")
Back to Journals » OncoTargets and Therapy » Volume 13
Saikosaponin D Inhibits Proliferation and Promotes Apoptosis Through Activation of MKK4–JNK Signaling Pathway in Pancreatic Cancer Cells
Authors Lai M, Ge Y, Chen M, Sun S, Chen J, Cheng R
Received 25 May 2020
Accepted for publication 20 August 2020
Published 24 September 2020 Volume 2020:13 Pages 9465—9479
DOI https://doi.org/10.2147/OTT.S263322
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Mengru Lai1 ,* Yuqing Ge2 ,* Meng Chen,1 Siya Sun,1 Jianzhen Chen,1 Rubin Cheng1
1College of Pharmaceutical Science, Zhejiang Chinese Medical University, Hangzhou 310053, People’s Republic of China; 2First Affiliated Hospital, Zhejiang Chinese Medical University, Hangzhou 310006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yuqing Ge
First Affiliated Hospital, Zhejiang Chinese Medical University, 54 Youdian Road, Hangzhou, Zhejiang Province 310006, People’s Republic of China
Tel +86-571-8662-0280
Email [email protected]
Rubin Cheng
College of Pharmaceutical Science, Zhejiang Chinese Medical University, 548 Binwen Road, Hangzhou, Zhejiang Province 310053, People’s Republic of China
Tel +86-571-6176-8180
Email [email protected]
Introduction: Pancreatic cancer remains one of the most lethal malignancies and has few treatment options. Saikosaponin D (SSD), a major bioactive triterpene saponin isolated from Bupleurum chinense, has been reported to exert cytotoxicity properties toward many cancer cells. However, the effects of SSD on pancreatic cancer have been little scrutinized.
Methods: Here, we investigated the effect of SSD on the proliferation and apoptosis of human pancreatic cancer BxPC3 and PANC1 cells and the mouse pancreatic cancer cell line Pan02. Cell viability was determined by MTT assays and cell apoptosis analyzed by DAPI staining and flow cytometry. Expression levels of apoptosis-regulating markers and activity of the MKK4–JNK signaling pathway were determined by Western blotting. The inhibitor SP600125 was applied to confirm the role of the JNK pathway in SSD efficiency.
Results: SSD significantly inhibited the proliferation of BxPC3, PANC1, and Pan02 cells in a concentration- and time-dependent manner. Flow-cytometry analysis indicated obvious apoptosis induction after SSD exposure. Furthermore, SSD significantly triggered cleavage of caspase 3 and caspase 9 proteins and increased the expression of FoxO3a. In addition, activity of the MKK4–JNK pathway was dramatically increased after treatment with SSD in BxPC3 cells. SSD obviously stimulated phosphorylation of JNK, cJun, and SEK1/MKK4 proteins within 30 minutes. The addition of SP600125 blocked the activation of SSD on the MKK4–JNK regulatory pathway and reversed the effects of SSD on proliferation inhibition and apoptosis induction in BxPC3 cells.
Conclusion: These results revealed that SSD was capable of suppressing tumor growth and promoting apoptosis of pancreatic cancer cells via targeting the MKK4–JNK signaling pathway, indicating the possibility of further developing SSD as a potential therapeutic candidate for pancreatic cancer.
Keywords: saikosaponin D, pancreatic cancer, proliferation inhibition, apoptosis induction, MKK4–JNK pathway
Introduction
Pancreatic cancer is one of the most lethal of solid malignancies, with 5-year survival not exceeding 5%.1 Surgery resection is still the primary therapy choice for pancreatic cancer if it is feasible. However, most pancreatic cancers are reported to have a complex and delayed diagnosis, due to the pancreas’s anatomic localization and the aspecific and intermittent nature of symptoms. As a result, >85% of diagnosed patients show metastasic infiltrations in proximal lymphatic nodes or the liver.2 First-line treatment for the unresectable pancreatic cancer is currently based on gemcitabine monotherapy or combination thereof with other agents.3 Despite recent major advances in therapeutic strategies for cancer, pancreatic cancer prognosis remains extremely poor, mainly because of its high resistance to apoptosis induction by chemotherapy or radiotherapy.4 Therefore, it is urgent to develop effective therapeutic strategies to improve the prognosis of this aggressive disease.
Saikosaponin D (SSD) is one of the main bioactive triterpene-saponin compounds derived from roots of Bupleurum chinense, and is a commonly used herbal drug for liver-disease treatment.5 SSD is regarded as the major effective ingredient in many traditional medicinal formulations, and has been shown to possess multiple pharmacological benefits.6 In recent years, SSD has attracted increasing attention in the field of cancer research. SSD exhibits significant growth suppression and apoptotic death induction on various human solid tumors, including hepatocellular carcinoma, breast cancer, prostate cancer, and lung cancer.7 In addition, SSD has diverse regulatory mechanisms in different cancer cells with unique genotypes. However, the effect of SSD and its underlying mechanisms on pancreatic cancer cells remain elusive and need further exploration.
MKK4 and its downstream mediator JNK, members of the MAPK family, play vital roles in chemically triggered cancer cell–cycle arrest and apoptotic processes.8 JNK phosphorylation activates substrates of transcription factors or proapoptotic proteins in an MKK4-dependent manner. Functional suppression of JNK activation has recently been considered as a pivotal cellular survival mechanism and contributes to cancer cells avoiding apoptosis.9 Low expression levels of phosphorylated MKK4 are closely associated with unfavorable prognosis in colorectal cancer patients, suggesting involvement in tumor progression and metastasis mediation.10 Inactivation of MKK4 can facilitate cancer-cell survival via inhibiting the JNK-mediated apoptosis pathway.11 In addition, MKK4 dysregulation occurs during the development of clinical cancer metastases, and endogenous expression of MKK4 can reduce metastasis of ovarian tumors in mice.12 High expression of the MKK4 protein is significantly correlated with longer overall survival and could predict favorable prognosis in resectable pancreatic ductal adenocarcinoma.13 Furthermore, activation of the MKK4–JNK signaling pathway would be a promising therapeutic approach for tumor cells to induce proliferation inhibition and metastasis prevention. Treatment with arsenite triggers the cell-apoptotic signals and activates the MKK4–JNK cell-death pathway dependent on GADD45α upregulation.14 Sophoridine exhibits effects of proliferation inhibition and mitochondrial-related apoptosis induction in pancreatic cancer cells via ROS-dependent JNK activation.15 Activation of the MKK4–JNK signaling cascade by curcumin also promotes cell apoptosis in human gastric cancer cells.16 As such, identifying bioactive modulators targeting the MKK4–JNK signaling pathway provides an alternative approach to contribute to chemotherapy advances for pancreatic cancer.
In the present study, we evaluated the effects of SSD on pancreatic cancer–cell proliferation, apoptosis, and expression of potential mediators. In addition, we further investigated the relationship of SSD and the MKK4–JNK signaling pathway in regulating growth inhibition and apoptotic death to clarify the underlying mechanisms of SSD in pancreatic cancer cells.
Methods
Main Reagents
SSD (molecular weight 780.98 Da, HPLC≥98%) was purchased from Yuanye Biotechnology (Shanghai, China), and its structural formula is shown in Figure 1A. SSD was dissolved in absolute DMSO and further diluted with culture medium at different concentrations. RPMI 1640 medium (2.05 mM L-glutamine without calcium nitrate) was obtained from Cellmax (Beijing, China) and FBS Hangzhou Sijiqing (Zhejiang, China). The MTT-assay kit and DAPI-staining kit were bought from Beyotime Biotechnology (Shanghai, China). An annexin V–FITC + PI double-staining kit from BD Biosciences (Franklin Lakes, NJ, USA) was used to determine cell-apoptosis rates. The JNK-specific inhibitor SP600125 (S1460) was obtained from Selleck Chemicals (Houston, TX, USA). Antibodies used in this study were purchased from Cell Signaling Technology (Danvers, MA, USA): cleaved caspase 3 (9664), p53 (2527), FoxO3a (12,829), pJNK (9251), p-cJun (2361), pSEK1/MKK4 (4514), pERK (4370), pATF2 (5112), and β-actin (3700). The cleaved caspase 9 antibody Asp353 (AF5240) was obtained from Affinity Biosciences (Cincinnati, OH, USA). Other reagents employed in this study are indicated separately wherever appropriate.
