")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Safety, tolerability, pharmacokinetics, and pharmacodynamics of novel glucokinase activator HMS5552: results from a first-in-human single ascending dose study
Authors Xu H, Sheng L , Chen W, Yuan F, Yang M, Li H, Li X , Choi J, Zhao G, Hu TH, Li Y, Zhang Y, Chen L
Received 25 January 2016
Accepted for publication 23 February 2016
Published 9 May 2016 Volume 2016:10 Pages 1619—1626
DOI https://doi.org/10.2147/DDDT.S105021
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Hongrong Xu,1,* Lei Sheng,1,* Weili Chen,1 Fei Yuan,1 Mengjie Yang,1 Hui Li,1 Xuening Li,1 John Choi,2 Guiyu Zhao,2 Tianxin Hu,2 Yongguo Li,2 Yi Zhang,2 Li Chen2
1Department of Clinical Pharmacology, Zhongshan Hospital, Fudan University, Shanghai, 2Department of Clinical Research & Development, Hua Medicine, Shanghai, People’s Republic of China
*These authors have contributed equally to this work
Background: HMS5552, a novel fourth-generation glucokinase (GK) activator, has demonstrated promising effects on glycemic control in preclinical models of type 2 diabetes. This single ascending dose study was conducted to investigate the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of HMS5552 during its first-in-human exposure.
Methods: Sixty healthy subjects were enrolled. In each of six dose-cohorts (5, 10, 15, 25, 35, and 50 mg), ten subjects were randomized with eight subjects receiving the same cohort-dose of HMS5552 and two receiving placebo. Plasma HMS5552 exposure, glucose, and insulin were measured repeatedly during fasting and after a standardized meal. Assessment included safety, PK, and PD endpoints.
Results: HMS5552 showed dose-proportional increases in area under the curve 0 to the last quantifiable concentration (AUC0–t) and maximum plasma concentration (Cmax). Slopes estimated by linear regression for AUC0–t and Cmax were ~1.0 (0.932 and 0.933, respectively). Geometric mean elimination half-life ranged from 4.48 to 7.51 hours and apparent clearance ranged from 11.5 to 13.1 L/h across all doses. No significant sex effect was observed in PK parameters. HMS5552 also demonstrated dose-related PD responses in terms of maximum glucose change from baseline (%) and mean glucose area under effect curve 0–4 hours change from baseline (%) (P<0.001). Fifteen adverse events were reported by nine subjects (ten with HMS5552 and five with the placebo). All adverse events were mild in intensity and resolved without any treatment.
Conclusion: This first-in-human single ascending dose study provided predicted PK of HMS5552 with dose-proportional increases in AUC0–t and Cmax, as well as dose-related glucose-lowering effects over the range of 5–50 mg in healthy subjects. HMS5552 at doses up to 50 mg in healthy subjects was safe and well-tolerated.
Keywords: HMS5552, glucokinase activator, type 2 diabetes, pharmacokinetics, pharmacodynamics
Introduction
Type 2 diabetes (T2DM) is a disease typically characterized by a combination of insulin resistance and β-cell dysfunction. Unfortunately, existing antidiabetic drugs used as monotherapy or even combinations of multiple agents have often been inadequate in maintaining glucose control over the long term,1 and have not addressed the problem of progressively deteriorating islet function.2 Therefore, it is urgent to develop additional therapies that not only leverage novel mechanisms-of-action to maintain recommended glycemic targets, but also offer the potential to improve or preserve the health of existing β-cells.
GK is a key enzyme that initiates the first step of glucose metabolism by phosphorylating glucose to glucose-6-phosphate.3 In pancreatic β-cells, GK acts as the central rate-limiting enzyme in glucose-stimulated insulin release (GSIR).4,5 In liver cells, GK regulates hepatic glucose utilization, glycolysis, and glycogenesis.6 GK is also present in pancreatic α-cells as well as enteroendocrine L-cells that secrete glucagon-like peptide 1,7 although its role in these cells has been less extensively studied. Importantly, GK shows a sigmoidal glucose dependency and enzyme cooperativity (Hill coefficient nH =1.7)8,9 with stronger activity at high glucose levels, and weaker activity at low glucose levels, thereby acting as a “glucose sensor” that helps facilitate glucose homeostasis.10
Hence, GK activators (GKAs) of the enzyme offer a promising class of drugs that would target multiple mechanisms for glycemic control. It has been shown in previous animal studies that GKAs regulate systemic blood glucose through the variety of mechanisms previously discussed, including directly enhancing insulin release (pancreatic β-cells), inhibiting production of endogenous glucose (hepatocytes), and by indirectly promoting glucagon-like peptide 1 release (enteroendocrine L-cells)9. Additionally, GKAs have been shown to have antiapoptotic effects on β-cells11–13 in preclinical studies that, if definitively confirmed in the clinic, would significantly influence current management of T2DM.
