most of the night. By next morning, the infant had transaminitis (aspartate aminotransferase [AST] in the 75–150 U/L range, normal 8–40 U/L), elevated coagulation markers including partial thromboplastin time (PTT) and international normalized ratio (INR), and mild pulmonary hemorrhage. At this time, his pH was 7.2, base excess -13, and he was still comatose. He began to have seizures, which was controlled with phenobarbital. By mid-afternoon he had a fatal cardiac arrhythmia and died. Genetic testing showed homozygous 985A>G in the ACADM gene confirming the diagnosis of MCAD deficiency.
Case 2
This male infant was the product of a second uncomplicated pregnancy for an unrelated Caucasian couple. He was delivered at term by C-section due to failure to progress, and was appropriate for gestational age. Over the following 24–48 hours, he became increasingly lethargic and had difficulty in arousing. While breastfeeding on day of life two, he became apneic, limp, and pulseless. The pediatrician witnessed the event and a code was called. Infant was found to be in asystole. Chest compressions were started and he was intubated. Emergent umbilical line was placed, and he received 14 rounds of epinephrine during resuscitation. Code reportedly lasted 30 minutes. Septic work-up was initiated, and infant was transferred to an outside hospital NICU for further management. NBS report was sent at 36 hours of life, which was before he had cardiac arrest. It showed elevated levels of C8 54.52 μmol/L (cutoff <0.30 μmol/L), C10 3.91 μmol/L (cutoff <0.40 μmol/L), and C10:1 0.99 μmol/L (cutoff <0.30 μmol/L), concerning for MCAD deficiency. He was again transferred to our NICU. Acid-base status prior to the arrival from the outside hospital NICU was reported as metabolic acidosis (pH 7.2, base excess 19, lactic acid 5 mmol/L [normal <2 mmol/L). The lowest glucose level was 42 mg/dL. The blood ammonia level was 70 μmol/L. His brain MRI showed findings consistent with hypoxic ischemic encephalopathy. He was treated with D10+1/4 NS, and NaHCO3 was administered to correct acidosis. A combination of studies including organic acids, acylcarnitine profiling, and molecular analysis confirmed the diagnosis of MCAD deficiency (homozygous 985A>G). He has been treated with frequent feeding, and avoidance of fasting and supportive rehabilitation therapies after his discharge. He is now 2 months old, without new episodes of illness related to MCAD deficiency.
Clinical presentation
As we see in the two cases that we described earlier, the most common presentation of MCAD deficiency is acute encephalopathy that is frequently complicated by hypoketotic hypoglycemia. The episodes vary in severity with some individuals presenting with mild changes in mental status, described as drowsiness, that can evolve to deep coma and sudden death. These metabolic crises usually occur in the setting of metabolic stress such as intercurrent illness, more commonly gastroenteritis.
Since the first published description of MCAD deficiency in 1976, the natural history of the disease has been transformed by the implementation of NBS. This first patient presented with unexplained episodes of lethargy and unconsciousness, and typical abnormalities in urine organic acid analysis including suberylglycine and C6–C10 dicarboxylic aciduria.4 In the 1980s, further characterization of the disease improved diagnostic methods and enhanced detection of affected individuals by molecular and biochemical methods. The first report associating sudden death and MCAD deficiency appeared in 1984.5 By the early 1990s, the calculated mortality rate was 20%–30% for cases identified clinically, usually presenting with severe hypoglycemia during the first 2 years of life.6 Prior to NBS, the most common clinical presentation was acute encephalopathy. Biochemical abnormalities such as hypoglycemia were documented in 42%–96% of patients with a severe metabolic decompensation, while analysis of ketone bodies was only reported in two of those studies.4,6–10
Although not typical, the initial episode may occur in the first week of life, with symptoms ranging from hypoglycemia to coma and death.11–13 In the neonatal period, inadequate caloric intake due to difficulties with breastfeeding is considered to play a role in triggering metabolic crisis. Most patients with MCAD deficiency present after the neonatal period with the median age at the initial episode reported as 1–1.5 years.6,8 Less commonly, symptomatic adolescent and adult cases have been reported presenting with a wider variable expressivity than formerly recognized. In addition to encephalopathy, hypoglycemia, and Reye-like symptoms, manifestations in adolescents and adults include rhabdomyolysis and a more severe cardiac phenotype with cardiac arrest and arrhythmia. Approximately, 50% of the adult patients showed significant elevations in creatine kinase in the range of 3,000–4,000 U/L.14–16 Although the majority of unscreened children survived their initial episode, many children who have experienced clinically unrecognized episodes suffer from long-term complications. Prior to wide use of NBS, approximately 40% of patients with MCAD deficiency who survive after diagnosis had abnormal developmental testing, including attention deficit, speech delay, and behavioral problems.6 Santos et al reported a case of undiagnosed maternal MCAD deficiency presenting with acute liver failure during pregnancy.17 A 29-year-old Caucasian primigravida felt unwell during pregnancy and had poor oral intake. In the 39th week, blood analysis showed signs of hepatopathy, and acute fatty liver was suspected leading to Caesarean section and delivery of a healthy boy. She quickly improved after delivery. Diagnosis was made through acylcarnitine analysis that revealed abnormalities in the mother consistent with MCAD deficiency. Metabolic follow-up analyses were normal in the baby. Acute liver failure in pregnancy might have occurred coincidentally with MCAD deficiency, but it cannot be ruled out as the underlying condition triggering an unusual presentation.17
Since there are no subtle specific symptoms or signs of MCAD deficiency that could help physicians suspect this disease before patients present with severe hypoglycemic coma and its consequences, NBS for MCAD deficiency is essential to detect affected individuals.3,18–26 The introduction of tandem mass spectrometry (MS/MS) NBS programs in the 1990s have helped to reduce metabolic crisis and death in MCAD deficiency.