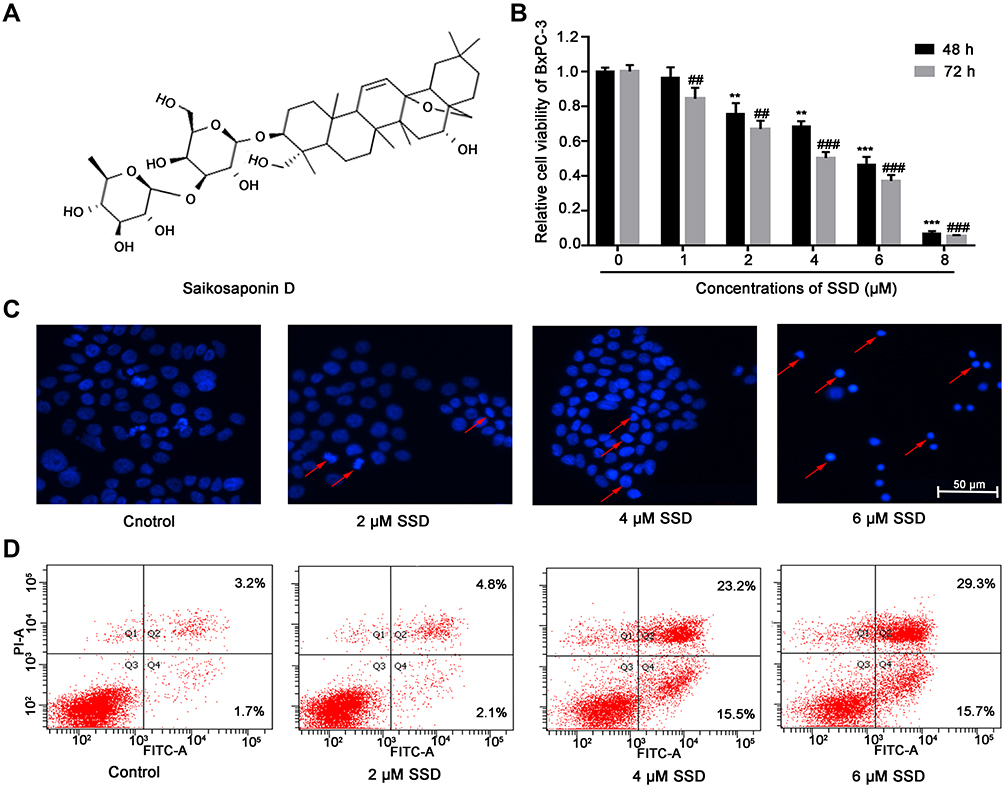 |
Figure 1 SSD treatment resulted in growth inhibition and apoptosis induction in BxPC3 pancreatic cancer cells. (A) Chemical structure of SSD. (B) BxPC3 cells were treated with indicated concentrations of SSD for 48 and 72 hours, after which cell viability was measured using MTT assays. Values represent mean ± SD of three independent experiments. **P<0.01, ***P<0.001 compared with cells without SSD treatment for 48 hours; ##P<0.01, ###P<0.001 compared with cells without SSD treatment for 72 hours. (C) BxPC3 cells were treated with different concentrations of SSD (0, 2, 4, and 6 μM) for 48 hours and then stained with DAPI. Fragmented or condensed nuclei were observed under fluorescence microscope, as indicated by the red arrows. (D) Flow-cytometry assay were performed to detect rates of apoptosis using annexin V–FITC + PI double-staining. BxPC3 cells were treated with different concentrations of SSD for 48 hours and then collected for apoptosis analysis. |
Cell Lines and Cell Culture
The human pancreatic cancer cell lines BxPC3 and PANC1 were purchased from the Institute of Biochemistry and Cell Biology at the Chinese Academy of Sciences (Shanghai, China). The mouse pancreatic cancer cell line Pan02 was purchased from Tongpai Biotechnology (Shanghai, China). Cells were cultured in RPMI 1640 medium containing 10% FBS and maintained at 37°C in a 5% CO2-humidified atmosphere.
Cell-Viability Assays
MTT assays followed the manufacturer’s recommendations to estimate cell viability after treatment with SSD. Pancreatic cancer cells were seeded in 96-well plates at a density of 104 cells per well in 100 μL RPMI 1640 medium supplemented with 10% FBS. After overnight incubation, the culture medium was removed and replaced by SSD diluted in medium to a final concentration of 1–8 μM for 48 hours and 72 hours. Cells were subsequently incubated with a final concentration of 5 mg/mL MTT for 4 hours, and formazan crystals were dissolved in 150 μL DMSO. Absorbance at 490 nm was measured on a microplate reader. As for JNK-inhibition analysis, the cells that had been seeded in 96-well plates were pretreated with 10 µM SP600125 for 1 hour, then treated with different concentrations of SSD. Next, absorbance was measured for cell viability. Independent experiments were repeated in triplicate.
DAPI Staining
Apoptotic cells were stained with DAPI solution as described previously.17 After treatment with various concentrations of SSD, culture media were discarded and BxPC3 cells fixed with 4% paraformaldehyde for 10 minutes at room temperature. Next, fixed cells were washed with PBS three times and stained with DAPI solution for 15 minutes at room temperature away from light. Then, cells were washed twice with PBS again and observed with fluorescence microscopy.
Flow-Cytometry Analysis
Apoptosis of pancreatic cancer cells was detected by annexin V–FITC + PI double-staining analysis. After treatment with different concentrations of SSD, cells were collected and washed twice with PBS. Then, cells were resuspended in 100 μL binding buffer containing 5 μL annexin V–FITC conjugate and 5 μL PI and incubated at room temperature for 20 minutes in the dark. After adding 300 μL annexin V–FITC binding buffer, cells were collected and quantitatively analyzed using flow cytometry (FACSCalibur; BD Biosciences).
Western Blotting Analysis
Pancreatic cancer cells were incubated in six-well plates at a density of 5×105 cells per well. After growth overnight, cells were treated with different concentrations of SSD. Next, total protein from cells was extracted using RIPA lysis buffer and PMSF according to the instructions (Beyotime Biotechnology). BCA-protein assays were carried out to quantify concentrations of cellular proteins, and equal amounts of denatured protein samples were used for subsequent experiments. Cellular proteins were separated by different concentrations of SDS-PAGE depending on the size of the target protein, and then electrotransferred onto PVDF membranes for about 2 hours. Next, membranes were blocked with 5% (w:v) nonfat dry milk and 0.1% Tween 20 in TBS at room temperature for at least 1 hour, then incubated with target antibodies overnight at 4°C. After being washed three times with TBST, membranes were incubated with antirabbit secondary antibody or antimouse secondary antibody for 1 hour at room temperature. An enhanced-chemiluminescence reagent (Thermo Fisher Scientific) was used to detect immunoreactive protein bands by gel imaging, while ImageJ software was used for grayscale analysis. Data were normalized to β-Actin.
Statistical Analysis
Results are shown as means ± SD of at least three independent experiments. All statistical analyses were done using GraphPad Prism 7.0 software. Data were determined by unpaired t-tests and one-way ANOVAs to compare between two independent groups. P<0.05 was considered statistically significant.
Results
SSD Decreased Cell Viability and Induced Cell Apoptosis in Pancreatic Cancer Cells
To investigate the antiproliferative effect of SSD, BxPC3 cells were incubated with different concentrations of SSD for 48 and 72 hours. Then, cell viability in each group was determined using MTT assays. The results revealed that SSD significantly inhibited growth rates of BxPC3 cells in a dosage- and time-dependent manner (Figure 1B). After treatment with 1 µM SSD for 48 hours in BxPC3 cells, the proliferative inhibitory rate was just 3.44%, while the inhibitory rate became 31.61% when the SSD concentration increased to 4 µM, which was 9.2-folds that of the 1 µM–treatment group. IC50 values of SSD on BxPC3 cells were 4.47 µM and 3.27 µM at 48 and 72 hours, respectively. DAPI-staining results indicated that BxPC3 cells exhibited typical morphological features of apoptotic cells following treatment with 4 and 6 µM SSD, including bright nuclear condensation and perinuclear apoptotic bodies (Figure 1C). Furthermore, SSD treatment triggered significantly increased cell-apoptosis rates in a dose-dependent manner (Figure 1D). Apoptosis in BxPC3 cells was only 4.9% in the control group. However, apoptosis rates had increased to 38.7% and 45.0% after treatment with 4 µM and 6 µM SSD for 48 hours: 7.9- and 9.2-fold that of the control group, respectively.
To further confirm the anticancer effects of SSD, proliferation and apoptosis rates were evaluated in PANC1 and Pan02 cells after treatment with various concentrations of SSD. MTT results demonstrated that SSD significantly decreased the growth of PANC1 and Pan02 cells in a dose- and time-dependent manner (Figure 2A and 2B). IC50 values of SSD in PANC1 and Pan02 cells at 48 hours were 5.18 and 4.76 µM, respectively. In addition, SSD treatment also promoted apoptosis in both PANC1 and Pan02 cells (Figure 2C and 2D). After treatment with 6 μM SSD, cell apoptosis in PANC1 and Pan02 cells was 63.6% and 25.1%, respectively (Figure 2C and 2D). These results were consistent with the cell viability–assay results. Our results demonstrated the proliferation-inhibition and apoptosis-promotion effects of SSD in pancreatic cancer cells.
