")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 13
Screening and Identifying Cisplatin-Related Gene Mutations in Lung Squamous Cell Carcinoma
Authors Li X, Wang Y, Hu S, Bai Y
Received 14 September 2020
Accepted for publication 1 December 2020
Published 18 December 2020 Volume 2020:13 Pages 757—766
DOI https://doi.org/10.2147/PGPM.S264545
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Martin H Bluth
Xiaohua Li,1,2,* Yuntao Wang,3,* Sheng Hu,4 Yifeng Bai2
1Department of Respiratory and Critical Care Medicine, Sixth People’s Hospital of Chengdu, Chengdu 610051, Sichuan, People’s Republic of China; 2Department of Oncology, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu 611731, People’s Republic of China; 3Department of Oncology, The Fifth People’s Hospital Affiliated to Chengdu University of Traditional Chinese Medicine the Second Clinical Medical College, Chengdu 611130, Sichuan, People’s Republic of China; 4Department of Respiratory and Intensive Care Medicine, The General Hospital of Western Theatre Command, Chengdu 610083, Sichuan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yifeng Bai
Department of Oncology, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu 611731, People’s Republic of China
Tel +86-18183298718
Email [email protected]
Introduction: Platinum-based chemotherapy is the cornerstone of treatment for patients with LUSC, but cisplatin resistance greatly restricts its clinical application. Therefore, it is particularly important to screen the predominant LUSC population using biomarkers.
Methods: Data for 15 LUSC cell lines were downloaded from the Genomics of Drug Sensitivity in Cancer (GDSC) Project database to screen for mutations related to cisplatin susceptibility. We conducted whole-exome sequencing (WES) of tumors from 58 LUSC patients from Sichuan Provincial People’s Hospital of University of Electronic Science and Technology. Subsequently, the clinical prognostic value of these mutations was verified by using The Cancer Genome Atlas (TCGA)-LUSC cohort and our cohort (n=58).
Results: Based on the cisplatin sensitivity data of GDSC-LUSC and survival analysis of TCGA-LUSC and Local-LUSC cohorts, we found that only mutation of IGF2R was associated with cisplatin sensitivity, better overall survival [OS; P=0.04, HR (95% CI): 0.42 (0.23– 0.78)] and progression-free survival [PFS; P =0.016, HR (95% CI): 0.26 (0.12– 0.59)]. However, there were no significant differences in the frequencies of gene mutations between the IGF2R-mutant (IGF2R-MT) and IGF2R-wild-type (IGF2R-WT) groups. Gene set enrichment analysis (GSEA) and single-sample GSEA (ssGSEA) indicated enhanced intracellular detoxification and decreased abnormal signaling activity to reverse cisplatin tolerance in the IGF2R-MT group.
Conclusion: The results suggest that IGF2R mutations are a potential biomarker for screening LUSC patients suitable for cisplatin treatment.
Keywords: cisplatin, lung squamous cell carcinoma, resistance, mutation, chemotherapy
Introduction
At present, lung cancer is a common malignant tumor. Indeed, lung cancer is the leading malignancy in terms of morbidity and mortality, and nonsmall cell lung cancer (NSCLC) accounts for approximately 85% of cases.1 Due to the lack of early symptoms, most NSCLC patients (including lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC) and large cell carcinoma patients) are diagnosed at a late stage and thus miss the opportunity to undergo surgery.2 LUSC is a common pathological type of NSCLC. Compared with LUAD, cisplatin-based chemotherapy is still the main treatment for advanced LUSC.3 However, due to drug tolerance, its curative effect is not ideal. Approximately 70% of NSCLC patients exhibit primary and/or secondary tolerance to cisplatin, and only 30% of them will benefit from this therapy.4 Due to the lack of effective prediction methods, chemotherapy overuse is widespread at present, which results not only in adverse reactions to chemotherapy but also delays in treatment. Therefore, screening of cisplatin resistance in LUSC patients is particularly important.
Cisplatin is the most commonly used platinum chemotherapy, targeting DNA. Cisplatin-DNA adducts can promote apoptosis and inhibit cell proliferation.5–7 Recently, the mechanisms of cisplatin resistance have been widely explored, such as the reduction in intracellular cisplatin accumulation, increase in intracellular detoxification ability, and increase in DNA damage repair (DDR) activity. Abnormal regulation-related molecules are related to cell signal transduction, apoptosis and anti-apoptosis imbalance.5–7
Cisplatin resistance is caused by a variety of factors,8,9 including gene mutations. Muller et al10 showed that p53 mutation upregulates expression of endogenous p53, binds to caspase-9 and ultimately inhibits apoptosis and mediates cisplatin resistance. BRCA2 mutations can increase the DNA damage repair ability of tumor cells and mediate cisplatin resistance.11 Mutations in DNA mismatch repair (MMR) genes, such as MLH1 or MSH2, have also been shown to be associated with acquired resistance to cisplatin.12,13 Therefore, specific gene mutations may play an important role in the regulation of cisplatin sensitivity in LUSC.
