")
Back to Journals » International Journal of General Medicine » Volume 13
Schnitzler Syndrome in a 27-Year-Old Man: Diagnostic and Therapeutic Dilemma in Adult Auto-Inflammatory Syndromes A Case Report and Literature Review
Authors Więsik-Szewczyk E , Felis-Giemza A , Dziuk M, Jahnz-Różyk K
Received 8 June 2020
Accepted for publication 14 August 2020
Published 25 September 2020 Volume 2020:13 Pages 713—719
DOI https://doi.org/10.2147/IJGM.S265482
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Ewa Więsik-Szewczyk,1 Anna Felis-Giemza,2 Mirosław Dziuk,3 Karina Jahnz-Różyk1
1Department of Internal Medicine, Pulmonology, Allergy and Clinical Immunology, Central Clinical Hospital of the Ministry of National Defense, Military Institute of Medicine in Warsaw, Warsaw, Poland; 2Department of Connective Tissue Diseases, National Institute of Geriatrics, Rheumatology and Rehabilitation, Warsaw, Poland; 3Department of Nuclear Medicine, Military Institute of Medicine in Warsaw, Warsaw, Poland
Correspondence: Ewa Więsik-Szewczyk
Department of Internal Medicine, Pulmonology, Allergy and Clinical Immunology, Military Institute of Medicine in Warsaw, Szaserów 128 01-141, Warsaw, Poland
Tel/Fax +48 261 816 581
Email [email protected]
Abstract: A 32-year-old-man, with a history of chronic urticaria from the age of 27, diagnosed with an adult-onset Still’s disease and received a low dose of glucocorticoids, methotrexate and tocilizumab. Despite the long-term combined treatments, he suffered from chronic urticaria, low-grade fever and bone pain. He was found to have high inflammatory markers, hypogammaglobulinemia, monoclonal IgM – kappa light chain in serum and increased radiotracer uptake in the whole bone scintigraphy. No pathological variants for monogenic autoinflammatory diseases were present in the genome exome sequencing. These investigations confirmed the diagnosis of Schnitzler syndrome, which is an exception before the age of 35. Switching from tocilizumab to interleukin 1 receptor inhibitor, anakinra led to a full clinical response and normalisation of inflammatory markers. Patients with a history of fever and chronic urticaria are routinely tested for monoclonal gammopathy in the context of malignancy, but it should also be considered as a sign of the autoinflammatory syndrome. The Schnitzler syndrome and the adult-onset Still’s disease share common features, so the diagnosis requires a thorough investigation to establish an optimal treatment. In the diagnostic algorithm, monoclonal gammopathy is usually considered red flag for malignancy but might be overlooked as a criterion of Schnitzler syndrome, particularly in young adults. We confirm that the interleukin 1 inhibitor should be the first line of therapy in Schnitzler syndrome, and in the presented case we found it more effective than the interleukin 6 blockade. The main goal of this paper is to increase awareness of Schnitzler syndrome among health care professionals. We aim to present features which can be helpful in differential diagnosis.
Keywords: chronic urticaria, monoclonal gammopathy, tocilizumab, anakinra, autoinflammation
Introduction
Schnitzler syndrome (SchS) is considered to be a rare disorder characterised by the presence of monoclonal IgM protein in serum, fever and chronic urticaria that is associated with considerable morbidity. SchS symptoms significantly impact a patient’s everyday life and in the long term may be followed up by AA amyloidosis. Moreover, up to 20% of SchS patients develop a clinically overt lymphoproliferative disorder such as multiple myeloma, marginal zone lymphomas, and Waldenstrom’s macroglobulinemia (WM).1 The syndrome is under-recognised and can be present in up to 1.5% patients with IgM monoclonal gammopathy.2 In the course of the disease patients are consulted by allergy, dermatology, haematology and rheumatology specialists for particular symptoms, while a common denominator – autoinflammation – is missed, and patients may be deprived of a highly effective therapy which inhibits interleukin 1 (IL-1).3
In this report, we describe the case of SchS with an early disease onset, which was initially diagnosed and treated as adult onset Still’s disease (AOSD). We present potential diagnostic and treatment pitfalls in the management of autoinflammatory syndromes in adults.