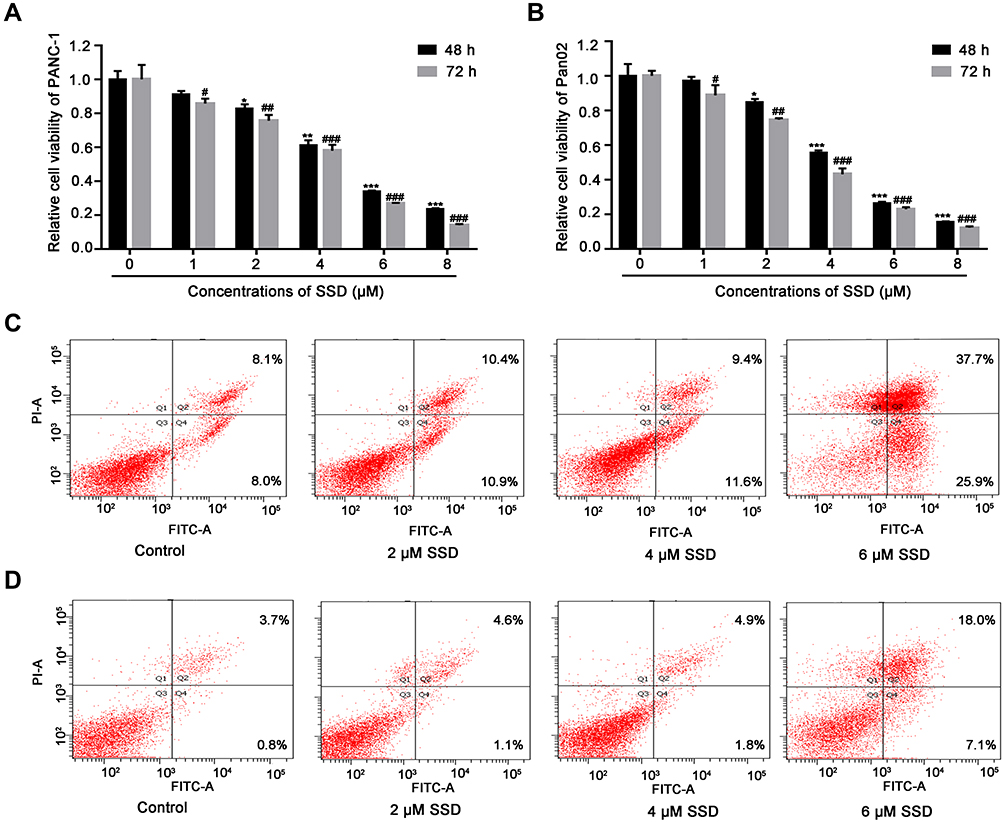 |
Figure 2 SSD treatment triggered growth inhibition and apoptosis induction in PANC1 and Pan02 cells. (A) Effect of SSD on proliferation in PANC1 cells. (B) Effect of SSD on proliferation in Pan02 cells. Cells were treated with indicated concentrations of SSD for 48 and 72 hours, after which cell viability was measured using MTT assays. Values represent means ± SD of three independent experiments.*P<0.05, **P<0.01, ***P<0.001 compared with cells without SSD treatment for 48 hours; #P<0.05, ##P<0.01, ###P<0.001 compared with cells without SSD treatment for 72 hours. (C) The effect of SSD on the cell apoptosis rate in PANC1 cells. (D) The effect of SSD on the cell apoptosis rate in Pan02 cells. Cells were treated with indicated concentrations of SSD for 48 hours and then collected for apoptosis analysis using annexin V–FITC + PI double-staining. |
SSD Activated Cleavage of Caspase Proteins and Increased Expression of FoxO3a in Pancreatic Cancer Cells
To understand the mechanisms of SSD involved in cell apoptosis, we carried out Western blotting experiments to determine expressions of potential apoptosis-regulatory proteins in pancreatic cancer cells. After treatment with different concentrations of SSD, caspase 3 and caspase 9 activity and protein-expression levels of p53 and FoxO3a were analyzed and quantified. As shown in Figure 3A, SSD significantly increased the cleavage of the proapoptotic proteins caspase 3 and caspase 9 and obviously enhanced the expression of FoxO3a in a dosage-dependent manner. Semiquantitative analysis indicated that cleavage of caspase 3 and caspase 9 in the 2 µM SSD–treatment group were 1.5- and 1.4-fold that of the control group, respectively (Figure 3B), while in the 4 µM SSD–treatment group protein expression levels of cleaved caspase 3 and caspase 9 became 2.1- and 1.8-fold that of the control group, respectively (Figure 3B). It is worth noting that SSD showed a slight regulatory effect on the total expression of mutant p53 protein in BxPC3 cells (Figure 3B).
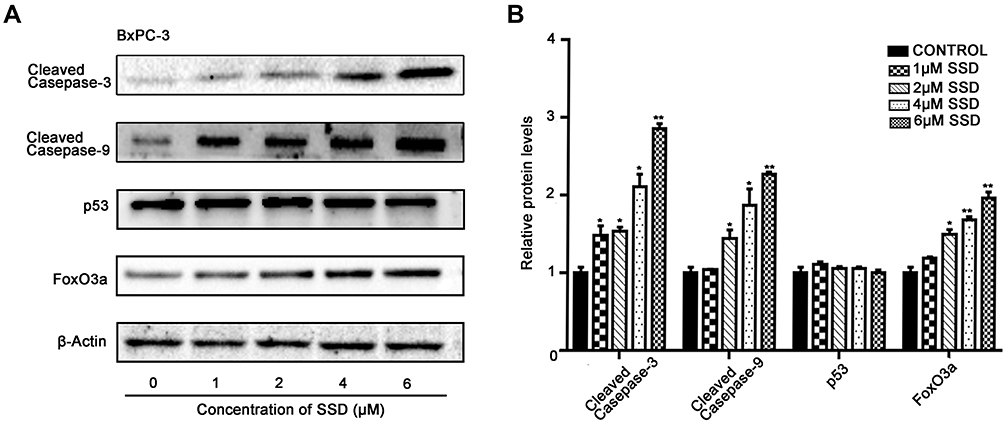 |
Figure 3 Effects of SSD on apoptosis-related proteins in BxPC3 cells. (A) BxPC3 cells were treated with indicated concentrations of SSD for 48 hours and expression levels of cleaved caspase 9, cleaved caspase 3, p53, and FoxO3a determined using Western blotting. β-actin served as the internal control. (B) Semiquantitative immunoblot-staining analysis of the indicated apoptosis-regulatory proteins on SSD treatment with an image analyzer. Grayscale-scan analysis of Western blot images was from three independent experiments, and fold changes included normalization to β-actin. *P<0.05, **P<0.01 denote significant differences compared with the control group for each protein. |
To further clarify the proapoptosis mechanisms of SSD in pancreatic cancer cells,expression levels of cleaved caspase 3, caspase 9, and FoxO3a were determined in PANC1 and Pan02 cells. The results demonstrated that SSD treatment significantly increased the activity of caspase proteins and promoted the expression of FoxO3a in PANC1 and Pan02 cells (Figure 4A and 4B). Compared with the control group, FoxO3a expression in the 2 µM SSD–treatment group and 4 µM SSD–treatment group increased by 54.6% and 65.7% in PANC1 cells, respectively (Figure 4C). Similar results were observed in Pan02 cells (Figure 4D). Treatment with 4 µM SSD led to a 68.3% increase in the expression level of FoxO3 and a 45.9% increase in caspase 9 cleavage in Pan02 cells (Figure 4D). These results suggested that SSD promoted apoptosis of pancreatic cancer cells through activating caspase 3 and caspase 9 proteins and increasing expression of FoxO3a in vitro.