Previous published experience with NBS and MCAD deficiency worldwide includes England, Australia, France, the Netherlands, Denmark, Portugal, Australia, Canada, and USA, among others.7,27–36 While comparison among different programs is difficult due to different detection rates, it is clear that NBS reduces clinical manifestations and death in the MCAD-deficient population.10,36,37 It has been reported that 15%–20% of the children with MCAD deficiency died suddenly during a first episode before NBS.38–42 Since the introduction of NBS for MCAD deficiency, death caused by the defect has become rare.37 Although death is now a rare occurrence, mortality is still reported, with a general risk likely to be approximately 4%–5%.31,36 The risk appears to be more notorious in the first few days of life due to either the performance of NBS or availability of NBS results in a timely fashion,13,43 as we see in the two patients reported in this article. Although the risk of sudden death is lower in the era of NBS, there still remains a fivefold higher risk of death or a severe event in the unscreened population.44 Favorable impact of NBS for MCAD deficiency has been consistently reported by NBS programs in the USA and the rest of the world.27–32,34–36 However, long-term outcome assessments after wide use of NBS is sparse. Hsu et al reported the developmental outcomes of 32 patients with MCAD deficiency detected through New England NBS program. Only one patient, who had a history of neonatal hypoglycemia, had mild expressive speech delay. The remaining 31 patients were all developmentally normal.31 These results are congruent with findings on neuropsychological outcomes from the Australian cohort.36
Overall, discriminatory data comparing developmental outcomes in children with episodes of severe metabolic crisis versus children with appropriate intervention and early glucose administration during fasting or illness are not available. Andresen et al from the Danish cohort proposed that although follow-up information on the patients was not collected systematically, it is worth noting that at least 21% (3/14) of their not-screened patients, diagnosed after their first metabolic decompensation, suffer from long-term sequelae.27 This experience is similar to the experience published by Lindner et al from the German cohort where developmental data were available in six patients with history of metabolic decompensations. In five of these patients, the results of neurological status and IQ tests were normal on follow-up. One patient showed normal intellectual and physical development, but noted to be slightly myoclonic on neurological examination. The only patient in the German cohort who showed severe neurological and intellectual impairment (IQ 74) never experienced a metabolic decompensation. However, he presented with severe neonatal onset cardiomyopathy, which seems to be part of a syndromic condition and unrelated to MCAD deficiency.32
Investigations on suspected MCAD deficiency
Continuous vigilance and high level of suspicion for MCAD deficiency are essential, since most of the neonatal patients may present symptomatically before results of comprehensive NBS are available. MCAD deficiency should be suspected in children/adults with undiagnosed encephalopathy (mild or severe), hypoglycemia, and documented liver disease. The biochemical diagnosis is based on plasma acylcarnitine profile characterized by increased levels of hexanoylcarnitine (C6), octanoylcarnitine (C8), decanoyl and decenoylcarnitines (C10 and C10:1) species, and prominent among them being octanoylcarnitine (C8). Urine organic acids analysis shows characteristic abnormalities including the presence of hexanoyl- and suberylglycine and C6–C10 dicarboxylic aciduria. Other investigations should include frequent blood sugar monitoring, acid-base markers, ammonia level, comprehensive metabolic panel, and liver function test. Cardiac arrest could occur; an echocardiogram and electrocardiography with telemetry should be studied.45 Sudden infant deaths should always be investigated for MCAD deficiency. MCAD deficiency is estimated to be responsible for 3%–6% of sudden infant death syndrome in nonscreened populations.46 The diagnosis can be confirmed by molecular analysis of ACADM gene, which encodes for the MCAD enzyme. Determination of MCAD enzyme activity can also be done in leukocytes or lymphocytes, with an HPLC-based assay using 3-phenylpropionyl-CoA as a substrate.47,48 In families with confirmed family history of a previous child affected with MCAD deficiency, appropriate precautions should be recommended including careful monitoring of oral intake and fasting avoidance alongside with glucose monitoring within the first hours of life until NBS result is available. These families should also see a biochemical geneticist for genetic counseling and discussion of prenatal diagnosis options.
Treatment
The mainstay of treatment for MCAD is avoidance of fasting. Patients can tolerate certain hours of fasting when they are well. The maximum safe fasting time in healthy MCAD patients have been reported as up to 8 hours in infants between ages 6 and 12 months; up to 10 hours during the second year of life, and up to 12 hours after age 2 years.49 After 1 year of age, 2 g/kg of uncooked cornstarch is usually recommended as a source of complex carbohydrate at bedtime, and it provides slow release of glucose and sufficient glucose supply overnight. Blood carnitine levels may be lower than normal in MCAD patients. We recommend measuring blood carnitine levels every 6 months and supplementing carnitine (10–25 mg/kg/day), if carnitine levels are persistently lower than normal. Patients do not need any fat restriction in their diet, and breastfeeding should be allowed as long as there is sufficient breast milk.50 All patients should carry an emergency room letter, which describes urgent management. This letter should be updated and include a detailed explanation of the management of acute metabolic decompensation, emphasizing the importance of intravenous glucose regardless of “normal” laboratory results and overnight in-hospital observation when oral intake is poor.