Case Presentation
The patient was a 32-year-old Caucasian man with chronic urticaria since the age of 27. At the age of 28, it was accompanied by a recurrent fever of over 39 degrees, lasting for 10–14 days. The initial frequency of the fever attacks increased from once per 4–6 months to once every month. His family history of chronic diseases was unremarkable. He was treated symptomatically with non-steroidal anti-inflammatory drugs (NSAID), antihistamine drugs and penicillin. When he was 31, generalised lymphadenopathy, hepatomegaly, peripheral arthralgia occurred. Laboratory tests revealed leukocytosis 27x10 3/uL with neutrophilia (88%), C-reactive protein (CRP) 14.2 mg/dl (0.1–1.0), monoclonal IgM antibody presence, ferritin 537,5 ng/mL (30–400). Anti-CCP antibodies, antinuclear antibodies (ANA), rheumatoid factor (RF), anti-neutrophil cytoplasmic antibodies (ANCA), thyroid peroxidase antibodies (TPOAb) and thyroglobulin antibodies (TgAb) were negative. C1q, C3 and C4 levels were normal. Lymph node histopathology examination and bone marrow aspirate biopsy showed reactive granulocytosis and excluded malignancy. The consulting haematologist ruled out monoclonal gammopathy and recommended further rheumatological and dermatological diagnostics. The skin specimen biopsy excluded necrotising vasculitis and showed perivascular lymphocyte and slight neutrophils infiltration. He was diagnosed with adult onset Still’s disease (AOSD) and received methylprednisolone pulses (500 mg intravenously for 3 consecutive days) followed by glucocorticoids (20 mg prednisolone orally once a day) and methotrexate (25 mg orally once a week), without any effect. Then, tocilizumab (8 mg/kg intravenously once a month) was added with partial response: CRP lowered but the patient continued to complain about pain in thoracic back, hips, sternum and ribs and chronic urticaria. Low-grade fever up to 38 degrees regularly reappeared about one week before the tocilizumab infusions. He was referred to a clinical immunologist for re-evaluation. On admission, his physical examination revealed peripheral lymphadenopathy and urticaria which covered his extremities, trunk, palms and soles (Figure 1). The skin lesions were associated with an unpleasant burning sensation. The clinical immunologist put into question the diagnosis of AOSD and referred the patient for further investigation. In total the patient received seventeen doses of intravenous tocilizumab in combination with 7.5 to 10 mg daily of prednisone. In his laboratory investigations leukocytosis with neutrophilia, CRP 3.1 mg/dl (ref 0–0.8), serum amyloid A (SAA) 24 mg/dl (ref <0.64) were present. Serum IgG level was 347 mg/dL (ref. 700–1600), IgA 18 mg/dL (ref. 70–400), IgM 1110 mg/dL (40–230), total IgE <2 IU/mL (10–135), monoclonal IgM – kappa serum-free light chain 892 mg/dl (680–1480), lambda serum-free light chain 180 mg/dL (360–840), kappa to lambda ratio 4.96 (1.35–2.65). The whole-body bone scintigraphy (99mTc-methylene diphosphonate) showed an increased uptake of radiotracer localisers in the ribs, in the right iliac bone, in the vertebrae (thoracic region) and in the distal part of the left femur (Figure 2). Exome sequencing was performed and variants in 289 genes, connected with autoinflammatory diseases and primary immune deficiencies, were analysed and did not reveal pathogenic variants. These findings led to the diagnosis of SchS and the switch from tocilizumab to an IL-1 receptor antagonist. Anakinra 100 mg subcutaneously once a day was started. Urticaria and the fever disappeared within 2 days, SAA normalised within 4 weeks. Glucocorticoids were stopped within the first month of anakinra. An unintentional pause in the anakinra treatment caused the re-occurrence of high fever, and generalised urticaria within 3 days. During a continuous anakinra treatment, the patient remains in clinical remission. After a one-year follow-up his SAA is stable within reference range, IgG is 792 mg/dL, IgA is 50 mg/dl (ref 40–400), IgM 907 g/dl (40–230), light serum kappa-free chain 1168 mg/dl (680–1480), light serum lambda-free chain 320 mg/dl (360–840), kappa to lambda ratio 3.65 (1.35–2.65).