In this study, gene mutations associated with cisplatin sensitivity were identified by analyzing the mRNA and cisplatin response data of LUSC cell lines in public databases, and clinical prognosis was analyzed by The Cancer Genome Atlas (TCGA)-LUSC cohort. We used gene set enrichment analysis (GSEA) and single-sample GSEA (ssGSEA) algorithms to further evaluate differences in cell function between mutant and wild-type groups. The results provide new targets for the diagnosis and treatment of patients with LUSC.
Materials and Methods
Clinical Samples and Cell Lines
The LUSC cell lines described in this study were derived from the GDSC database,27 with cisplatin sensitivity, gene expression and whole exome sequencing (WES) data available (Table S1). Nonsynonymous mutations (mutation frequency > 10%) were used for downstream analysis. The unit of cisplatin sensitivity data was the half-maximal inhibitory concentration (IC50) value. Using 3 µM as the cutoff, the samples from the GDSC database were divided into cisplatin-sensitive and cisplatin-resistant groups to verify the effect on the survival of patients with LUSC. The TCGAbiolinks R package28 was used to download the somatic mutation, RNA-seq and clinical data of the cohort TCGA-LUSC. The detailed analysis process of this study is depicted in Figure 1A. We used the Mann–Whitney U-test to examine the difference between the 896 mutations (mutation frequency > 10%) and cisplatin’s IC50 values, and mutations with an adjusted P value less than 0.05 were included in downstream analysis. Finally, we obtained one gene mutation that was associated with cisplatin sensitivity and clinical prognosis. We retrospectively collected 58 formalin-fixed paraffin-embedded (FFPE) tumor samples with matched germline specimens and performed whole-exome sequencing (WES). The human LUSC tumor specimens, WES, and data processing are detailed in the Supplementary Methods.
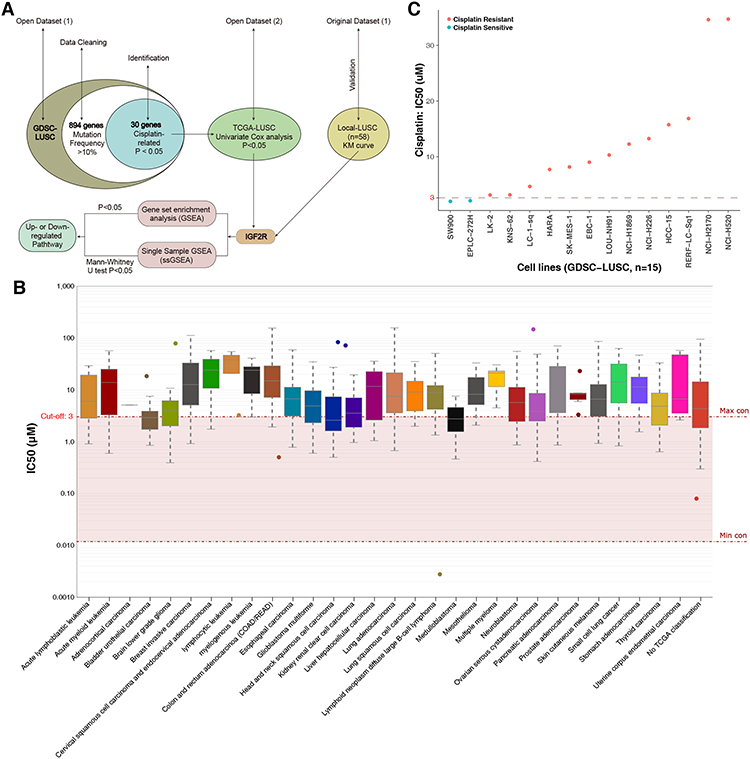 |
Figure 1 (A) The workflow of the bioinformatics analysis. (B) IC50 distribution of cisplatin by tissue type. (C) Scatter plot of the IC50 distribution of cisplatin in fifteen LUSC cell lines in the GDSC database. Abbreviations: IC50, half-maximal inhibitory concentration; LUSC, lung squamous cell carcinoma; GDSC, Genomics of Drug Sensitivity in Cancer. |
Results
IC50 Value Distribution of Cisplatin by Tissue Type
The sensitivity or resistance of tumor cell lines to certain drugs can be assessed by the half-maximal inhibitory concentration (IC50) value. The 15 LUSC cell lines analyzed in this study were derived from the GDSC database (https://www.cancerrxgene.org/compound/Cisplatin/1496/overview/ic50tissue=LUSC). mRNA expression and WES data were used for downstream analysis (Table S1). The IC50 cutoff of the cisplatin drug-sensitive cell line was 3 µM; most of the malignant tumor tissues (including LUSC) were cisplatin resistant, with only a small number (eg, brain lower-grade glioma and medulloblastoma; Figure 1B) showing no resistance. Figure 1C illustrates the cisplatin response values of the 15 LUSC cell lines, of which 86.7% (13/15) were resistant and 13.3% (2/15) sensitive.