During acute decompensation (IV), glucose (a bolus of 2 mL/kg 25% dextrose) should be initiated immediately to correct hypoglycemia and followed by intravenous glucose administration of 10% dextrose solution to provide glucose infusion rates between 10 and 12 mg glucose/kg/min. It is important to maintain frequent glucose monitoring in order to tailor individual glucose needs. If indicated, higher dextrose concentrations should be used via central line access.51 During acute illnesses including acute gastroenteritis and vomiting, intravenous fluid (IVF) with 10% dextrose should be started as early as possible, before the onset of hypoglycemia.6,9,49,52,53 Early institution of treatment with intravenous dextrose appears to be remarkably effective in reducing morbidity and mortality.9,10 MCAD patients should never receive intravenous lipid solution when they are sick and need total parenteral nutrition.
Molecular aspects
MCAD deficiency is an autosomal recessive disorder caused by mutations in the ACADM gene on chromosome 1p31.54 ACADM gene consists of 12 exons that encode 421 amino acids.55 The use of NBS for early detection of MCAD deficiency has revealed a more varied mutational and biochemical spectrum of MCAD deficiency than the studies done on clinically ascertained populations.56 More than 100 different mutations of all types – missense, nonsense, splicing, and small insertions/deletions – are now known in the ACADM gene and are distributed to all the exons. Gross alterations, such as deletions and insertions involving more than 10–20 nucleotides, are rare, with only a single example reported so far.57 Prior to the institution of population-based NBS programs, clinically diagnosed MCAD patients with northern European background most often carried a common single base change mutation (c.985A>G ), accounting for up to 90% of abnormal alleles. Approximately, 80% of these patients were homozygous for this mutation, and 18% were heterozygous for c.985A>G.7,58–63
In order to explore the founder effect hypothesis, Leal et al in 2014 combined genotyping and screening data from 43 publications reporting the frequency of c.985A>G spanning over 10 million individuals, and they found significant variation in the frequency of the mutation across regions supporting a reported founder effect.58 The proportion of c.985A>G homozygotes were highest in western Europe with 4.1 (95% confidence interval [CI]: 2.8–5.6) per 100,000 individuals than the New World (3.2, 95% CI: 2.0–4.7), southern (1.2, 95% CI: 0.6–2.0), and eastern European regions (0.9, 95% CI: 0.5–1.7). No cases with the common mutation were identified in Asian and Middle Eastern regions.58
Prior to NBS, the incidence of MCAD deficiency was approximately in the range 1 in 30,000 to 1 in 135,000.28,64 Population-based NBS has revealed a much higher incidence of MCAD deficiency in newborns, ranging from 1 in 10,000 to 1 in 20,000 in northern European populations.60,65 Taking all the reported cases detected by NBS in the USA from 2001 to 2010, the incidence of MCAD was 1 in 17,759.66 In Massachusetts, approximately 1 out of 15,000 infants screened was diagnosed with MCAD, almost identical to the 1:14,000 frequency of phenylketonuria.31 In Pennsylvania and Bavaria, the MCAD frequency from the introduction of NBS has been estimated to be even greater at 1:8,000 and 1:9,000, respectively.37,67 Additionally, in countries such as the USA and the UK, where people of northwestern European descent make up a significant proportion of the population, the c.985A>G carrier frequency ranges from 1:40 to 1:100, whereas the mutation is virtually absent in the people of Japan.68 Despite the absence of the c.985A>G mutation in other populations, MCAD deficiency caused by other mutations has been reported in the children of Asia and Europe, including Portugal and Denmark, among others.12,27,35,58
Newborn screening for MCAD
MCAD deficiency may be diagnosed only after the child has expired or after permanent adverse neurological damage has occurred. Prior to the wide use of NBS, more than half of all the children were diagnosed after the age of 1 year.7 The goal of NBS is to identify affected infants before they have an acute episode and implement simple interventions such as avoidance of fasting as the mainstay of treatment. The availability of simple intervention such as avoidance of fasting makes MCAD deficiency an excellent candidate for early detection by NBS programs. NBS is conceived as a public health policy, and the understanding of cost-effectiveness for each of the conditions screened becomes a critical issue. Several studies have examined the cost-effectiveness of NBS for MCAD deficiency and have demonstrated that screening helps to both improve health and save cost.69–72
In the USA, the recommendations made by the American College of Medical Genetics in 2006 provided the foundation for the screening programs of most states to include MCAD in their mandated NBS panels. Till date, MCAD screening is performed in all 50 states in the USA. NBS programs from different countries including Germany, the UK, Austria, Portugal, Denmark, and France published their NBS data on MCAD deficiency in the past years (Table 1).