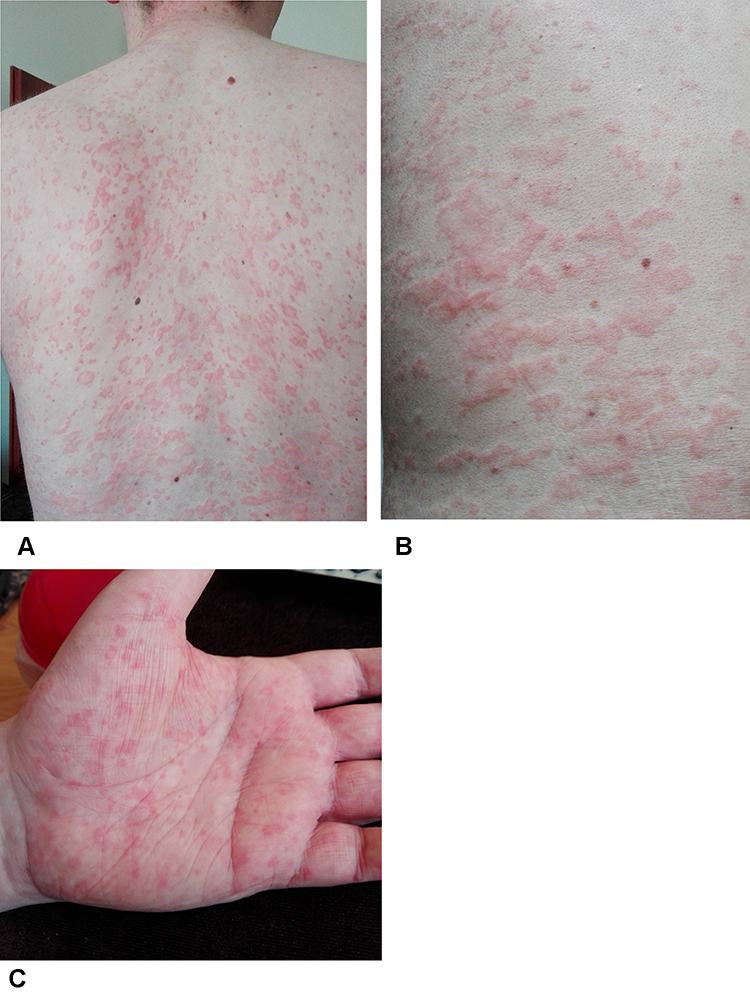 |
Figure 1 The trunk (A and B) and palm (C) of the hand covered in urticarial rash observed in a Schnitzler syndrome patient during treatment with tocilizumab. |
 |
Figure 2 Whole-body bone scintigraphy (99mTc- methylene diphosphonate) in a Schnitzler syndrome (SchS) patient. Arrows indicate an increased radiotracer uptake. |
Discussion
We describe the young male patient with chronic urticaria, monoclonal IgM gammopathy and systemic inflammation. Although he initially fulfilled the criteria of SchS according to literature4 some exceptional features were present: his young age, the urticaria of the palms and soles [Figure 1], and the generalised lymphadenopathy, so a more thorough differential diagnosis work-up was needed. We have shown that due to low awareness of the disease, SchS can be missed in an initial differential algorithm of chronic urticaria. The consequence might be a choice of an ineffective or partially effective treatment. What is more, we have shown that in the presented case the IL-1 receptor antagonist, anakinra, in a standard dose of 100 mg subcutaneously once a day, was more beneficial than long-term tocilizumab treatment.