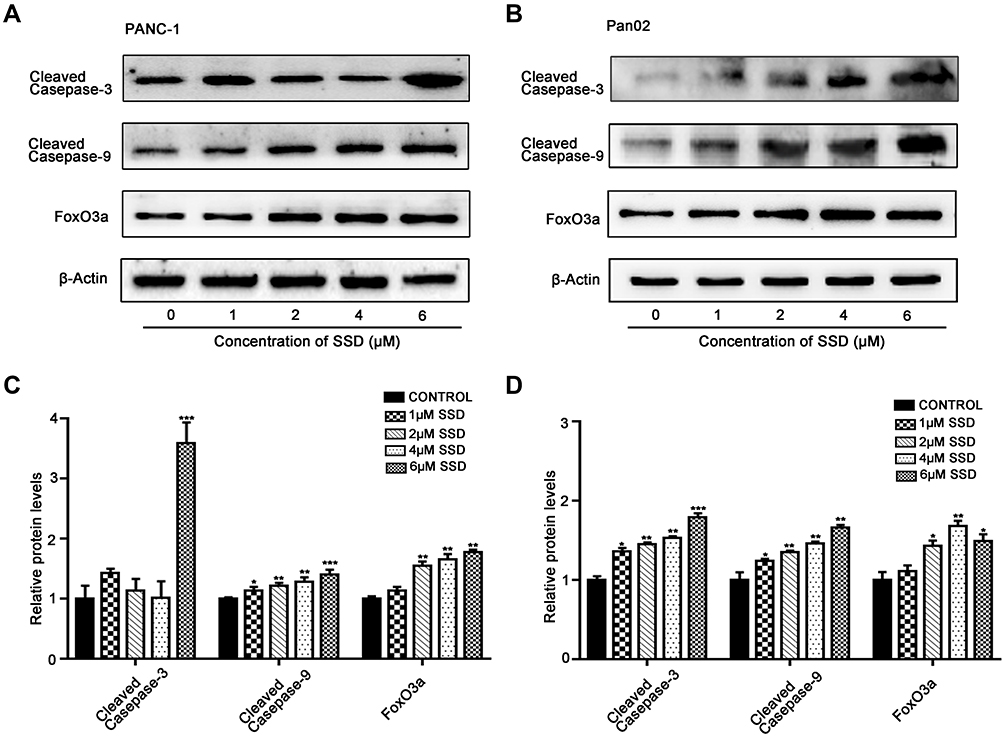 |
Figure 4 Effects of SSD on apoptosis-regulatory proteins in PANC1 and Pan02 cells. (A) SSD treatment caused activation of caspase proteins and increased expression of FoxO3a in PANC1 cells. (B) SSD treatment caused activation of caspase proteins and increased expression of FoxO3a in Pan02 cells. Cells were treated with indicated concentrations of SSD for 48 hours and levels cleaved caspase 9, cleaved caspase 3, and FoxO3a expression determined using Western blotting. β-actin served as the internal control. (C) Semiquantitative analysis of Western blotting results in PANC1 cells. (D) Semiquantitative analysis of Western blotting results in Pan02 cells and semiquantitative immunoblotting analysis of indicated proteins on SSD treatment with an image analyzer. Grayscale scan analysis of Western blot images was from three independent experiments, and fold changes included normalization to β-actin. *P<0.05, **P<0.01, ***P<0.001 compared with the control group for each protein |
SSD Induced Activation of MKK4–JNK Signaling Pathway in BxPC3 Cells
Given the central roles of the MKK4–JNK signaling pathway in apoptosis induction and tumor progression,18 we determined changes in master regulators involved in this pathway after treatment with SSD. As shown in Figure 5A, SSD increased the phosphorylation of JNK, SEK1/MKK4, and ATF2 in a concentration-dependent way in BxPC3 cells. After treatment with 2 µM SSD for 48 hours, protein levels of pJNK, pSEK1/MKK4, and pATF2 had increased 1.5-, 2.6-, and 1.7-fold, respectively, over the control group, while 4 µM SSD treatment upregulated the activities of JNK, SEK1/MKK4, and ATF2 proteins by 1.8-, 3.7-, and 2.4-fold, respectively (Figure 5B). In addition, the expression of phosphorylation of ERK was obviously increased by low concentrations of SSD (Figure 5A). However, 6 μM SSD treatment significantly inhibited the expression of pERK, indicating complex regulatory mechanisms involving SSD and the ERK-signaling pathway in pancreatic cancer cells.15,19
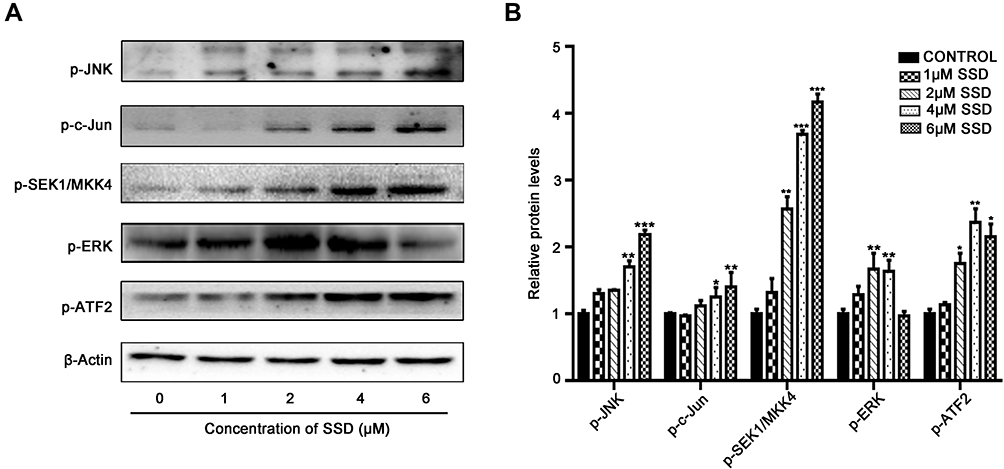 |
Figure 5 Effects of SSD on activiy of MKK4–JNK pathway in BxPC3 cells. (A) BxPC3 cells were treated with indicaed concentrations of SSD, and expression levels of phosphorylation of JNK, cJun, SEK1/MKK4, ERK, and ATF2 were analyzed using Western blotting. β-actin served as internal control. (B) Semiquantitative immunoblot-staining analysis of indicated MKK4–JNK signaling-pathway proteins on SSD treatment by grayscale-scan analysis. *pP<0.05, **pP<0.01, ***pP<0.001 compared with control group for each protein. Data expressed as means ± SD, n=3. |
The time-course activities of the MKK4–JNK signaling pathway were further analyzed after SSD treatment. As shown in Figure 6A, SSD triggered rapid activation in this pathway in BxPC3 cells. Semiquantitative analysis suggested that after the treatment with SSD for just 30 minutes, expression levels of pJNK and pSEK1/MKK4 had increased 1.2- and 1.5-fold over the control group, respectively (Figure 6B). Phosphorylation of the JNK protein reached its maximum at 4 hours, whileexpression of pSEK1/MKK4 peaked at 2 hours. Furthermore, SSD exposure obviously activated the phosphorylation of cJun, suggesting a potential regulatory target for SSD in BxPC3 cells. Compared with the ERK-signaling pathway, the MKK4–JNK signaling pathway was more sensitive to SSD treatment in PANC1 cells (Figure 6A). These data indicated that activation of the MKK4–JNK pathway was a primary event during SSD-induced proliferation inhibition and apoptosis in these cells.
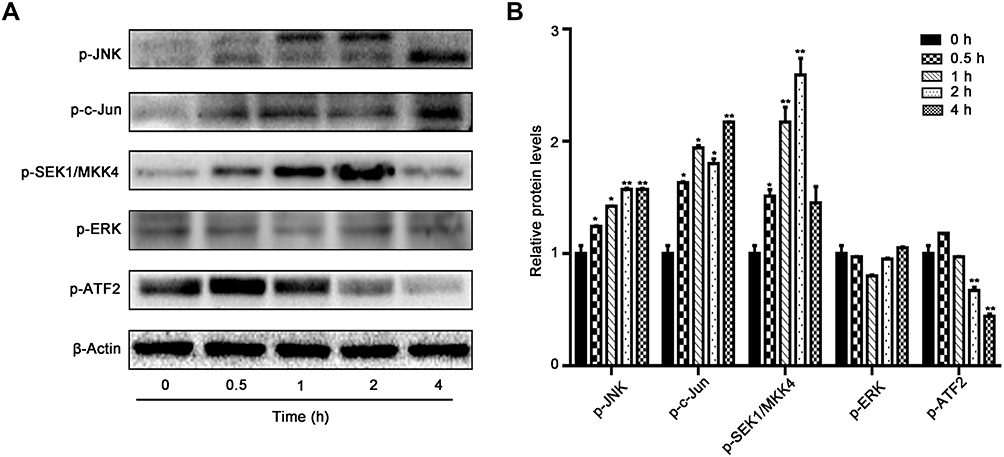 |
Figure 6 Effects of time-course activity on MKK4–JNK signaling pathway in BxPC3 cells. (A) BxPC3 cells were treated with 4 μM SSD for different periods and changes in protein levels of phosphorylation of JNK, cJun, SEK1/MKK4, ERK, and ATF2 determined using Western blotting. (B) Semiquantitative immunoblot-staining analysis of indicated proteins on different time courses with an image analyzer. Grayscale-scan analysis of Western blot images was from three independent experiments, and fold changes include normalization to β-actin. *pP<0.05, **pP<0.01 compared with the control group (0 hours) for each protein. |
Blocking of JNK Activation Suppressed SSD‑Induced Apoptosis and Reversed SSD-Induced Proliferative Inhibition
In order to further confirm the regulatory effect of the JNK pathway during SSD treatment in pancreatic cancer, the specific JNK inhibitor SP600125 was applied to block JNK activation by SSD exposure. As shown in Figure 7A, SSD increase the phosphorylation of JNK, SEK1/MKK4, ERK, and ATF2 effectively, consistent with previous results. After treatment with SP600125, the upregulated expression of pJNK, pERK, and pATF2 on SSD had significantly decreased. In the absence of SP600125, 2 µM SSD treatment increased phosphorylated protein levels of JNK, ERK, and ATF2 1.06-, 1.11-, and 1.30-folds over the control group, respectively (Figure 7B), while after exposure to SP600125, activation of JNK, ERK, and ATF2 proteins on SSD was obviously inhibited (Figure 7B). The results suggested that pre-treatment with SP600125 effectively prevented the JNK activation induced by SSD in BxPC3 cells. It was also interesting to note that SP600125 showed only a slightly inhibitory effect on activation of the SEK1/MKK4 protein by SSD and exhibited almost no effect on p-cJun levels in BxPC3 cells (Figure 7A). These results demonstrated that SSD regulated the activities of SEK1/MKK4 and cJun independently of JNK activation in PANC1 cells.