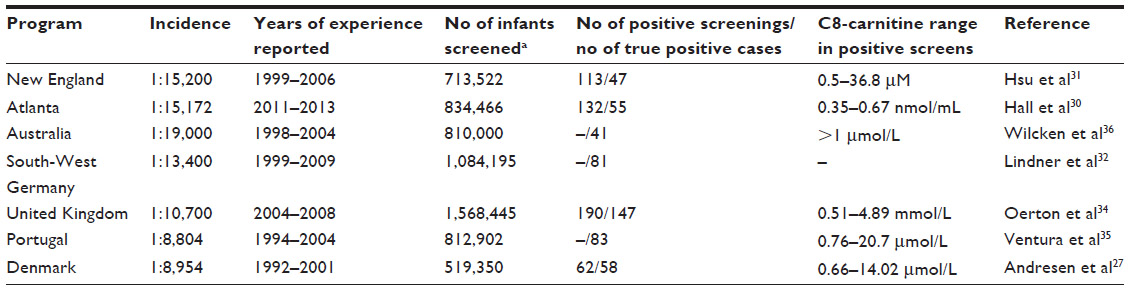 | Table 1 MCAD deficiency incidence and newborn screening (NBS) C8 levels in screen-positive cases |
While state laws dictate which conditions need to be included in the NBS panels, state laboratories must decide how to perform the screening. Screening for MCAD deficiency is universally achieved by measuring octanoylcarnitine (C8) as the primary marker using MS/MS. Several factors can influence C8 concentrations in neonates with MCAD deficiency, including timing of sample collection, feeding/fasting state, and secondary carnitine deficiency in blood. Chace et al showed that C8 levels were significantly higher in MCAD patients younger than <3 days of age compared to the ones older than 8 days.73 This shows us that if C8 levels are higher in newborns in the first days of life, probably due to the stress of birth, it goes down once they start eating and become more anabolic. In order to avoid false negative results, the screening should be performed in the first days of life. It is also known that C8 levels may be higher than cutoff values in MCAD carriers.74,75
As expanded NBS panels have been adopted on a widespread basis, screening programs have observed that a disproportionate number of false positive screens occur in premature and very low birth weight infants who are often in the NICU.76,77 For example, Hall et al from the Atlanta cohort proposed that C8/C8:1 ratio could be an effective marker to differentiate true positive cases from false positives.30 Other reports and our own unpublished data have demonstrated that newborns with higher initial C8 values were most likely to receive a diagnosis of MCAD deficiency and were also more likely to be homozygous for the common p.K324E mutation.31 Many NBS programs use acylcarnitine ratios such as C8/10 as a marker besides individual C8 levels to increase sensitivity and specificity of the NBS test. C8/C10 ratio may remain elevated in true positive MCAD deficiency cases, even after the initial C8 level decreases.31 The C8/C10 ratio may also be a useful parameter in the assessment of patients who have novel MCAD genotypes.27,35,73,78,79
Challenges after diagnosis of MCAD through NBS
Although NBS has been proven to be efficient in detecting MCAD deficiency in the neonatal period, sudden death is still reported in these populations. Feeding difficulties, insufficient breast milk, and/or increased stress during delivery may increase the risk of metabolic crisis in the first days of life, as we described in two cases mentioned earlier, before NBS is reported. Patients with MCAD deficiency are at high risk for metabolic crisis and sudden death even after diagnosis of the disease through NBS. NBS follow-up requires 1) frequent communication and collaboration between primary care providers and metabolic centers; 2) effective communication and education with specific information to the caregivers and families with respect to the potential complications and possibility of life-threatening events during episodes of acute illness and the need for prompt medical attention during illness; and 3) social support for the family when it is needed.
The distinction among subpopulations and risk for sudden death based on initial C8 levels in NBS has been explored in the past. For example, Korman et al13 reported the NBS C8 values in two siblings with MCAD deficiency; the sibling with a C8 level of 11.8 μmol/L died suddenly at 2 days of age, whereas the sibling with a C8 level of 0.55 μmol/L was asymptomatic at 6 years of age. State-based examples include the New York State experience in which infants with MCAD deficiency who had symptoms of an acute metabolic episode had NBS C8 levels ranging from 6.8 to 37.4 μmol/L.31,80 These data suggest that C8 level in the NBS specimen may be a strong indicator of risk for sudden death in MCAD deficiency and that this level might begin at 6 μmol/L.43 From the molecular perspective, at least two cases of sudden death were heterozygous for the c.985A>G mutation and one was homozygous for a novel splicing mutation.13 Accordingly, lack of homozygosity for the c.985A>G mutation does not eliminate the risk of sudden death in MCAD deficiency when the C8 level in the newborn is noticeably increased. Determination of residual MCAD enzyme activity may also help understanding ACADM variant genotypes and may contribute to risk stratification. In 2012, Touw et al examined 84 patients from 76 families. Twenty-two percent of the subjects had a variant ACADM genotype. In patients with classical ACADM genotypes, residual MCAD enzyme activity