SchS was first described by a French dermatologist, Liliane Schnitzler.5 Its cardinal features include urticarial rash, recurrent fever, monoclonal IgM or, more rarely, IgG gammopathy and elevated acute inflammatory markers. Urticaria is often the first symptom, preceding the fever.6,7
The disease occurs worldwide, although the overall incidences remain unknown. The published literature involves case reports, case series and one paper which collected the data from 281 patients.1,4,8–11, The pathogenesis involves IL−1, interleukin 6 (IL−6), interleukin 17 (IL−17) activation, but the relation to monoclonal gammopathy remains obscure.12–15
The diagnosis is based on Lipsker’s or Strasbourg’s criteria with high sensitivity and specificity.16–18
SchS is included in the autoinflammatory syndromes group, which all are diagnosed per exclusionism, so the differentiation among them can be a challenge.19 A comparison of main features of discussed autoinflammatory syndromes is shown in Table 1. In the presented case, the initial diagnosis of AOSD was supported by Yamagouchi’s criteria: fever, joint pain, leucocytosis with neutrophilia, hepatomegaly, negative RF and ANA.20 Yet, it was premature. The most important part from the Yamaguchi’s criteria is the requirement of exclusion of other rheumatic conditions, malignancies, and infections. However, the autoinflammatory syndrome had not been considered initially. All autoinflammatory diseases share fever as the leading symptom and reveal high acute phase reactants, so an accurate diagnostic work-up is needed. The most helpful laboratory parameter indicating SchS is the presence of monoclonal IgM, while highly elevated ferritin (5 times the upper reference level) suggests AOSD.21 In the case of our patient, monoclonal IgM led to the search for lymphoproliferative malignancy, but was not considered an indicator of the autoinflammatory syndrome. A bone scintigraphy showed signs of increased bone metabolism are present in 80% of patients with SchS. The bone scans appear to be more sensitive for diagnosis and may correlate with clinical activity better than a positron-emission tomography (PET) scan. Moreover, bone scans may be well positioned to distinguish SchS relapse from other aetiologies of