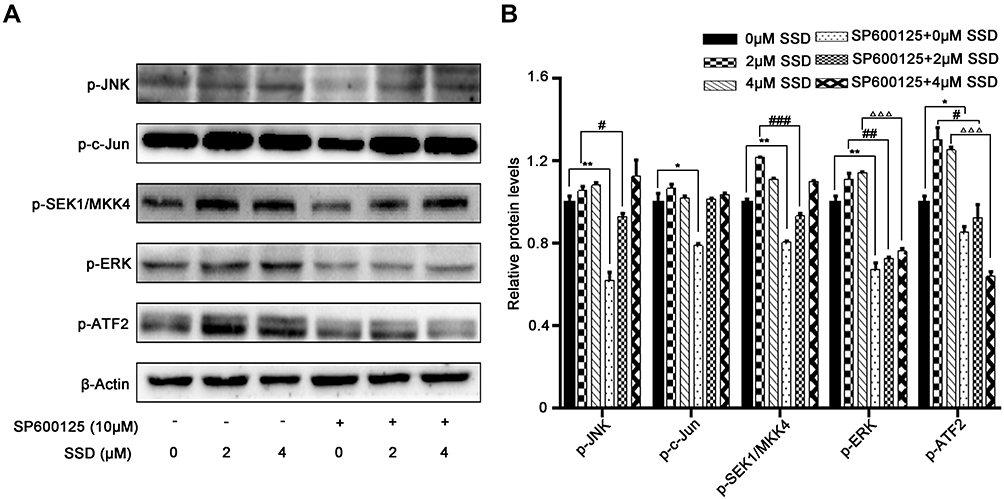 |
Figure 7 JNK inhibition suppressed SSD-induced activation of MKK4–JNK pathway in BxPC3 cells. (A) BxPC3 cells were pretreated with SP600125 for 1 hour, followed by treatment with or without SSD for 4 hours. Expression of phosphorylation of JNK, cJun, SEK1/MKK4, ERK, and ATF2 was analyzed using Western blotting. (B) Semiquantitative immunoblot-staining analysis of indicated MKK4–JNK signaling-pathway proteins on SSD treatment by grayscale-scan analysis. *P<0.05, **P<0.01 compared with control group; #P<0.05, ##P<0.01, ###P<0.001 compared with 2 μM SSD–treated group without SP600125 exposure; ΔΔΔP<0.001 compared with 4 μM SSD–treated group without SP600125 exposure. Data expressed as means ± SD, n=3. |
The apoptosis-induction and proliferation-inhibition effects of SSD were further determined in the presence of SP600125 to confirm the role of JNK activation in the antitumor efficacy of SSD. As shown in Figure 8A, blocking activation of the JNK pathway significantly reduced the apoptosis-induction effect mediated by SSD in BxPC3 cells (Figure 8A). SSD-induced typical apoptotic morphological features decreased obviously after treatment with SP600125 in BxPC3 cells, including bright nuclear condensation and perinuclear apoptotic bodies (Figure 8A). In addition, apoptotic cell fractions induced by SSD were significantly inhibited through blocking JNK activation (Figure 8B and 8C). Apoptosis fractions of BxPC3 cells were 9.8% and 12.6% after treatment with 2 and 4 µM SSD, respectively (Figure 8C). However, in the presence of the JNK inhibitor, apoptosis rates of BxPC3 cells at the same SSD concentrations reduced to 7.2% and 8.6%, respectively (Figure 8C). Furthermore, inhibiting JNK activation also substantially reversed SSD-induced cell-proliferation inhibition in BxPC3 cells, in accordance with its role in cell apoptosis (Figure 8D). The proliferation-inhibition rate of 4 µM SSD reduced from 26.53% to 16.37% after the addition of SP600125 (Figure 8D). These data clearly indicated that activation of the JNK pathway was required for apoptosis induction and proliferation inhibition by SSD in BxPC3 cells.
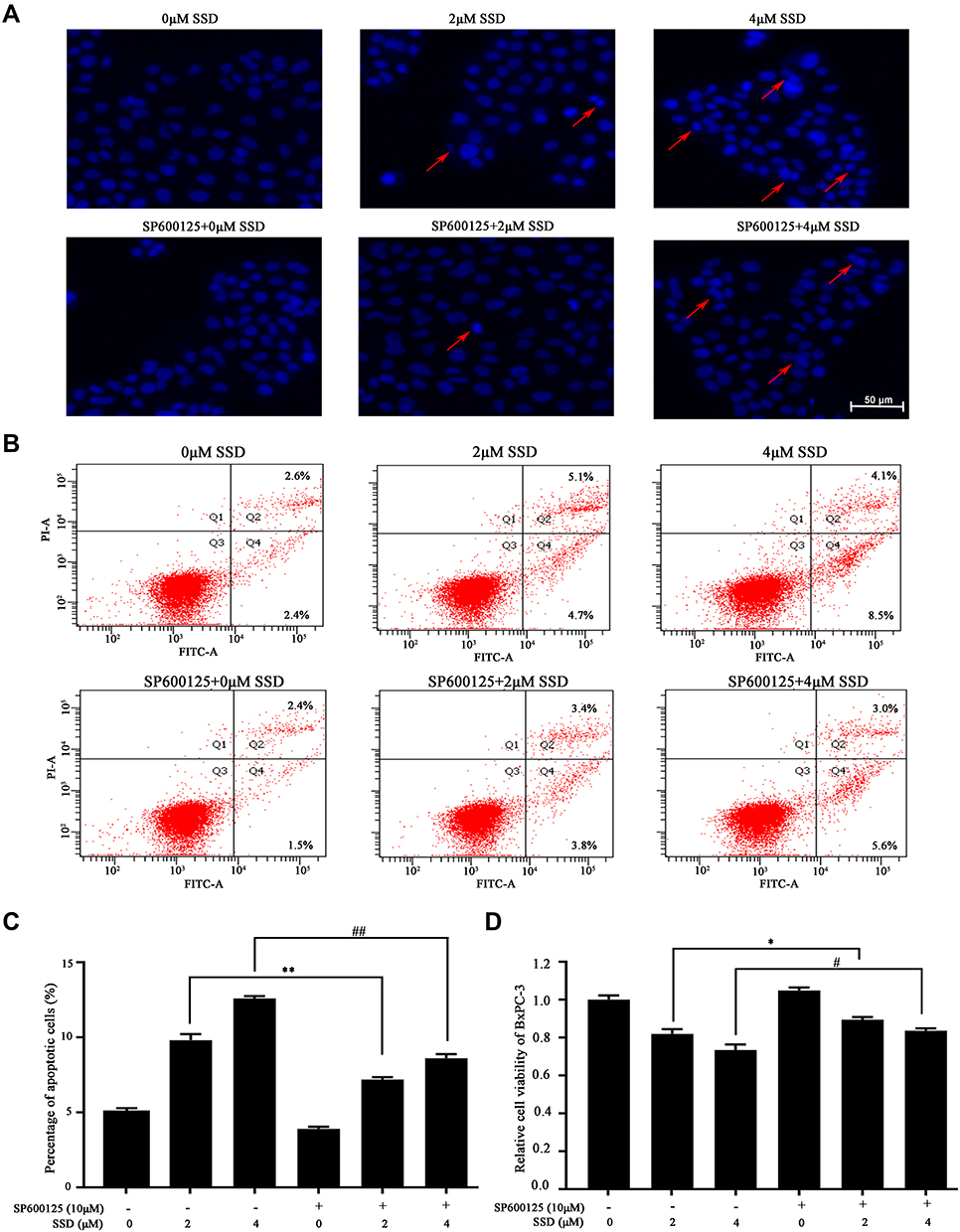 |
Figure 8 JNK inhibition by SP600125 reversed SSD-caused proliferative inhibition and apoptosis induction in BxPC3 cells. (A) Apoptotic morphology was observed after DAPI fluorescence staining. BxPC3 cells were pretreated with SP600125 for 1 hour, followed by treatment with or without SSD for another 20 hours. Red arrows indicate cells with typical apoptotic morphology. (B) Apoptosis was quantified using the annexin V–FITC + PI double-staining flow cytometry. (C) Quantified data of rate of apoptosis. (D) Cell viability of BxPC3 cells was determined by MTT in the absence or presence of SP600125. Values represent means ± SD of three independent experiments. *P<0.05, **P<0.01 compared with 2 μM SSD–treated group without SP600125 exposure; #P<0.05, ##P<0.01 compared with 4 μM SSD–treated group without SP600125 exposure. |
Finally, the clinical significance and prognostic value of the MKK4–JNK signaling pathway in pancreatic cancer were evaluated based on the gene-expression data from the Cancer Genome Altas. mRNA-expression levels of MKK4, JNK1, and JNK2 proteins exhibited no differences between pancreatic cancer tissue and normal paracancerous tissue (Figure 9A). However, low expression of MKK4 was found to be significantly associated with poor survival, suggesting a protective role of the MKK4 gene in pancreatic cancer progression (Figure 9B), while the JNK1/JNK2 genes showed no significant correlation with overall survival rate (Figure 9B). In addition, a strong positive relationship between MKK4 expression and JNK1/JNK2 was observed in PANC1 cells (Figure 9C and 9D). These results indicated that activation of the MKK4–JNK signaling pathway would be a promising therapeutic approach. Our results demonstrated that SSD increased the activity of the MKK4–JNK regulatory pathway to suppress cell growth and induce apoptosis in PANC1 cells, providing a potential chemotherapeutic agent for future drug development.