HMS5552 is a fourth-generation GKA that was discovered a decade after the first 2003 publication in Science introduced GKAs,14 by the same research labs in the USA, EU, and the People’s Republic of China. The compound has a structurally novel amino-acid-based chemical scaffold and is currently in Phase II clinical trials. Here, we present the results of a randomized, double-blind, placebo-controlled, and single ascending dose (SAD) Phase Ia study of HMS5552 (NCT01952535) to evaluate the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) following oral administration in healthy adult subjects.
Materials and methods
This trial was performed at Zhongshan Hospital, Fudan University, People’s Republic of China, and was conducted in compliance with hospital protocol. The study was approved by the Ethics Committee of Zhongshan Hospital, Fudan University and was carried out in accordance with the Good Clinical Practice and the Declaration of Helsinki. Written informed consents were obtained from all subjects before admission to the study. Clinical trial registration ID: NCT01952535.
Subjects
Subjects (18–45 years of age; body mass index 18–24 kg/m2) were eligible to participate in the study if they were in good general health based on medical history, physical examination, vital signs, electrocardiogram (ECG), fasting plasma glucose (FPG), oral glucose tolerance test, and laboratory measurements (serum biochemistry, hematology, hepatitis, HIV, and urinalysis). Subjects were excluded from the study if they met one of the following criteria: had a history of alcohol abuse; had a history of psychotropic drug or tobacco use; had electrocardiographic abnormality (QT interval corrected for heart rate using Bazett’s method [QTcB] >450 milliseconds or QTcB >30 milliseconds) as determined by ECG; had used an investigational drug within 3 months; or had any clinically relevant abnormalities; had refused to accept effective contraception in the next 3 months after completion of the study. Female subjects were eligible to be enrolled if they were of non-childbearing potential and were excluded if they were lactating or pregnant, as determined by positive human chorionic gonadotropin test at screening or prior to dosing (day −1).
Study design
This study was designed as a single-center, randomized, double-blind, placebo-controlled, parallel-group, and dose-escalation Phase Ia trial of HMS5552 administered to healthy subjects. After eligibility had been confirmed at screening, subjects were assigned to six different dose-cohorts (5, 10, 15, 25, 35, and 50 mg). Within each dose-cohort, ten subjects were randomized to receive either active or matching placebo treatment with a ratio of 4:1 based on a randomized list provided by an independent statistician. Each subject was assigned to only one dose treatment (5, 10, 15, 25, 35, 50 mg, or matching placebo). Subjects were screened between day −28 and day −7 and admitted to the clinic on day −2. Each subject was fasted overnight (at least 8 hours) on Day-1 (baseline) before taking the morning dose of the drug or placebo with 240 mL of water on day 1. Additional water intake was not allowed for 1 hour before and after dose administration. All subjects were served with standardized meals during this study. Per protocol, the first meal was served 4 hours after dose administration in the morning of day 1. Finally, the subjects were discharged on day 4.
Safety assessments
Safety evaluation was conducted throughout the course of the study, which included physical examination, vital signs (eg, blood pressure, heart rate, respiratory rate, and ear temperature), 12-lead ECG, clinical laboratory tests (ie, hematology, serum biochemistry, and urinalysis), adverse events (AEs), and a global assessment of tolerability by the investigator.
Vital signs were assessed at screening, at admission (day −2), prior to dosing (day −1), and after dosing (day 1 to day 4). Twelve-lead ECGs were recorded in triplicate on each of day −1 to day 3. Hematology, serum biochemistry, and urinalysis were measured at screening, on day −1 and, on day 3. AEs were graded in accordance with the Common Terminology Criteria for Adverse Events version 4.02.