IGF2R Mutation is Associated with Increased Cisplatin Sensitivity and Better Clinical Prognosis
For the GDSC-LUSC dataset, the Mann–Whitney U-test was used to compare differences in cisplatin sensitivity between mutant and wild-type cell lines. A total of 30 genes related to cisplatin sensitivity (Table S2) were screened, and we used TCGA-LUSC mutation data and survival data to further explore the prognostic value of these genes. Ultimately, we found only the IGF2R mutation to be related to cisplatin response and survival, mainly manifesting as significantly increased cisplatin sensitivity and better overall survival (OS) [log-rank P = 0.04, hazard ratio (HR) (95% confidence interval (CI)): 0.42 (0.23–0.78); Figure 2A and B]. Next (Kaplan-Meier) KM analysis was used to evaluate associations with IGF2R mutations and the prognosis of LUSC. In the Local-LUSC cohort (n=58), LUSC-MT was associated with a better PFS, with a significance (log-rank P = 0.016; HR: 0.26, 95%Cl: 0.12–0.59; Figure 2C). Subsequently, we analyzed the potential association between clinical phenotypes and survival of LUSC using univariable-Cox models. In TCGA-LUSC/Local-LUSC cohorts, only alterations in IGF2R were associated with significantly favorable OS/PFS; other clinical features conferred no survival benefit (Figure 2D). We identified no difference in TNM stage between IGF2R-MT and IGF2R-WT in TCGA-LUSC or Local-LUSC cohorts (Figure 2E).
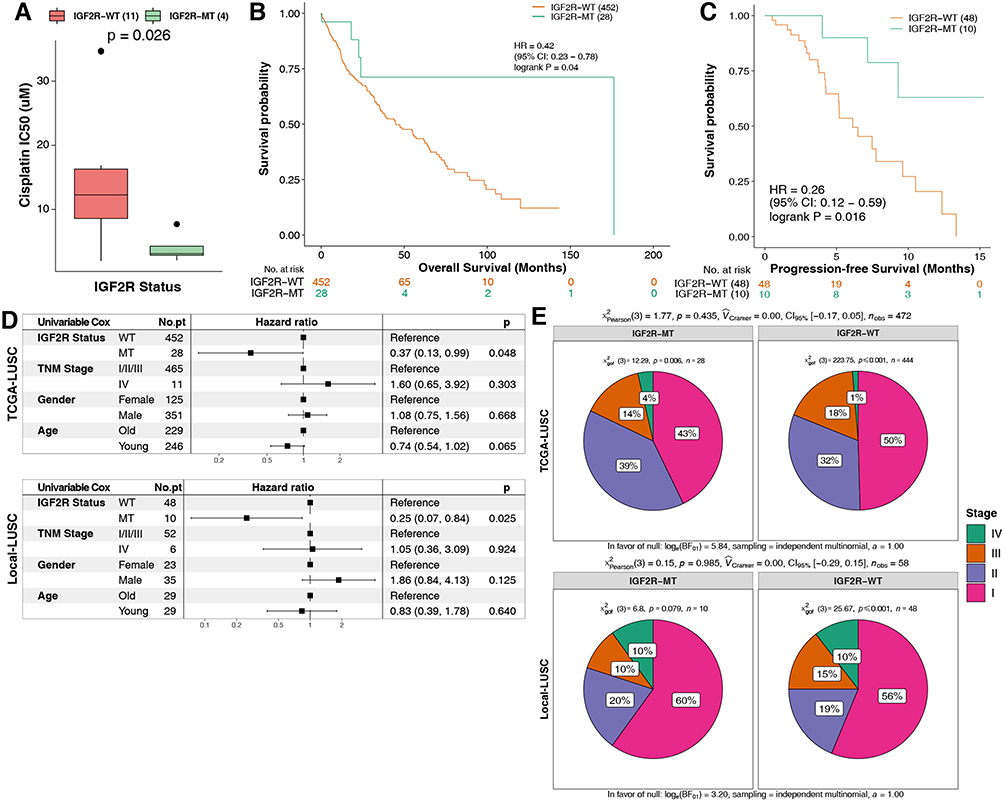 |
Figure 2 (A) IC50 values of cisplatin in GDSC-LUSC cell lines with or without IGF2R mutations. (B) The Kaplan-Meier method was used to assess the overall survival of patients with IGF2R mutations (green) and IGF2R wild-type (orange) in the dataset of 480 patients with LUSC (TCGA database). (C) For PFS, the Kaplan-Meier method revealed IGF2R mutations (green) and wild-type IGF2R (orange) in the dataset of 58 patients with LUSC (the Local-LUSC cohort). (D) The association between several clinical features, IGF2R status and prognosis using the univariable Cox model in TCGA-LUSC/Local-LUSC cohorts. (E) Comparison of the differences in TNM stage between the IGF2R-MT and IGF2R-WT groups in TCGA-LUSC/Local-LUSC cohorts. Abbreviations: IC50, half-maximal inhibitory concentration; LUSC, lung squamous cell carcinoma; GDSC, Genomics of Drug Sensitivity in Cancer; TCGA, The Cancer Genome Atlas. |
LUSC Mutational Landscape
In the GDSC-LUSC dataset (Figure 3A), we identified the top 20 mutations, among which the top 5 were TP53 (100% vs 91%), TTN (100% vs 64%), CSMD3 (100% vs 55%), HYDIN (75% vs 45%), and LRP1B (50% vs 55%). However, there were no significant differences in the frequencies of mutations in the 20 genes between the IGF2R-mutant (IGF2R-MT) and IGF2R-wild-type (IGF2R-WT) groups (Fisher’s exact test). The main mutation type in the GDSC-LUSC cohort was missense mutations, whereas frameshift and nonsense mutations accounted for a small proportion. The mutation frequencies of the top 20 genes in the cohort TCGA-LUSC and the top 5 mutations (TP53 (86% vs 80%), TTN (82% vs 72%), CSMD3 (57% vs 42%), MUC16 (50% vs 40%) and RYR2 (32% vs 37%)) were slightly different from those in the cohort GDSC-LUSC. The