was significantly lower (median 0%, range 0%–8%) when compared to subjects with variant ACADM genotypes (range 0%–63%; four cases with 0%, remainder 20%–63%). Patients with fatal neonatal presentations before diagnosis displayed residual MCAD enzyme activities <1%. After diagnosis and initiation of treatment, residual MCAD enzyme activities <10% were associated with an increased risk of hypoglycemia and carnitine supplementation. Subjects with variant ACADM genotypes and residual MCAD enzyme activities <10% should be considered to have the same risks as patients with classical ACADM genotypes. Parental instructions and an emergency regimen should remain as principles of the treatment in any type of MCAD deficiency, as the effect of intercurrent illness on residual MCAD enzyme activity has not been completely elucidated. There are, however, arguments in favor of abandoning the general advice to avoid prolonged fasting in subjects with variant ACADM genotypes and >10% residual MCAD enzyme activity.47,48
Despite the issues during the neonatal period and childhood, future challenges are emerging with respect to the anticipation of serious episodes and deterioration of outcome in screened individuals entering adolescence and adulthood. As they appear entirely healthy, despite having a risk of disease, this risk and the need for preventative measures may be easily undervalued and forgotten by the adolescent and young adults who remain at risk for severe metabolic decompensation. Maintaining close follow-up may be achieved by increasing the frequency of clinical visits, specifically to repeat educational goals, and provide guidance by the metabolic disease team.44
Conclusion
In conclusion, NBS using MS/MS is cost-effective for early detection of MCAD deficiency. The extended use of NBS has advanced further understanding of the molecular basis and biochemical features of patients with MCAD deficiency, with a substantial reduction in mortality and morbidity. MCAD deficiency should always be included in the differential diagnosis for neonatal hypoglycemia, particularly in infants with severe metabolic acidosis, liver dysfunction, and hyperammonemia. Although NBS has proven effective particularly in the presymptomatic individuals, severe metabolic crisis and sudden death have not been entirely prevented.
Disclosure
The authors report no conflicts of interest in this work.
References
Scriver CR. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001. | |
Fukao T, Mitchell G, Sass JO, Hori T, Orii K, Aoyama Y. Ketone body metabolism and its defects. J Inherit Metab Dis. 2014;37(4):541–551. | |
Stanley CA, Hale DE, Coates PM, et al. Medium-chain acyl-CoA dehydrogenase deficiency in children with non-ketotic hypoglycemia and low carnitine levels. Pediatr Res. 1983;17(11):877–884. | |
Gregersen N, Lauritzen R, Rasmussen K. Suberylglycine excretion in the urine from a patient with dicarboxylic aciduria. Clinica chimica acta. Int J Clin Chem. 1976;70(3):417–425. | |
Howat AJ, Bennett MJ, Variend S, Shaw L. Deficiency of medium chain fatty acylcoenzyme A dehydrogenase presenting as the sudden infant death syndrome. Br Med J. 1984;288(6422):976. | |
Iafolla AK, Thompson RJ Jr, Roe CR. Medium-chain acyl-coenzyme A dehydrogenase deficiency: clinical course in 120 affected children. J Pediatr. 1994;124(3):409–415. | |
Pollitt RJ, Leonard JV. Prospective surveillance study of medium chain acyl-CoA dehydrogenase deficiency in the UK. Arch Dis Child. 1998;79(2):116–119. | |
Touma EH, Charpentier C. Medium chain acyl-CoA dehydrogenase deficiency. Arch Dis Child. 1992;67(1):142–145. | |
Wilson CJ, Champion MP, Collins JE, Clayton PT, Leonard JV. Outcome of medium chain acyl-CoA dehydrogenase deficiency after diagnosis. Arch Dis Child. 1999;80(5):459–462. | |
Wilcken B, Hammond J, Silink M. Morbidity and mortality in medium chain acyl coenzyme A dehydrogenase deficiency. Arch Dis Child. 1994;70(5):410–412. | |
Andresen BS, Bross P, Jensen TG, et al. A rare disease-associated mutation in the medium-chain acyl-CoA dehydrogenase (MCAD) gene changes a conserved arginine, previously shown to be functionally essential in short-chain acyl-CoA dehydrogenase (SCAD). Am J Hum Genet. 1993;53(3):730–739. | |
Ensenauer R, Winters JL, Parton PA, Kronn DF, Kim JW, Matern D, et al. Genotypic differences of MCAD deficiency in the Asian population: novel genotype and clinical symptoms preceding newborn screening notification. Genet Med. 2005;7(5):339–343. | |