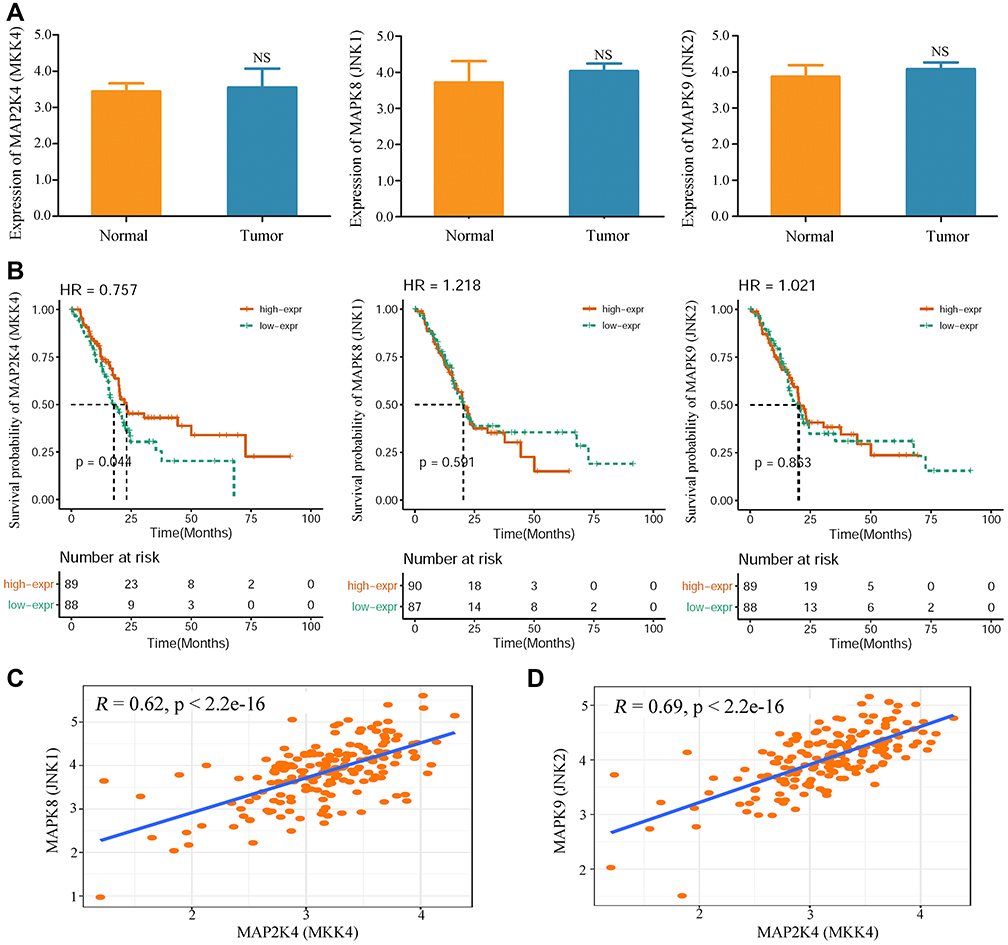 |
Figure 9 Clinical significance of MKK4–JNK signaling pathway in pancreatic cancer patients. (A) Expression levels of MAP2K4 gene (MKK4 protein), MAPK8 gene (JNK1 protein), and MAPK9 gene (JNK2 protein) in tumor tissue and normal tissue of pancreatic cancer patients. No significant differences were observed in expression levels of MKK4, JNK1, or JNK2 between tumor tissue and normal tissue. P-values of MKK4, JNK1, and JNK2 were 0.0523, 0.1607, and 0.3716, respectively. (B) Influence of MKK4 and JNK1/2 expression levels on overall survival in pancreatic cancer patients. Kaplan–Meier curves for patients with high and low gene expression were constructed based on data from TCGA cohort (n=178). Log-rank test on MKK4 -gene analysis showed pP=0.044 overall. (C) The relationship between MKK4 and JNK1 expression was analyzed in pancreatic cancer tissue. (D) The relationship between MKK4 and JNK2 expression was determined. |
Discussion
Pancreatic cancer has long been acknowledged as a fatal malignancy with gradually increasing incidence and extremely dismal prognosis, partly attributed to robust apoptosis resistance to current chemotherapy methods.20 In this study, we found that SSD possessed potent antitumor and obvious apoptosis-induction activity against pancreatic cancer. Exposure to SSD significantly activated the MKK4–JNK signaling pathway, while blocking the JNK pathway with SP600125 reduced the regulatory effects of SSD in pancreatic cancer cells. Taken together, the data at hand demonstrated that SSD inhibited proliferation and caused apoptosis of pancreatic cancer cells via activation of the MKK4–JNK signaling pathway.
SSD has been proven to be a potential therapeutic strategy for several cancers. For instance, SSD effectively inhibits cell proliferation and increased cell-apoptosis rates in hepatocellular carcinoma cells.21 Treatment with SSD also causes growth suppression and apoptosis-dependent death in triple-negative breast cancer cells.22 SSDenhances the sensitivity of gefitinib in human non–small cell lung cancer cells via regulating the STAT3–BCL2 signaling pathway.23 Furthermore, SSD has been reported to enhance cell radiosensitivity of liver cancer cells.24 In this study, our results indicated that SSD triggered apoptotic death of pancreatic cancer cells through increasing caspase 3/9 protein cleavage and enhancing FoxO3a expression (Figures 3 and 4). Further results indicated that the anticancer effect of SSD was dependent on activation of the MKK4–JNK pathway in pancreatic cancer cells (Figure 8). In addition to pancreatic cancer cells, recent reports have indicated that SSD ameliorates pancreatic fibrosis and inhibits the activation of pancreatic stellate cells, which play a crucial role in the progression and therapeutic resistance of pancreatic cancer.25 More importantly, SSD inhibits the proliferation and migration of hepatic stellate cells, which are morphologically and functionally similar to pancreatic stellate cells.26 These results suggest that SSD has potential regulatory effects on both pancreatic cancer cells and pancreatic stellate cells, which are important components of the tumor microenvironment. It would be interesting to determine further the effect of SSD on cross talk and interaction between pancreatic stellate cells and cancer cells and investigate whether SSD treatment might be useful for the destruction of the pancreatic cancer–supportive microenvironment.
Gemcitabine has been approved by FDA as the standard first-line chemotherapeutic agent for the treatment of locally advanced and metastatic pancreatic cancers. However, gemcitabine monotherapy offers only a 1.5-month increase in median survival, requiring continuous research into novel active agents and development of more effective therapeutic strategies for pancreatic cancer patients.27 Combination therapy based on gemcitabine and other active antitumor agents could provide a modest improvement in survival.28 For example, compared with gemcitabine alone, gemcitabine and harmine combination therapy provides enhanced therapeutic benefit in pancreatic cancer.29 Consequently, it is of great interest to identify new therapeutic agents to increase the chemosensitivity of gemcitabine and develop novel combination-therapy approaches. Herbal medicines and plant-derived compounds are becoming increasingly important sources of cancer-chemopreventive or adjuvant chemotherapy.30 Oridonin can overcome gemcitabine resistance in PANC1/Gem cells by regulating GSTπ and LRP1–ERK–JNK signaling, inducing cell apoptosis.31 Our previous results have also demonstrated that cryptotanshinone and imatinib have a cooperative role in inducing cell apoptosis, suggesting a combination therapy for the treatment of chronic myeloid leukemia patients.32 In this study, we proved that SSD was effective in the induction of apoptosis and activation of the MKK4–JNK pathway in pancreatic cancer cells. Furthermore, SSD has been confirmed to increase the chemosensitivity of resistant cancer cells via facilitating mitochondrial fission.33 Therefore, a combination-treatment strategy based on SSD and gemcitabine in pancreatic cancer is promising and needs to be explored further in follow-up studies.