main mutation type in TCGA-LUSC was also missense mutations, with no significant differences in the frequencies of mutations in the top 20 genes (Figure 3B). In Local-LUSC (Figure 3C; Supplementary Table S3), the mutation frequencies of several genes were higher in IGF2R-MT than in IGF2R-WT, such as MUC16 (60% vs 29%; P < 0.05); TP53 (60% vs 29%; P < 0.05); KMT2C (50% vs 13%; P < 0.01); PRUNE2 (40% vs 13%; P < 0.05); SYNE1 (60% vs 8%; P < 0.001); ZFHX3 (50% vs 11%; P < 0.01); FAT4 (40% vs 11%; P < 0.05); NEB (50% vs 8%; P < 0.01); and PCNT (50% vs 8%; P < 0.01). We then applied the maftools package to visualize the mutation sites of IGF2R in TCGA-LUSC and Local-LUSC cohorts. We mainly identified alterations in IGF2R located in the CIMR and FN2 domains in Local-LUSC, while many IGF2R mutations were evenly distributed throughout the gene in TCGA-LUSC (Figure 3D).
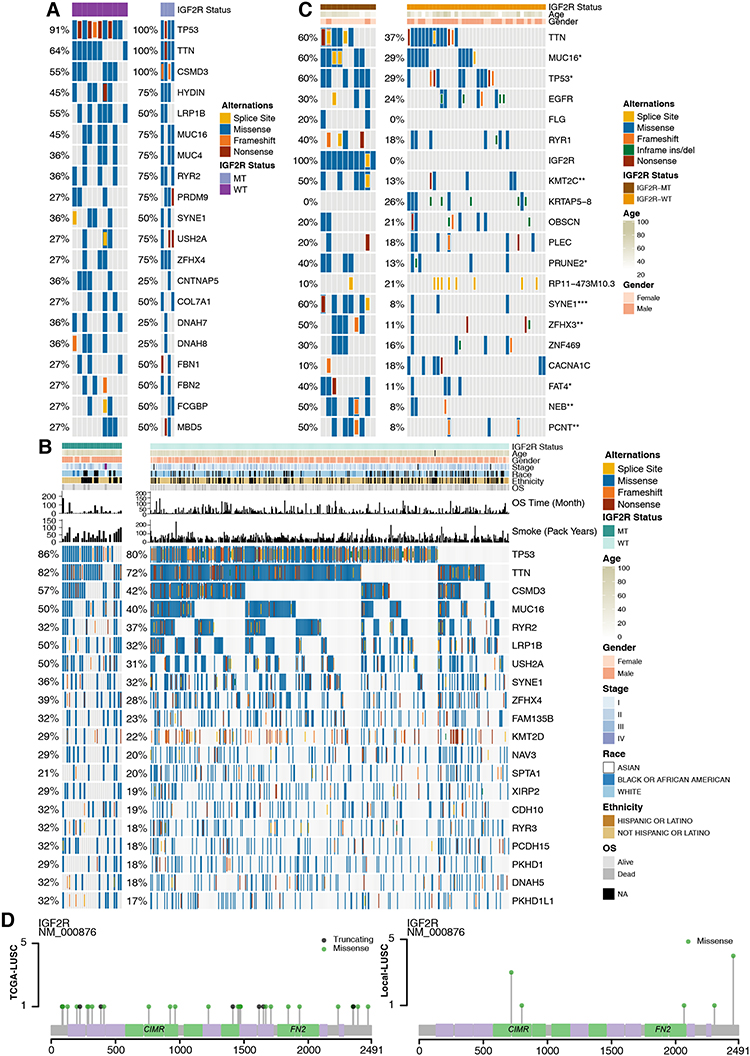 |
Figure 3 Genomic alterations in LUSC. (A) Fifteen LUSC cell lines are arranged from left (with IGF2R mutations) to right (without IGF2R mutations). Alterations in the LUSC cell line genes are annotated for each sample according to the color panel below the image. (B) Tumor samples from the cohort TCGA-LUSC are arranged from left (with IGF2R mutations) to right (without IGF2R mutations). Alterations in TCGA-LUSC candidate genes are annotated for each sample according to the color panel below the image. The clinical information for each candidate gene is plotted on the top panel. (C) Alterations in Local-LUSC candidate genes are annotated for each sample according to the color panel below the image. Clinical information for each candidate gene is plotted in the top panel. (D) Lolipop plot of mutation sites of IGF2R in the cohorts TCGA-LUSC and Local-LUSC. Abbreviations: LUSC, lung squamous cell carcinoma; GDSC, Genomics of Drug Sensitivity in Cancer; TCGA, The Cancer Genome Atlas. |
IGF2R-WT is Enriched in Intracellular Detoxification, Abnormal Signaling Pathways, and Drug Accumulation Regulation
To elucidate the effect of IGF2R-WT on cisplatin resistance in LUSC, we used GSEA to assess differences in signaling between IGF2R-WT and IGF2R-MT. In the GDSC-LUSC cohort, IGF2R-WT was significantly enriched in the NADP metabolic process and glucose 6-phosphate metabolic process (Figure 4A). Additionally, abnormal signatures, such as the regulation of macroautophagy, MAPK3 (ERK1) activation, VEGFR2-mediated cell proliferation and unfolded protein response (UPR), were significantly upregulated in IGF2R-WT cells (Figure 4B). Similarly, mutant and wild-type groups of the cohort TCGA-LUSC showed distinct functional signaling. For example, pathways including RHO GTPases that activate NADPH oxidases, β-catenin–independent WNT and interferon signaling were significantly increased in IGF2R-WT LUSC (Figure 5A and B). According to ssGSEA, the NADP metabolic process correlated significantly and positively with cisplatin sensitivity (P = 0.019, Spearman R = 0.61; Figure 5C). Additionally, activity of the signal transduction pathway related to cisplatin resistance in IGF2R-WT LUSC cell lines was significantly higher than that in IGF2R-MT cell lines (Figure 5D).