bone, joint, or muscle pain.22 In our case, the scintigraphy showed an increased radiotracer uptake, which correlated with bone pain, but the alkaline phosphatase was normal. Urticaria is a common symptom in SchS and has to be differentiated from chronic spontaneous urticaria (CSU). In CSU individual lesions usually appear, enlarge and then resolve within 24 hours and are accompanied by severe pruritus. A rash in SchS is more painful and burning, often lasting for longer than 24–48 hours. The skin biopsy should be obtained. Infiltration of neutrophils without evidence of vasculitis further supports the diagnosis of autoinflammatory syndromes.
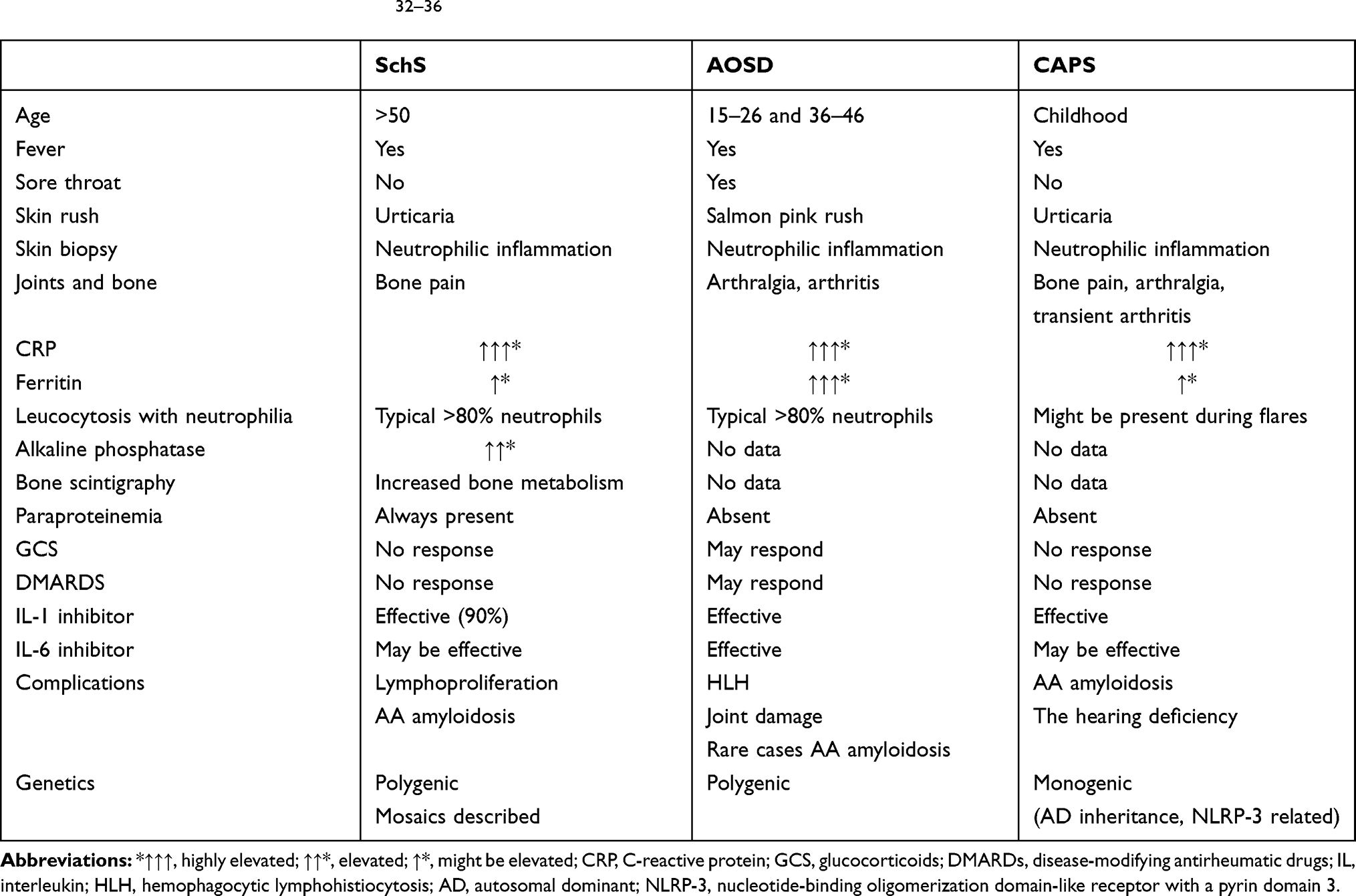 |
Table 1 Comparison of Main Features of Schnitzler Syndrome (SchS), an Adult-Onset Still’s Disease (AOSD) and Cryopyrin-Associated Periodic Syndrome (CAPS)32–36 |
The skin biopsy showed no evidence of vasculitis and only a slight neutrophilic inflammation which occurred in 50% of patients;4 however, it was helpful to exclude spontaneous chronic urticaria and urticarial vasculitis.14,23 Our patient fulfilled both Lipsker’s and Strasbourg’s diagnostic criteria for SchS. However, he was only 27 at the disease’s onset. The presence of monoclonal gammopathy is very rare before 40 and the average age at the onset of SchS is 51–54 years.4,11,24 Until now, a SchS diagnosis before the age of 35 has been an exception.10 In our opinion, due to phenotypical similarities to NLRP3 related diseases, a late onset of monogenic autoinflammatory disease has to be included in the diagnostic work-up in such cases. No pathological variants in exome sequencing were found. Genetic testing for monogenic autoinflammatory syndromes in such cases is an open question.25 Somatic mosaicism of NLRP3 mutations in the myeloid lineage has been reported in two patients with variant SchS; however, the further study of 21 patients with SchS did not support somatic mosaicism in NLRP3, TNFRSF1A, NLRC4, or NOD2 genes. NGS analysis of 32 genes associated with inherited autoinflammatory diseases failed to reveal any common susceptibility factors for SchS.11 In addition, we searched for MYD88 mutations, which may predict the development of Waldenstrom’s macroglobulinemia, and the results were negative.