Korman SH, Gutman A, Brooks R, Sinnathamby T, Gregersen N, Andresen BS. Homozygosity for a severe novel medium-chain acyl-CoA dehydrogenase (MCAD) mutation IVS3-1G > C that leads to introduction of a premature termination codon by complete missplicing of the MCAD mRNA and is associated with phenotypic diversity ranging from sudden neonatal death to asymptomatic status. Mol Genet Metab. 2004;82(2):121–129. | |
Boles RG, Boesel C, Rinaldo P. Sudden death beyond SIDS. Pediatr Pathol Lab Med. 1996;16(4):691–693. | |
Raymond K, Bale AE, Barnes CA, Rinaldo P. Medium-chain acyl-CoA dehydrogenase deficiency: sudden and unexpected death of a 45 year old woman. Genet Med. 1999;1(6):293–294. | |
Ruitenbeek W, Poels PJ, Turnbull DM, et al. Rhabdomyolysis and acute encephalopathy in late onset medium chain acyl-CoA dehydrogenase deficiency. J Neurol Neurosurg Psychiatr. 1995;58(2):209–214. | |
Santos L, Patterson A, Moreea SM, Lippiatt CM, Walter J, Henderson M. Acute liver failure in pregnancy associated with maternal MCAD deficiency. J Inherit Metab Dis. 2007;30(1):103. | |
Del Valle JA, Garcia MJ, Merinero B, et al. A new patient with dicarboxylic aciduria suggestive of medium-chain Acyl-CoA dehydrogenase deficiency presenting as Reye’s syndrome. J Inherit Metab Dis. 1984;7(2):62–64. | |
Bougneres PF, Rocchiccioli F, Kolvraa S, Hadchouel M, Lalau-Keraly J, Chaussain JL, et al. Medium-chain acyl-CoA dehydrogenase deficiency in two siblings with a Reye-like syndrome. J Pediatr. 1985;106(6):918–921. | |
Treem WR, Witzleben CA, Piccoli DA, et al. Medium-chain and long-chain acyl CoA dehydrogenase deficiency: clinical, pathologic and ultrastructural differentiation from Reye’s syndrome. Hepatology. 1986;6(6):1270–1278. | |
Brus F, Smit GP, Knoester H, Reijngoud DJ. [Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency in 2 patients with symptoms of Reye syndrome]. Tijdschr Kindergeneeskd. 1988;56(3):132–137. | |
Matsubara Y, Ikeda Y, Tanaka K. [Medium chain acyl-CoA dehydrogenase deficiency]. Tanpakushitsu Kakusan Koso. 1988;33(5):560–563. | |
Nobukuni Y, Yokoo T, Ohtani Y, et al. Neonatal onset of medium-chain acyl-CoA dehydrogenase deficiency in two siblings. Brain Dev. 1988;10(2):129–134. | |
Egidio RJ, Francis GL, Coates PM, Hale DE, Roesel A. Medium-chain acyl-CoA dehydrogenase deficiency. Am Fam Physician. 1989;39(5):221–226. | |
Treem WR, Stanley CA, Goodman SI. Medium-chain acyl-CoA dehydrogenase deficiency: metabolic effects and therapeutic efficacy of long-term L-carnitine supplementation. J Inherit Metab Dis. 1989;12(2):112–119. | |
Bennett MJ, Pollitt RJ, Taitz LS, Variend S. Medium-chain acyl-CoA dehydrogenase deficiency: a useful diagnosis five years after death. Clin Chem. 1990;36(9):1695–1697. | |
Andresen BS, Lund AM, Hougaard DM, et al. MCAD deficiency in Denmark. Mol Genet Metab. 2012;106(2):175–188. | |
Derks TG, Boer TS, van Assen A, et al. Neonatal screening for medium-chain acyl-CoA dehydrogenase (MCAD) deficiency in The Netherlands: the importance of enzyme analysis to ascertain true MCAD deficiency. J Inherit Metab Dis. 2008;31(1):88–96. | |
Feillet F, Ogier H, Cheillan D, Aquaviva C, Labarthe F, Baruteau J, et al. Medium-chain acyl-CoA-dehydrogenase (MCAD) deficiency: French consensus for neonatal screening, diagnosis, and management. Arch Pediatr. 2012;19(2):184–193. | |
Hall PL, Wittenauer A, Hagar A. Newborn screening for medium chain acyl-CoA dehydrogenase deficiency: performance improvement by monitoring a new ratio. Mol Genet Metab. 2014;113(4):274–277. | |
Hsu HW, Zytkovicz TH, Comeau AM, et al. Spectrum of medium-chain acyl-CoA dehydrogenase deficiency detected by newborn screening. Pediatrics. 2008;121(5):e1108–e1114. | |
Lindner M, Gramer G, Haege G, et al. Efficacy and outcome of expanded newborn screening for metabolic diseases – report of 10 years from South-West Germany. Orphanet J Rare Dis. 2011;6:44. | |
Lindner M, Hoffmann GF, Matern D. Newborn screening for disorders of fatty-acid oxidation: Experience and recommendations from an expert meeting. J Inherit Metab Dis. 2010;33(5):521–526. | |
Oerton J, Khalid JM, Besley G, et al. Newborn screening for medium chain acyl-CoA dehydrogenase deficiency in England: prevalence, predictive value and test validity based on 1.5 million screened babies. J Med Screen. 2011;18(4):173–181. | |
Ventura FV, Leandro P, Luz A, et al. Retrospective study of the medium-chain acyl-CoA dehydrogenase deficiency in Portugal. Clin Genet. 2014;85(6):555–561. | |
Wilcken B, Haas M, Joy P, et al. Outcome of neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency in Australia: a cohort study. Lancet. 2007;369(9555):37–42. | |