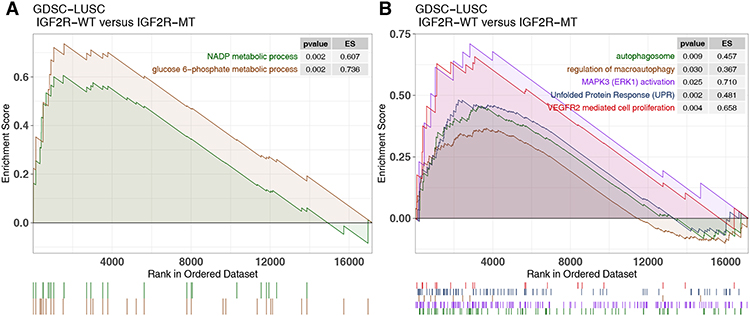 |
Figure 4 Transcriptome traits of GDSC-LUSC cell lines with or without IGF2R mutations. (A and B). Gene set enrichment analysis of hallmark gene sets downloaded from MSigDB. All transcripts were ranked by log2 (fold change) between IGF2R-WT and IGF2R-MT LUSC cell lines. Each run was performed with 1000 permutations. Abbreviations: IGF2R-WT, IGF2R wild-type; IGF2R-MT, IGF2R mutant; GDSC, Genomics of Drug Sensitivity in Cancer; LUSC, lung squamous cell carcinoma. |
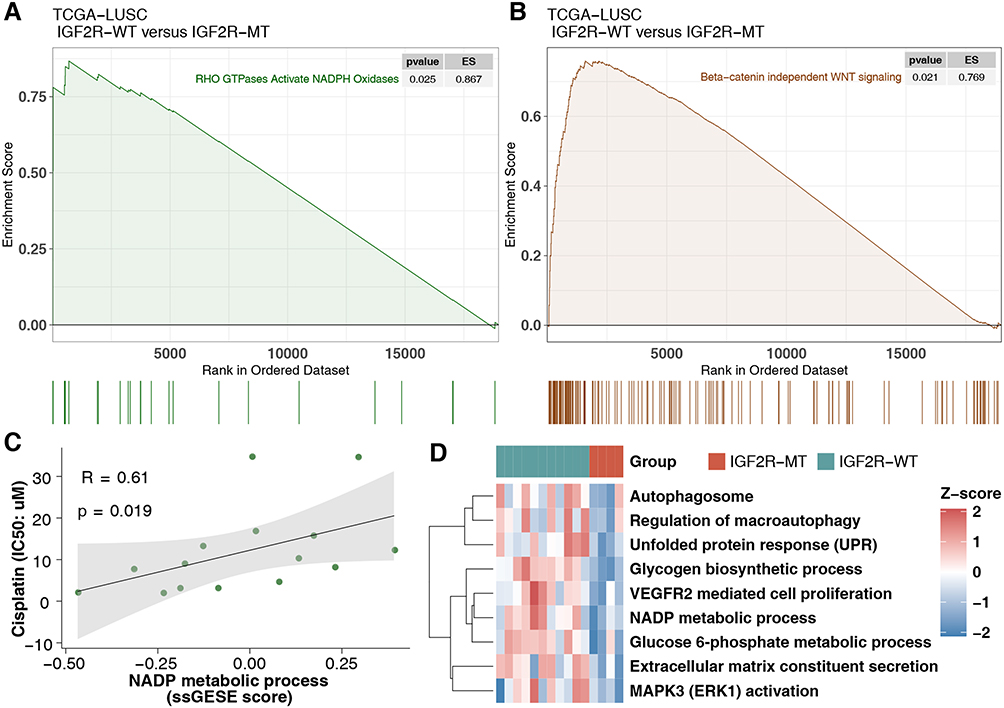 |
Figure 5 Transcriptome traits of TCGA-LUSC patients with or without IGF2R mutations. (A and B) Gene set enrichment analysis of hallmark gene sets downloaded from MSigDB. All transcripts were ranked by log2 (fold change) between IGF2R-WT and IGF2R-MT LUSC patients. Each run was performed with 1000 permutations. (C) Correlation between the ssGSEA enrichment score and IC50 values of 15 LUSC cell lines in GDSC (Spearman method). (D) Heatmap of ssGSEA scores, as estimated using gene sets from MSigDB, for IGF2R-WT and IGF2R-MT in the GDSC-LUSC dataset. Abbreviations: IGF2R-WT, IGF2R wild-type; IGF2R-MT, IGF2R mutant; ssGSEA, single-cell gene set enrichment analysis; TCGA, The Cancer Genome Atlas; LUSC, lung squamous cell carcinoma. |
Discussion
Squamous cell carcinoma of the lung is a common pathological type of NSCLC. The treatment of LUSC remains mainly surgery and adjuvant chemotherapy. Although platinum-based chemotherapy is the primary treatment for LUSC, the efficacy of chemotherapy is decreasing with the emergence of drug resistance. Drug resistance is ubiquitous, and resistance mechanisms are extremely complex, involving multiple factors and multiple genes.14 Therefore, screening biomarkers for cisplatin resistance in LUSC is particularly important. With the discovery of mutations related to cisplatin sensitivity, this study screened such genes and clinical prognosis and also attempted to elucidate the mechanism and role of mutations in