The next unique issue in the presented case was hypogammaglobulinemia. In rheumatic and hematologic conditions hypogammaglobulinemia can be primary or secondary and can be a warning sign for immunodeficiency.26 In our case, hypogammaglobulinemia was not clinically significant, we did not observe increased susceptibility for any common or atypical infection during the immunosuppressive or tocilizumab treatment. In our opinion, the hypogammaglobulinemia was related to tocilizumab, as IgG and IgA normalised after the tocilizumab was ceased.
The most effective treatment in SchS is IL-1 inhibition. The majority of patients respond to anakinra or canakinumab.27–29 A lack of response to IL-1 blocking therapies should lead to reconsideration of the diagnosis of SchS, while a rapid response to anakinra could also become a diagnostic criterion. The inhibition of IL-6 can be effective, but data is limited to rare cases.7,30 In the case of our patient, we did not observe a clinical response to the Il-6 blockade – only CRP lowered, but urticaria, low-grade fever and bone pain persisted. A difference in response to tocilizumab and anakinra was previously observed in AOSD patients.31 Another explanation is the need to use higher doses of tocilizumab or more frequent administration – for example, every two weeks; however, it would be burdensome for the patient, as it is administered in hospital, and leads to absence at work, which is inconvenient in long-term treatment and raises concerns of a higher cost.
The open question is how long the Il-1 blockade should be conducted. Our experience and data from literature showed that even a short break in anakinra led to the recurrence of symptoms.
The next point to discuss is the prognosis of SchS. At the early observation stage, SchS was considered a benign condition; however, a further follow-up revealed long-term complications.4,7,10,17 Lymphoproliferative disease occurred in 12% of 281 patients after a median 8 years from the diagnosis.4 These were particularly Walderstrom’s macroglobulinemia [WM] (in 21 patients of 35 reported cases) and single cases of non-Hodgkin lymphoma. One patient had acute myeloid leukaemia.4 It is not clear if good control of inflammation has any impact on the risk of progression to lymphoproliferation, and, if in this group, the risk factors are similar to a monoclonal gammopathy of unknown significance (MGUS).24 In MGUS the risk of progression to multiple myeloma or another plasma-cell or lymphoid disorder is higher if the ratio of the kappa to lambda chain is abnormal and monoclonal protein IgM is ≥1.5 g/dl. A deficiency of two other main immunoglobulin classes is also disadvantageous.24 In our case, IgA and IgG normalised, and the IgM threshold was stable. Another complication might be AA amyloidosis that justifies the careful control of SAA during treatment.3
Conclusion
SchS and AOSD share common features and the differential diagnosis requires a thorough investigation to establish the optimal treatment. Interleukin 1 inhibitor is the first-line therapy in SchS and in the presented case we found it more effective than tocilizumab. Patients with a history of recurrent fever and chronic urticaria are routinely tested for monoclonal gammopathy. However, when malignancy is excluded, the presence of monoclonal protein can represent a sign of SchS. Due to the potential development of lymphoproliferative disorder, SchS requires a long-term haematological follow-up.
Ethics Statement
Based on the regulations of Clinical Hospital of the Ministry of National Defense, Military Institute of Medicine in Warsaw an institutional review board approval is not required for case reports.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bashir M, Bettendorf B, Hariman RA. Rare but fascinating disorder: case collection of patients with Schnitzler syndrome. Case Rep Rheumatol. 2018;7041576. doi:10.1155/2018/7041576
2. Jain T, Offord CP, Kyle RA, Dingli D. Schnitzler syndrome: an under-diagnosed clinical entity. Haematologica. 2013;98(10):
3. Palladini G, Merlini G. The elusive pathogenesis of Schnitzler syndrome. Blood. 2018;131(9):944–946. doi:10.1182/blood-2018-01-824862
4. de Koning HD. Schnitzler’s syndrome: lessons from 281 cases. Clin Transl Allergy. 2014;
5. Schnitzler L, Schubert B, Boasson M, et al. Urticaire chronique, lésions osseuses, macroglobulinémie IgM: maladie de Waldenström. Bull Soc Fr Dermatol Syphiligr. 1974;81:363.