Nennstiel-Ratzel U, Arenz S, Maier EM, et al. Reduced incidence of severe metabolic crisis or death in children with medium chain acyl-CoA dehydrogenase deficiency homozygous for c.985A>G identified by neonatal screening. Mol Genet Metab. 2005;85(2):157–159. | |
Lecoq I, Mallet E, Bonte JB, Laroche D, Travert G. [Screening of A985 to G mutation of medium-chain acyl-CoA dehydrogenase (MCAD) gene in Normandy. Evaluation of the role of MCAD deficiency in sudden infant death]. C R Seances Soc Biol Fil. 1995;189(2):295–301. | |
Roe CR, Millington DS, Maltby DA, Kinnebrew P. Recognition of medium-chain acyl-CoA dehydrogenase deficiency in asymptomatic siblings of children dying of sudden infant death or Reye-like syndromes. J Pediatr. 1986;108(1):13–18. | |
Santer R, Gregersen N, Tanaka K, Hinck-Kneip C, Krawinkel M, Schaub J. The prevalence of the G985 allele of medium-chain acyl-CoA dehydrogenase deficiency among sudden infant death victims and healthy newborns in northern Germany. Eur J Pediatr. 1995;154(6):497. | |
Santer R, Schmidt-Sommerfeld E, Leung YK, Fischer JE, Lebenthal E. Medium-chain acyl CoA dehydrogenase deficiency: electron microscopic differentiation from Reye syndrome. Eur J Pediatr. 1990;150(2):111–114. | |
Bennett MJ, Rinaldo P, Millington DS, Tanaka K, Yokota I, Coates PM. Medium-chain acyl-CoA dehydrogenase deficiency: postmortem diagnosis in a case of sudden infant death and neonatal diagnosis of an affected sibling. Pediatr Pathol. 1991;11(6):889–895. | |
Yusupov R, Finegold DN, Naylor EW, Sahai I, Waisbren S, Levy HL. Sudden death in medium chain acyl-coenzyme a dehydrogenase deficiency (MCADD) despite newborn screening. Mol Genet Metab. 2010;101(1):33–39. | |
Schatz UA, Ensenauer R. The clinical manifestation of MCAD deficiency: challenges towards adulthood in the screened population. J Inherit Metab Dis. 2010;33(5):513–520. | |
Yusuf K, Jirapradittha J, Amin HJ, Yu W, Hasan SU. Neonatal ventricular tachyarrhythmias in medium chain acyl-CoA dehydrogenase deficiency. Neonatology. 2010;98(3):260–264. | |
Olpin SE. The metabolic investigation of sudden infant death. Ann Clin Biochem. 2004;41(Pt 4):282–293. | |
Touw CM, Smit GP, de Vries M, et al. Risk stratification by residual enzyme activity after newborn screening for medium-chain acyl-CoA dehyrogenase deficiency: data from a cohort study. Orphanet J Rare Dis. 2012;7:30. | |
Touw CM, Smit GP, Niezen-Koning KE, et al. In vitro and in vivo consequences of variant medium-chain acyl-CoA dehydrogenase genotypes. Orphanet J Rare Dis. 2013;8:43. | |
Derks TG, van Spronsen FJ, Rake JP, van der Hilst CS, Span MM, Smit GP. Safe and unsafe duration of fasting for children with MCAD deficiency. Eur J Pediatr. 2007;166(1):5–11. | |
Saudubray JM, Van den Berghe G, Walter J. Inborn Metabolic Diseases: Diagnosis and Treatment. 5th ed. Berlin: Springer; 2012: pp. xxv, 656. | |
Saudubray JM, Martin D, de Lonlay P, et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis. 1999;22(4):488–502. | |
Derks TG, Reijngoud DJ, Waterham HR, et al. The natural history of medium-chain acyl CoA dehydrogenase deficiency in the Netherlands: clinical presentation and outcome. J Pediatr. 2006;148(5):665–670. | |
Morris AA, Leonard JV. Early recognition of metabolic decompensation. Arch Dis Child. 1997;76(6):555–556. | |
Matsubara Y, Kraus JP, Yang-Feng TL, Francke U, Rosenberg LE, Tanaka K. Molecular cloning of cDNAs encoding rat and human medium-chain acyl-CoA dehydrogenase and assignment of the gene to human chromosome 1. Proc Nat Acad Sci U S A. 1986;83(17):6543–6547. | |
Zhang ZF, Kelly DP, Kim JJ, et al. Structural organization and regulatory regions of the human medium-chain acyl-CoA dehydrogenase gene. Biochemistry. 1992;31(1):81–89. | |
Maier EM, Liebl B, Roschinger W, et al. Population spectrum of ACADM genotypes correlated to biochemical phenotypes in newborn screening for medium-chain acyl-CoA dehydrogenase deficiency. Hum Mutat. 2005;25(5):443–452. | |
Morris AA, Taylor RW, Lightowlers RN, Aynsley-Green A, Bartlett K, Turnbull DM. Medium chain acyl-CoA dehydrogenase deficiency caused by a deletion of exons 11 and 12. Hum Mol Genet. 1995;4(4):747–749. | |
Leal J, Ades AE, Wordsworth S, Dezateux C. Regional differences in the frequency of the c.985A>G ACADM mutation: findings from a meta-regression of genotyping and screening studies. Clin Genet. 2014;85(3):253–259. | |
Matsubara Y, Narisawa K, Miyabayashi S, et al. Identification of a common mutation in patients with medium-chain acyl-CoA dehydrogenase deficiency. Biochem Biophys Res Commun. 1990;171(1):498–505. | |
Andresen BS, Dobrowolski SF, O’Reilly L, et al. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet. 2001;68(6):1408–1418. | |