sensitivity to the drug (Figure 6). To provide a theoretical basis for improving cisplatin resistance in LUSC patients, we found that IGF2R-MT was associated with significantly increased cisplatin sensitivity, prolonged OS [log-rank P = 0.04, HR (95% CI): 0.42 (0.23–0.78)] and better PFS [log-rank P = 0.016, HR (95% CI): 0.26 (0.12–0.59); Figure 2C; Local-LUSC].
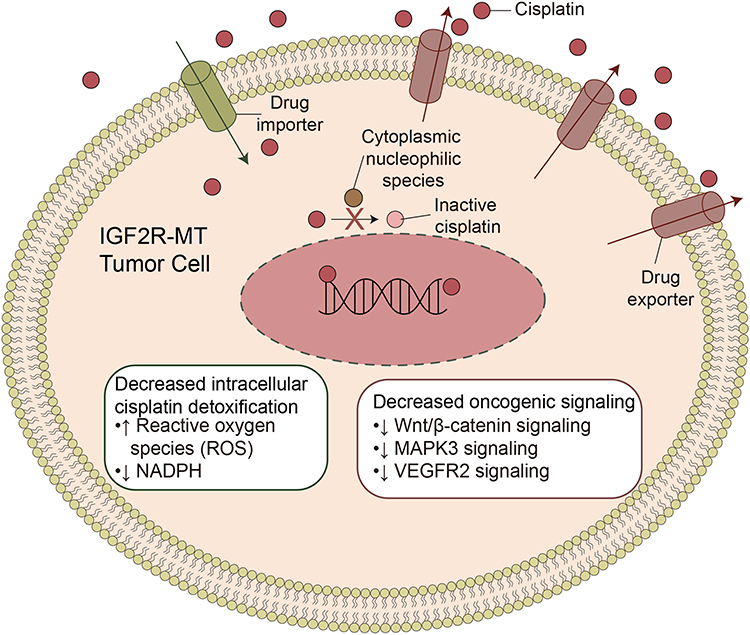 |
Figure 6 Potential mechanism of IGF2R-WT mediating cisplatin resistance in LUSC. Abbreviations: IGF2R-WT, IGF2R wild-type; LUSC, lung squamous cell carcinoma. |
The anticancer activity of cisplatin involves the formation of stable cisplatin-DNA adducts in the nucleus that interfere with DNA replication and transcription.7 Cisplatin-resistant tumors can be activated by increasing the binding of cisplatin with other biological macromolecules, especially glutathione (GSH) and metallothionein, resulting in loss of its activity.7,15 For example, NADPH is one of the raw materials for the regeneration of GSH and thioredoxin. In the redox process, harmful molecules are transformed into harmless ones through biotransformation.6 An increase in GSH16,17 and a decrease in intracellular ROS18,19 have been associated with cisplatin resistance. Our GSEA suggested significant increases in NADP and glucose 6-phosphate metabolic processes in the IGF2R-WT group and that RHO GTPases activate NADPH oxidase activity, though the activity of IGF2R-MT in the above pathways was significantly downregulated. This finding suggests that IGF2R mutations reduce the metabolism of NADPH and NADP, downregulating the synthesis of GSH and eventually promoting cisplatin resistance. IGF2R serves as a multifunctional receptor and is involved in the IGF pathway.20 Stimulation of the pathway contributes to carcinogenesis or tumor progression in multiple tumors.20,21 Inhibitors targeting the IGF pathway, such as anti-IGF1R antibodies,22 are being investigated as candidates for cancer treatment in ongoing clinical trials.