The well-documented tumor-suppressor gene P53 is frequently mutated and represents a crucial initiating event in the development of pancreatic cancer.34 P53 mutations are closely associated with decreased time to recurrence and contribute significantly to poor prognosis and chemotherapeutic resistance in pancreatic cancer patients.35 The BxPC3 cell linecarries a unique Y220C P53Y220C mutation, which creates an extended surface crevice in the DNA-binding domain, destabilizes p53, and causes denaturation and aggregation.36 Compared with other mutations, P53Y220C in BxPC3 cells has been found to maintain transactivation activity of the P21 gene.37 Our results indicated that SSD showed no obvious regulatory effect on p53 total protein in BxPC3 cells (Figure 3). However, Kumar et al suggested that the active triterpenoid nimbolide from Azadirachta indica induced higher levels of apoptotic cell death through reducing mutant p53 in pancreatic cancer cells.35 On the other hand, mesothelin has been found to regulate growth and apoptosis via a p53-independent signaling pathway in pancreatic cancer cells harboring mutated p53 protein.38 Wu et al demonstrated that dihydrosanguinarine exhibited completely opposite effects on the expression of total p53 protein in different pancreatic cancer cells,39 indicating that the regulation mechanism of p53 depends on both drugs and genotypes of cancer-cell lines. The MEK–ERK signaling pathway also plays important roles in regulatiing the expression and distribution of mutant p53 protein in pancreatic cancer cells. Asperolide A has been proven to induce p53–p21 stabilization through activation of the MEK–ERK signaling pathway in lung carcinoma cells.40 However, because of the feedback-loop control of the ERK cascade, the ERK-signaling pathway exhibited bidirectional regulatory effects in tumor progression. For instance, curcumin has been reported to suppress H2O2-induced pancreatic cancer invasion and migration via inhibiting the ERK pathway,19 while sophoridine has been demonstrated to trigger apoptosis of human pancreatic cancer cells through ROS-dependent ERK activation.15 In this study, we found that concentration was an important factor in the regulatory effect of SSD on ERK activity in BxPC3 cells (Figure 5). Different concentrations of SSD even showed opposite effects on expression levels of pERK. Furthermore, SSD treatment failed to cause obvious changes in the activity of ERK during the first 6 hours, indicating a indirect regulatory role in pancreatic cancer cells (Figure 6). Taken together, these results suggest complex and multiple mediating mechanisms of SSD on the functions of p53 and the ERK-signaling pathway, and further investigations on this issue are required.
MKK4 is a critical mediator of stress-activated protein-kinase signals, and plays vital roles in the development and progression of various human cancers. As a midstream member of the MAPK family, MKK4 is the crucial transducer upstream of JNK signaling through directly phosphorylating specific Tyr residues located in the activation loop of the JNK protein in response to various extracellular stimuli.41 MKK4 activation leads to phosphorylation and activation of the downstream JNK-signaling pathway, which is a well-known therapeutic target for apoptosis induction in pancreatic cancer.42 MKK4 has been indicated to be a tumor metastasis–suppressor gene in prostate and ovarian cancers.9 Reduced expression of MKK4 is closely associated with cancer metastasis and may contribute to the progression and poor prognosis in colorectal cancer patients.10 Lu et al demonstrated that cytoplasmic MKK4 expression was significantly downregulated in tumor tissue compared with nontumor tissue in pancreatic ductal adenocarcinoma, and high cytoplasmic MKK4 levels were closely associated with longer cancer-specific survival.13 On the other hand, activation of MKK4 would be a promising approach to increase JNK signaling and induce cell apoptosis in pancreatic cancer. Zhang et al suggested that triptonide inhibits pancreatic cancer–cell tumorigenicity and tumor growth via activating an MKK4-mediated tumor-suppressive signaling pathway.43 Capsaicin causes cell apoptosis and pancreatic tumor-growth suppression through activating MKK4 other related downstream effector by ASK1.44 Our results also demonstrated that SSD was an effective activator of MKK4 protein in BxPC3 cells. Compared with the control group, 4 µM SSD induced 2.7-fold higher expression of pMKK4 protein (Figure 5). Moreover, SSD triggered activation of the MKK4-signaling pathway within 30 minutes, suggesting SSD upregulated MKK4 phosphorylation directly (Figure 6). MKK4-protein activity was also closely associated with chemosensitivity to gemcitabine in human pancreatic cancer cells. Kreutzer et al demonstrated that cellular depletion of CK2α leads to enhanced phosphorylation of MKK4 and JNK proteins and increased sensitivity to gemcitabine in PANC1 cells.45 Due to the the frequently mutated and loss of function of the MKK4–JNK signaling node in pancreatic cancer, identifying effective and selective activators for the MKK4–JNK pathway would be an alternative approach for pancreatic cancer therapy.46
Conclusion
In the present study, we demonstrated that SSD inhibited proliferation and induced apoptosis of pancreatic cancer cells through upregulating the key regulatory MKK4–JNK pathway. These findings also suggest that the MKK4–JNK pathway may be a potential target in the treatment of pancreatic cancer. SSD may be developed into a leading candidate drug for pancreatic cancer therapy.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81673755) and Zhejiang Provincial Natural Science Foundation of China (LY17H290010).
Author Contributions
All authors made contributions to acquisition and analysis of data, took part in drafting the article or revising it critically, gave final approval to the version to be published, and agree to be accountable for all aspects of the work. All authors have read and approved the final manuscript to be published.
Disclosure
The authors declare no conflicts of interest. This article does not contain any studies with human participants or animals performed by any of the authors.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi:doi:10.3322/caac.20107
2. Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7(3):163–172. doi:doi:10.1038/nrclinonc.2009.236
3. Burris HA
4. Ito M, Makino N, Ueno Y. Glucose intolerance and the risk of pancreatic cancer. Transl Gastrointest Cancer. 2013;2(4):223–229. doi:doi:10.3978/j.issn.2224-4778.2013.09.01
5. Lee J, Yang DH, Suh JH, et al. Species discrimination of Radix Bupleuri through the simultaneous determination of ten saikosaponins by high performance liquid chromatography with evaporative light scattering detection and electrospray ionization mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(32):3887–3895. doi:doi:10.1016/j.jchromb.2011.10.040
6. Ikegami F, Sumino M, Fujii Y, Akiba T, Satoh T. Pharmacology and toxicology of Bupleurum root-containing Kampo medicines in clinical use. Hum Exp Toxicol. 2006;25(8):481–494. doi:doi:10.1191/0960327106het654oa
7. Li XQ, Song YN, Wang SJ, Rahman K, Zhu JY, Zhang H. Saikosaponins: a review of pharmacological effects. J Asian Nat Prod Res. 2018;20(5):399–411. doi:doi:10.1080/10286020.2018.1465937
8. Tournier C, Hess P, Yang DD, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;88(5467):870–874. doi:doi:10.1126/science.288.5467.870
9. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12(1):14–21. doi:doi:10.1016/S0959-437X(01)00258-1
10. Wang PN, Huang J, Duan YH, et al. Downregulation of phosphorylated MKK4 is associated with a poor prognosis in colorectal cancer patients. Oncotarget. 2017;8(21):34352–34361. doi:doi:10.18632/oncotarget.16128
11. Cuenda A. Mitogen-Activated Protein Kinase Kinase 4 (MKK4). Int J Biochem Cell B. 2000;32(6):581–587. doi:doi:10.1016/s1357-2725(00)00003-0