Ferreira AC, Orlandi MP, Oliveira VC, et al. A985G mutation incidence in the medium-chain acyl-CoA dehydrogenase (MCAD) gene in Brazil. Genet Mol Research. 2009;8(2):487–493. | |
Gregersen N, Andresen BS, Bross P, et al. Molecular characterization of medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: identification of a lys329 to glu mutation in the MCAD gene, and expression of inactive mutant enzyme protein in E. coli. Hum Genet. 1991;86(6):545–551. | |
Tanaka K, Yokota I, Coates PM, et al. Mutations in the medium chain acyl-CoA dehydrogenase (MCAD) gene. Hum Mutat. 1992;1(4):271–279. | |
Derks TG, Duran M, Waterham HR, Reijngoud DJ, Ten Kate LP, Smit GP. The difference between observed and expected prevalence of MCAD deficiency in The Netherlands: a genetic epidemiological study. Eur J Hum Genet. 2005;13(8):947–952. | |
Liebl B, Nennstiel-Ratzel U, Roscher A, von Kries R. Data required for the evaluation of newborn screening programmes. Eur J Pediatr. 2003;162(Suppl 1):S57–S61. | |
Therrell BL Jr, Lloyd-Puryear MA, Camp KM, Mann MY. Inborn errors of metabolism identified via newborn screening: ten-year incidence data and costs of nutritional interventions for research agenda planning. Mol Genet Metab. 2014;113(1–2):14–26. | |
Ziadeh R, Hoffman EP, Finegold DN, et al. Medium chain acyl-CoA dehydrogenase deficiency in Pennsylvania: neonatal screening shows high incidence and unexpected mutation frequencies. Pediatr Res. 1995;37(5):675–678. | |
Tanaka K, Gregersen N, Ribes A, et al. A survey of the newborn populations in Belgium, Germany, Poland, Czech Republic, Hungary, Bulgaria, Spain, Turkey, and Japan for the G985 variant allele with haplotype analysis at the medium chain Acyl-CoA dehydrogenase gene locus: clinical and evolutionary consideration. Pediatr Res. 1997;41(2):201–209. | |
Haas M, Chaplin M, Joy P, Wiley V, Black C, Wilcken B. Healthcare use and costs of medium-chain acyl-CoA dehydrogenase deficiency in Australia: screening versus no screening. J Pediatr. 2007;151(2):121–126, 126. e1. | |
Hamers FF, Rumeau-Pichon C. Cost-effectiveness analysis of universal newborn screening for medium chain acyl-CoA dehydrogenase deficiency in France. BMC Pediatr. 2012;12:60. | |
van der Hilst CS, Derks TG, Reijngoud DJ, Smit GP, TenVergert EM. Cost-effectiveness of neonatal screening for medium chain acyl-CoA dehydrogenase deficiency: the homogeneous population of The Netherlands. J Pediatr. 2007;151(2):115–120, 120. e1–e3. | |
Venditti LN, Venditti CP, Berry GT, et al. Newborn screening by tandem mass spectrometry for medium-chain Acyl-CoA dehydrogenase deficiency: a cost-effectiveness analysis. Pediatrics. 2003;112(5):1005–1015. | |
Chace DH, Hillman SL, Van Hove JL, Naylor EW. Rapid diagnosis of MCAD deficiency: quantitative analysis of octanoylcarnitine and other acylcarnitines in newborn blood spots by tandem mass spectrometry. Clin Chem. 1997;43(11):2106–2113. | |
Blois B, Riddell C, Dooley K, Dyack S. Newborns with C8-acylcarnitine level over the 90th centile have an increased frequency of the common MCAD 985A>G mutation. J Inherit Metab Dis. 2005;28(4):551–556. | |
Lehotay DC, LePage J, Thompson JR, Rockman-Greenberg C. Blood acylcarnitine levels in normal newborns and heterozygotes for medium-chain acyl-CoA dehydrogenase deficiency: a relationship between genotype and biochemical phenotype? J Inherit Metab Dis. 2004;27(1):81–88. | |
Pourfarzam M, Morris A, Appleton M, Craft A, Bartlett K. Neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency. Lancet. 2001;358(9287):1063–1064. | |
Sahai I, Bailey JC, Eaton RB, Zytkovicz T, Harris DJ. A near-miss: very long chain acyl-CoA dehydrogenase deficiency with normal primary markers in the initial well-timed newborn screening specimen. J Pediatr. 2011;158(1):172; author reply: 3. | |
Okun JG, Kolker S, Schulze A, et al. A method for quantitative acylcarnitine profiling in human skin fibroblasts using unlabelled palmitic acid: diagnosis of fatty acid oxidation disorders and differentiation between biochemical phenotypes of MCAD deficiency. Biochim Biophys Acta. 2002;1584(2–3):91–98. | |
Van Hove JL, Zhang W, Kahler SG, et al. Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: diagnosis by acylcarnitine analysis in blood. Am J Hum Genet. 1993;52(5):958–966. | |
Arnold GL, Saavedra-Matiz CA, Galvin-Parton PA, et al. Lack of genotype-phenotype correlations and outcome in MCAD deficiency diagnosed by newborn screening in New York State. Mol Genet Metab. 2010;99(3):263–268. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.