In addition to the destruction of oxidative/antioxidant processes by tumor cells, activation of some abnormal signaling pathways (including autophagy (macroautophagy), heat shock proteins (HSPs), PI3K/AKT, etc.) is involved in cisplatin resistance.7,23 For example, studies have shown that various components of the autophagy mechanism and some molecular chaperones of the HSP family reduce the cytotoxicity of cisplatin in tumor cells.24 Studies have also shown that inhibitors of AKT1, mTOR, PI3K and MAPK signaling can significantly increase cisplatin’s cytotoxicity in breast cancer.23 Cisplatin activates MAPK signaling and the production of multidrug resistance,25 and the Wnt/β-catenin pathway is closely related to the MAPK pathway. When reducing the expression level of β-catenin, the activity of ERK is inhibited, which indicates that cisplatin-mediated multidrug resistance is closely related to Wnt/β-catenin signaling. The downstream target genes of the Wnt/β-catenin pathway also significantly affect cisplatin resistance.26 For example, based on the enrichment score in GSEA, autophagosomes, MAPK3 (ERK1) activation, VEGFR2-mediated cell proliferation, UPR and beta-catenin–independent WNT signaling were significantly enriched in the IGF2R-WT group. In contrast, IGF2R mutations increased the sensitivity of tumor cells to cisplatin by reducing autophagy, MAPK3, VEGFR, and WNT/β−catenin signaling activity, mediating cisplatin sensitivity.
There are some limitations in this study. First, the screening of cisplatin-sensitive/sensitive and -resistant genes came from the was based on mutation data of for tumor cell lines. However, in vitro, the continuous passage of tumor cell lines allow allows them to adapt to the environment of petri dishes environment, which lacks lacking a tumor microenvironment including immune cells, fibroblasts, and the extracellular matrix and so on matrix, among others. Second, we used only the GSEA and ssGSEA algorithms to evaluate the differences in cell signatures between the mutant group and wild-type group and did not use employ cell experiments or animal experiments for subsequent validation. Third, based on the averaging of bulk transcriptional data, this study covered the explored differences between single cells but lacked an analysis of tumor heterogeneity. Moreover, in the validation of the association between genes and survival, only one cohort TCGA-LUSC cohort was included included. Finally, the limited number of local LUSC patients may have resulted in this study the identification of different mutation frequencies among the three cohorts. Therefore, more samples and prospective studies are needed for subsequent analysis and validation.
In conclusion, IGF2R mutations are potential biomarker for screening cisplatin sensitivity in patients with LUSC. IGF2R mutations are associated with cisplatin sensitivity and better survival prognoses, while wild-type IGF2R promotes the development of cisplatin resistance in LUSC. Additionally, mechanistic analysis suggests that IGF2R mutations increase the intracellular detoxification of drugs and reduce the activity of abnormal signaling pathways, which may reverse cisplatin tolerance and significantly improve clinical benefit in LUSC populations.
Pathway Enrichment Analysis
The Limma R package29 was used to normalize the mRNA data for the GDSC-LUSC and TCGA-LUSC cohorts. The clusterProfiler R package30 was employed for GSEA, including Gene Ontology (GO) term, Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome analyses. The GSVA R package31 was applied for ssGSEA, including gene sets downloaded from the Molecular Signatures Database (MSigDB).32
Statistical Analysis
The pan-cancer cisplatin sensitivity distribution was downloaded from the GDSC website. The Mann–Whitney U-test was used to compare cisplatin sensitivity between mutant and wild-type LUSC cell lines. Differences in the mutation frequencies (top 20) of wild-type and mutant-type LUSC (GDSC-LUSC; TCGA-LUSC and Local-LUSC) were compared using Fisher’s exact test. The KM method and Log rank test were used for survival analysis. P < 0.05 was considered statistically significant, and all statistical tests were two-tailed. All statistical and visual analyses were carried out in R software (version 3.6.1). In addition, the Complexheatmap R package33 (Version 2.2.0) was employed to visualize the heatmap and ggpubrR34 (2.2.0) to visualize the boxplot.
Abbreviations
LUSC, lung squamous cell carcinoma; GDSC, Genomics of Drug Sensitivity in Cancer; TCGA, The Cancer Genome Atlas; MT, mutant; WT, wild-type; GSEA, Gene set enrichment analysis; ssGSEA, single-sample GSEA; NSCLC, non-small cell lung cancer; LUAD, lung adenocarcinoma; DDR, DNA damage repair; MMR, Mutations in DNA mismatch repair; WES, whole exome sequencing; IC50, the half maximal inhibitory concentration; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; MSigDB, Molecular Signatures Database; OS, overall survival; HR, hazard ratio; CI, confidence interval; UPR, unfolded protein response; HSP, heat shock proteins.
Data Sharing Statement
All the data generated or analyzed during this study are included in this published article and its Supplementary Methods File.
Ethics Statement
The patients/participants provided their written informed consent to participate in this study and the research presented here has been performed in accordance with the Declaration of Helsinki and has been approved by the ethics committee of Sichuan Provincial People’s Hospital of University of Electronic Science and Technology.