6. Gusdorf L, Lipsker D. Schnitzler syndrome: a review. Curr Rheumatol Rep. 2017;19(8):46. doi:10.1007/s11926-017-0673-5
7. Davis MDP, van der Hilst JCH. Mimickers of urticaria: urticarial vasculitis and autoinflammatory diseases. J Allergy Clin Immunol Pract. 2018;6(4):1162–1170. doi:10.1016/j.jaip.2018.05.006
8. Kim YS, Song YM, Bang CH, et al. Schnitzler syndrome: a case report and review of literature. Ann Dermatol. 2018;30(4):483–485. doi:10.5021/ad.2018.30.4.483
9. Sokumbi O, Drage LA, Peters MS. Clinical and histopathologic review of Schnitzler syndrome: the Mayo clinic experience (19722011). J Am Acad Dermatol. 2012;67(6):1289–1295. doi:10.1016/j.jaad.2012.04.027
10. de Koning HD, Bodar EJ, van der Meer JW, Simon A. Schnitzler Syndrome Study Group. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum. 2007;37(3):137–148. doi:10.1016/j.semarthrit.2007.04.001
11. Rowczenio DM, Pathak S, Arostegui JI, et al. Molecular genetic investigation, clinical features, and response to treatment in 21 patients with Schnitzler syndrome. Blood. 2018;131(9):
12. Noster R, de Koning HD, Maier E, Prelog M, Lainka E, Zielinski CE. Dysregulation of proinflammatory versus anti-inflammatory human TH17 cell functionalities in the autoinflammatory Schnitzler syndrome. J Allergy Clin Immunol. 2016t;138(4):1161–1169.e6. doi:10.1016/j.jaci.2015.12.1338.
13. Pathak S, Rowczenio DM, Owen RG, et al. Exploratory study of MYD88 L265P, Rare NLRP3 variants, and clonal hematopoiesis prevalence in patients with Schnitzler syndrome. Arthritis Rheumatol. 2019;71(12):2121–2125. doi:10.1002/art.41030
14. Bonnekoh H, Scheffel J, Wu J, Hoffmann S, Maurer M, Krause K. Skin and systemic inflammation in Schnitzler’s syndrome are associated with neutrophil extracellular trap formation. Front Immunol. 2019;10:546. doi:10.3389/fimmu.2019.00546
15. Migliorini P, Italiani P, Pratesi F, Puxeddu I, Boraschi D. Cytokines and soluble receptors of the interleukin-1 family in Schnitzler syndrome. Scand J Rheumatol. 2019;48(3):235–238. doi:10.1080/03009742.2018.1550210
16. Lipsker D, Veran Y, Grunenberger F, Cribier B, Heid E, Grosshans E. The Schnitzler syndrome. Four new cases and review of the literature. Medicine. 2001;80(1):37–44. doi:10.1097/00005792-200101000-00004
17. Simon A, Asli B, Braun-Falco M, et al. Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy. 2013;68(5):562–568. doi:10.1111/all.12129
18. Gusdorf L, Asli B, Barbarot S, et al. Schnitzler syndrome: validation and applicability of diagnostic criteria in real-life patients. Allergy. 2017;72(2):177–182. doi:10.1111/all.13035
19. Gusdorf L, Lipsker D. Neutrophilic urticarial dermatosis: an entity bridging monogenic and polygenic autoinflammatory disorders, and beyond. J Eur Acad Dermatol Venereol. 2020;34(4):685–690. doi:10.1111/jdv.15984
20. Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19(3):424–430.