12. Yamada SD, Hickson JA, Hrobowski Y, et al. Mitogen-activated Protein Kinase Kinase 4 (MKK4) acts as a metastasis suppressor gene in human ovarian carcinoma. Cancer Res. 2002;62(22):6717–6723. doi:doi:10.1002/cncr.10960
13. Lu J, Zhou L, Yang G, et al. Clinicopathologic and prognostic significance of MKK4 and MKK7 in resectable pancreatic ductal adenocarcinoma. Hum Pathol. 2019;86:143–154. doi:10.1016/j.humpath.2018.11.026
14. Song L, Li J, Zhang D, et al. IKKbeta programs to turn on the GADD45alpha-MKK4-JNK apoptotic cascade specifically via p50 NF-kappaB in arsenite response. J Cell Biol. 2006;175(4):607–617. doi:doi:10.1083/jcb.200602149
15. Xu Z, Zhang F, Bai C, et al. Sophoridine induces apoptosis and S phase arrest via ROS-dependent JNK and ERK activation in human pancreatic cancer cells. J Exp Clin Cancer Res. 2017;36(1):124. doi:doi:10.1186/s13046-017-0590-5
16. Liang T, Zhang X, Xue W, Zhao S, Zhang X, Pei J. Curcumin induced human gastric cancer BGC-823 cells apoptosis by ROS-mediated ASK1-MKK4-JNK stress signaling pathway. Int J Mol Sci. 2014;15(9):15754–15765. doi:doi:10.3390/ijms150915754
17. Daniel B, DeCoster MA. Quantification of sPLA2-induced early and late apoptosis changes in neuronal cell cultures using combined TUNEL and DAPI staining. Brain Res Brain Res Protoc. 2004;13(3):144–150. doi:doi:10.1016/j.brainresprot.2004.04.001
18. Wang W, Wang YQ, Meng T, et al. MCL-1 degradation mediated by JNK activation via MEKK1/TAK1-MKK4 contributes to anticancer activity of new tubulin inhibitor MT189. Mol Cancer Ther. 2014;13(6):1480–1491. doi:doi:10.1158/1535-7163.MCT-13-0629
19. Cao L, Liu J, Zhang L, Xiao X, Li W. Curcumin inhibits H2O2-induced invasion and migration of human pancreatic cancer via suppression of the ERK/NF-κB pathway. Oncol Rep. 2016;36(4):2245–2251. doi:doi:10.3892/or.2016.5044
20. Shi X, Liu S, Kleeff J, Friess H, Büchler MW. Acquired resistance of pancreatic cancer cells towards 5-fluorouracil and gemcitabine is associated with altered expression of apoptosis-regulating genes. Oncology. 2002;62(4):354–362. doi:doi:10.1159/000065068
21. Ren M, McGowan E, Li Y, et al. Saikosaponin-d suppresses COX2 through p-STAT3/C/EBPβ signaling pathway in liver cancer: a novel mechanism of action. Front Pharmacol. 2019;10:623. doi:doi:10.3389/fphar.2019.00623
22. Wang J, Qi H, Zhang X, et al. Saikosaponin D from Radix Bupleuri suppresses triple-negative breast cancer cell growth by targeting β-catenin signaling. Biomed Pharmacother. 2018;108:724–733. doi:doi:10.1016/j.biopha.2018.09.038
23. Tang JC, Long F, Zhao J, et al. The effects and mechanisms by which Saikosaponin-D enhances the sensitivity of human non-small cell lung cancer cells to gefitinib. J Cancer. 2019;10(26):6666–6672. doi:doi:10.7150/jca.30361
24. Tian YD, Lin S, Yang PT, et al. Saikosaponin-d increases the radiosensitivity of hepatoma cells by adjusting cell autophagy. J Cancer. 2019;10(20):4947–4953. doi:doi:10.7150/jca.30286
25. Cui LH, Li CX, Zhuo YZ, Yang L, Cui NQ, Zhang SK. Saikosaponin d ameliorates pancreatic fibrosis by inhibiting autophagy of pancreatic stellate cells via PI3K/Akt/mTOR pathway. Chem Biol Interact. 2019;300:18–26. doi:doi:10.1016/j.cbi.2019.01.005
26. Chen MF, Huang CC, Liu PS, Chen CH, Shiu LY. Saikosaponin a and saikosaponin d inhibit proliferation and migratory activity of rat HSC-T6 cells. J Med Food. 2013;16(9):793–800. doi:doi:10.1089/jmf.2013.2762.
27. Li J, Wientjes MG, Au JL. Pancreatic cancer: pathobiology, treatment options, and drug delivery. AAPS J. 2010;12(2):223–232. doi:doi:10.1208/s12248-010-9181-5
28. Higuchi T, Miyake K, Sugisawa N, et al. The combination of olaratumab with gemcitabine and docetaxel arrests a chemotherapy-resistant undifferentiated soft-tissue sarcoma in a patient-derived orthotopic xenograft mouse model. Cancer Chemother Pharmacol. 2019;83(6):1075–1082. doi:doi:10.1007/s00280-019-03824-3
29. Wu L, Zhang J, Rao M, Zhang Z, Zhu H, Zhang C. Harmine suppresses the proliferation of pancreatic cancer cells and sensitizes pancreatic cancer to gemcitabine treatment. Onco Targets Ther. 2019;12:4585–4593. doi:doi:10.2147/OTT.S205097
30. Li L, Leung PS. Use of herbal medicines and natural products: an alternative approach to overcoming the apoptotic resistance of pancreatic cancer. Int J Biochem Cell Biol. 2006;53:224–236. doi:doi:10.1016/j.biocel.2014.05.021
31. Wang B, Shen C, Li Y, et al. Oridonin overcomes the gemcitabine resistant PANC-1/Gem cells by regulating GST pi and LRP/1 ERK/JNK signalling. Onco Targets Ther. 2019;12:5751–5765. doi:doi:10.2147/OTT.S208924
32. Ge Y, Yang B, Xu X, Dai Q, Chen Z, Cheng R. Cryptotanshinone acts synergistically with imatinib to induce apoptosis of human chronic myeloid leukemia cells. Leuk Lymphoma. 2015;56(3):730–738. doi:doi:10.3109/10428194.2014.928934
33. Tsuyoshi H, Wong VKW, Han Y, Orisaka M, Yoshida Y, Tsang BK. Saikosaponin-d, a calcium mobilizing agent, sensitizes chemoresistant ovarian cancer cells to cisplatin-induced apoptosis by facilitating mitochondrial fission and G2/M arrest. Oncotarget. 2017;8(59):99825–99840. doi:doi:10.18632/oncotarget.21076
34. Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363(9414):1049–1057. doi:doi:10.1016/S0140-6736(04)15841-8
35. Kumar S, Inigo JR, Kumar R, et al. Nimbolide reduces CD44 positive cell population and induces mitochondrial apoptosis in pancreatic cancer cells. Cancer Lett. 2018;413:82–93. doi:doi:10.1016/j.canlet.2017.10.029
36. Bullock AN, Henckel J, Fersht AR. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene. 2000;19(10):1245–1256. doi:doi:10.1038/sj.onc.1203434
37. Zhou R, Shanas R, Nelson MA, Bhattacharyya A, Shi J. Increased expression of the heterogeneous nuclear ribonucleoprotein K in pancreatic cancer and its association with the mutant p53. Int J Cancer. 2010;126(2):395–404. doi:doi:10.1002/ijc.24744
38. Zheng C, Jia W, Tang Y, Zhao H, Jiang Y, Sun S. Mesothelin regulates growth and apoptosis in pancreatic cancer cells through p53-dependent and -independent signal pathway. J Exp Clin Cancer Res. 2012;31(1):84. doi:doi:10.1186/1756-9966-31-84
39. Wu SZ, Xu HC, Wu XL, et al. Dihydrosanguinarine suppresses pancreatic cancer cells via regulation of mut-p53/WT-p53 and the Ras/Raf/Mek/Erk pathway. Phytomedicine. 2019;59:152895. doi:doi:10.1016/j.phymed.2019.152895
40. Lv C, Sun W, Sun H, et al. Asperolide A, a marine-derived tetranorditerpenoid, induces G2/M arrest in human NCI-H460 lung carcinoma cells, is mediated by p53-p21 stabilization and modulated by Ras/Raf/MEK/ERK signaling pathway. Mar Drugs. 2013;11(2):316–331. doi:doi:10.3390/md11020316
41. Kishimoto H, Nakagawa K, Watanabe T, et al. Different properties of SEK1 and MKK7 in dual phosphorylation of stress-induced activated protein kinase SAPK/JNK in embryonic stem cells. J Biol Chem. 2003;278(19):16595–16601. doi:doi:10.1074/jbc.M213182200
42. Kim JH, Kim MJ, Choi KC, Son J. Quercetin sensitizes pancreatic cancer cells to TRAIL-induced apoptosis through JNK-mediated cFLIP turnover. Int J Biochem Cell Biol. 2016;78:327–334. doi:doi:10.1016/j.biocel.2016.07.033
43. Zhang B, Meng M, Xiang S, et al. Selective activation of tumor-suppressive MAPKP signaling pathway by triptonide effectively inhibits pancreatic cancer cell tumorigenicity and tumor growth. Biochem Pharmacol. 2019;166:70–81. doi:doi:10.1016/j.bcp.2019.05.010
44. Pramanik KC, Srivastava SK. Apoptosis signal-regulating kinase 1-thioredoxin complex dissociation by capsaicin causes pancreatic tumor growth suppression by inducing apoptosis. Antioxid Redox Signal. 2012;17(10):1417–1432. doi:doi:10.1089/ars.2011.4369
45. Kreutzer JN, Ruzzene M, Guerra B. Enhancing chemosensitivity to gemcitabine via RNA interference targeting the catalytic subunits of protein kinase CK2 in human pancreatic cancer cells. BMC Cancer. 2010;10:440. doi:doi:10.1186/1471-2407-10-440.
46. Davies CC, Harvey E, McMahon RF, et al. Impaired JNK signaling cooperates with KrasG12D expression to accelerate pancreatic ductal adenocarcinoma. Cancer Res. 2014;74(12):3344–3356. doi:doi:43.10.1158/0008-5472.CAN-13-2941
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.