Author Contributions
XHL, YTW wrote the manuscript; YFB designed the research; XHL, YTW and SH performed the research; XHL, YTW, SH and YFB Writing-review and editing. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (grant number 81301910).
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. United States.
2. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. England. doi:10.1038/nature25183
3. Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389:299–311. England. doi:10.1016/S0140-6736(16)30958-8
4. Socinski MA. Cytotoxic chemotherapy in advanced non-small cell lung cancer: a review of standard treatment paradigms. Clin Cancer Res. 2004;10:4210s–4214s. United States.
5. Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. England. doi:10.1038/onc.2011.384
6. Chen S-H, Chang J-Y. New insights into mechanisms of cisplatin resistance: from tumor cell to microenvironment. Int J Mol Sci. 2019;20.
7. Galluzzi L, Vitale I, Michels J, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257.
8. Shen D-W, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64:706–721. doi:10.1124/pr.111.005637
9. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. England. doi:10.1038/nrm2970
10. Muller PAJ, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–317. doi:10.1016/j.ccr.2014.01.021
11. Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi:10.1038/nature06633
12. Vaisman A, Varchenko M, Umar A, et al. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998;58:3579–3585.
13. Duckett DR, Drummond JT, Murchie AIH, et al. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc Natl Acad Sci. 1996;93:6443–6447. doi:10.1073/pnas.93.13.6443
14. Chen ZJ, Le HB, Zhang YK, Qian LY, Sekhar KR, Li WD. Lung resistance protein and multidrug resistance protein in non-small cell lung cancer and their clinical significance. J Int Med Res. 2011;39:1693–1700. England. doi:10.1177/147323001103900511
15. Catanzaro D, Gaude E, Orso G, et al. Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget. 2015;6:30102–30114. doi:10.18632/oncotarget.4945
16. Yang Y, Li H, Hou S, Hu B, Liu J, Wang J. The noncoding RNA expression profile and the effect of lncRNA AK126698 on cisplatin resistance in non-small-cell lung cancer cell. PLoS One. 2013;8:e65309. doi:10.1371/journal.pone.0065309
17. Jamali B, Nakhjavani M, Hosseinzadeh L, Amidi S, Nikounezhad N, Shirazi H. Intracellular GSH alterations and its relationship to level of resistance following exposure to cisplatin in cancer cells. Iran J Pharm Res. 2015;14:513–519.
18. Okuno S, Sato H, Kuriyama-Matsumura K, et al. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br J Cancer. 2003;88:951–956. doi:10.1038/sj.bjc.6600786
19. Wangpaichitr M, Sullivan EJ, Theodoropoulos G, et al. The relationship of thioredoxin-1 and cisplatin resistance: its impact on ROS and oxidative metabolism in lung cancer cells. Mol Cancer Ther. 2012;11:604–615. doi:10.1158/1535-7163.MCT-11-0599
20. Iida Y, Salomon MP, Hata K, et al. Predominance of triple wild-type and IGF2R mutations in mucosal melanomas. BMC Cancer. 2018;18:1054. doi:10.1186/s12885-018-4977-2
21. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–330. doi:10.1056/NEJMoa1412082
22. Pollak M. The insulin receptor/insulin-like growth factor receptor family as a therapeutic target in oncology. Clin Cancer Res. 2012;18:40–50. doi:10.1158/1078-0432.CCR-11-0998
23. Wong SW, Tiong KH, Kong WY, et al. Rapamycin synergizes cisplatin sensitivity in basal-like breast cancer cells through up-regulation of p73. Breast Cancer Res Treat. 2011;128:301–313. Netherlands. doi:10.1007/s10549-010-1055-0
24. Karasawa T, Sibrian-Vazquez M, Strongin RM, Steyger PS. Identification of cisplatin-binding proteins using agarose conjugates of platinum compounds. PLoS One. 2013;8:e66220. doi:10.1371/journal.pone.0066220
25. Akita H, Doki Y, Miyata H, et al. Clinical significance of the second cycle response to cisplatin-based chemotherapy as preoperative treatment for esophageal squamous cell carcinoma. J Surg Oncol. 2006;93:401–409. United States. doi:10.1002/jso.20501
26. Wang Z, Ma L-J, Kang Y, Li X, Zhang X-J. Dickkopf-3 (Dkk3) induces apoptosis in cisplatin-resistant lung adenocarcinoma cells via the Wnt/β-catenin pathway. Oncol Rep. 2015;33:1097–1106. Greece. doi:10.3892/or.2014.3704
27. Yang W, Soares J, Greninger P, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955–61. doi:10.1093/nar/gks1111
28. Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44:e71–e71. doi:10.1093/nar/gkv1507
29. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47–e47. doi:10.1093/nar/gkv007
30. Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J Integr Biol. 2012;16:284–287. doi:10.1089/omi.2011.0118
31. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013;14:7. England. doi:10.1186/1471-2105-14-7
32. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545–15550. doi:10.1073/pnas.0506580102
33. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. England. doi:10.1093/bioinformatics/btw313.
34. Kassambara A. ggpubr: “ggplot2” based publication ready plots. R package version 0.2; 2018. Available from: https://CRAN.R-project.org/package=ggpubr.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.