21. Bilgin E, Hayran M, Erden A, et al. Proposal for a simple algorithm to differentiate adult-onset Still’s disease with other fever of unknown origin causes: a longitudinal prospective study. Clin Rheumatol. 2019;38(6):1699–1706. doi:10.1007/s10067-019-04455-y
22. Alix L, Néel A, Cador B, et al. Diagnostic value of 18-F fluorodeoxyglucose PET/CT and bone scan in Schnitzler syndrome. Autoimmunity. 2019;52(78):264–271. doi:10.1080/08916934.2019
23. Bonnekoh H, Scheffel J, Maurer M, Krause K. Use of skin biomarker profiles to distinguish Schnitzler syndrome from chronic spontaneous urticaria: results of a pilot study. Br J Dermatol. 2018;178(2):561–562. doi:10.1111/bjd.15705
24. Kyle RA, Larson DR, Therneau TM, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018;378(3):241–249. doi:10.1056/NEJMoa1709974
25. Chuamanochan M, Weller K, Feist E, et al. State of care for patients with systemic autoinflammatory diseases – results of a tertiary care survey. World Allergy Organ J. 2019;12(3):100019. doi:10.1016/j.waojou.2019.100019
26. Napiórkowska-Baran K, Zalewska J, Jeka S, et al. Determination of antibodies in everyday rheumatological practice. Reumatologia. 2019;57(2):91–99. doi:10.5114/reum.2019.84814
27. Néel A, Henry B, Barbarot S, et al. Long-term effectiveness and safety of interleukin-1 receptor antagonist (anakinra) in Schnitzler’s syndrome: a French multicenter study. Autoimmun Rev. 2014;13(10):1035–1041. doi:10.1016/j.autrev.2014.08.031
28. Krause K, Tsianakas A, Wagner N, et al. Efficacy and safety of canakinumab in Schnitzler syndrome: a multicenter randomized placebo-controlled study. J Allergy Clin Immunol. 2017;139(4):1311–1320. doi:10.1016/j.jaci.2016.07.041
29. Krause K, Bonnekoh H, Ellrich A, et al. Long-term efficacy of canakinumab in the treatment of Schnitzler syndrome. J Allergy Clin Immunol. 2020;
30. Claus J, Vanderschueren S. Variable responses to Tocilizumab in four patients with Schnitzler syndrome. J Clin Immunol. 2019;39(4):370–372. doi:10.1007/s10875-019-00644-1
31. Vercruysse F, Barnetche T, Lazaro E, et al. Adult-onset Still’s disease biological treatment strategy may depend on the phenotypic dichotomy. Arthritis Res Ther. 2019;21(1):53. doi:10.1186/s13075-019-1838-6
32. Gerfaud-Valentin M, Maucort-Boulch D, Hot A, et al. Adult-onset still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine. 2014;93(2):91–99. doi:10.1097/MD.0000000000000021
33. Colafrancesco S, Priori R, Valesini G, et al. Response to Interleukin-1 Inhibitors in 140 Italian patients with adult-onset still’s disease: a multicentre retrospective observational study. Front Pharmacol. 2017;8:369. doi:10.3389/fphar.2017.00369
34. Zhou S, Qiao J, Bai J, Wu Y, Fang H. Biological therapy of traditional therapy-resistant adult-onset Still’s disease: an evidence-based review. Ther Clin Risk Manag. 2018;24(14):167–171. doi:10.2147/TCRM.S155488
35. Ruscitti P, Cipriani P, Liakouli V, et al. Managing adult-onset Still’s disease: the effectiveness of high-dosage of corticosteroids as first-line treatment in inducing the clinical remission. Results from an observational study. Medicine. 2019;98(15):e15123. doi:10.1097/MD.0000000000015123
36. Levy R, Gérard L, Kuemmerle-Deschner J, et al.; for PRINTO and Eurofever. Phenotypic and genotypic characteristics of cryopyrin-associated periodic syndrome: a series of 136 patients from the Eurofever Registry. Ann Rheum Dis. 2015;74(11):2043–2049. doi:10.1136/annrheumdis-2013-204991.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.