Determination of HMS5552 concentrations
Blood samples for determination of HMS5552 were collected at the following time points after a single oral administration of HMS5552 on day 1: pre-dose (0), 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 12, 16, 24, 36, 48, and 72 hours. Meanwhile, urine collection was carried out at pre-dose (0), and at the time intervals of 0–4, 4–8, 8–12, 12–24, 24–36, 36–48, and 48~72 hours. The collected blood samples were immediately centrifuged at 3,500 rpm for 10 minutes at 4°C within 1 hour before being aliquoted into two polypropylene tubes. The aliquots were stored first at −20°C, then at −80°C until further analyses were conducted. As for urine samples, the volume of urine samples was recorded before adding 1% (volume ratio) of 50 mg/mL Triton X-100 solution; after mixing completely, the samples were aliquoted into two polypropylene tubes and stored first at −20°C, then at −80°C until further analyses.
HMS5552 concentrations in plasma and urine were determined using a validated liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) assay. The method was validated using plasma or urine samples of HMS5552 in the range of 0.500–500 ng/mL with a lower limit of quantification as 0.500 ng/mL. In brief, the assays were performed as follows: a 100 μL aliquot of each sample, quality control sample, and calibrator was diluted with 50 μL of 13C-labeled internal standard and tert-butyl methyl ether (600 μL for plasma sample, 800 μL for urine sample), shaken at room temperature for 15 minutes and then centrifuged at 4,000 rpm, 5°C for 10 minutes. After centrifugation, a 400 μL aliquot of the supernate was moved into a 96-well plate, dried by nitrogen at 40°C followed by 50% methanol (250 μL for plasma sample, 400 μL for urine sample), and then analyzed by LC-MS/MS after shaking for 10 minutes.
Metabolite identification in plasma and urine
Metabolites of HMS5552 in plasma and urine were identified following a single oral administration of 50 mg to healthy subjects. The metabolites were identified by LC-MSn (n=1~2). The area under the curve (AUC) pooling plasma sample (0~24 hours), and selected time points for both plasma samples (0.5, 1, 2, 4, and 8 hours) and urine samples (0–4, 4–8, 8–12, 12–24, 24–36, and 36–48 hours) were detected on a UPLC/Q-TOF mass spectrometer (Waters Corporation, Milford, MA, USA).
PD methods
Plasma samples for measuring glucose and insulin were taken at the following time points after a single oral administration of HMS5552 on day 1: pre-dose (−0.5, −0.25, and 0 hours), 0.25, 0.5, 1, 2, 3, 4, 4.25, 4.5, 5, and 6 hours. Plasma glucose concentrations were determined by an enzymatic oxidation method (GLU Glucose GOD-PAP, Roche Diagnostics, Mannheim, Germany), while insulin was analyzed by radioimmunoassay (kit catalog number H1-14K, Merck Millipore, Billerica, MA, USA).
Plasma glucose levels were used to calculate the following PD parameters: maximum glucose change from baseline (%), mean glucose area under effect curve 0–4 hours (AUEC0–4), mean glucose AUEC0–4 change from baseline (%). Baseline values were obtained on day −1.
PK assessment
Plasma HMS5552 concentrations were used to calculate the following PK parameters: the area under the plasma concentration-time curve from 0 to infinity (AUC0–∞), the area under the plasma concentration-time curve from 0 to the last measured time point (AUC0–t), the maximum plasma concentration (Cmax) and the time to reach (Tmax), the apparent terminal elimination half-life (t1/2), apparent total plasma clearance (CL/f), and apparent volume of apparent distribution during the terminal phase (V/f). The Cmax and Tmax were obtained from the concentration-time data. Urinary concentrations and urine volumes were used to measure the amount of drug excreted in urine from 0 to 72 hours (Ae0–72) and the fraction of unchanged dose excreted in urine from 0 to 72 hours (fe0–72). PK parameters were calculated for the subjects via a non-compartmental method using WinNonlin (version 6.2.1; Pharsight Corporation, Mountain View, CA, USA). AUCs were calculated by the trapezoidal rule.
Statistical analysis
The PK parameters were summarized with geometric mean and standard deviations, except Tmax, for which median values and standard deviations are reported. Dose-proportionality at six dose levels for HMS5552 was assessed. A linear relationship between the logarithmically transformed PK parameters (AUC0–t and Cmax) and dose was fitted by using a power model:
ln(Y) = β0 + β * ln(Dose) + error | (1) |
where Y represents AUC0–t or Cmax and β presents the dose-proportionality coefficient. Slope β and its 90% confidence intervals (CIs) were calculated to assess the dose-proportionality.
Placebo data across groups were pooled in the analysis. All statistical analyses were performed using SPSS software (version 19.0; IBM Corporation, Armonk, NY, USA). The PK properties of HMS5552 were compared not only between single doses but also between female and male subjects using Student's t-test (α=0.05).
Results
Subject disposition and demographics
Sixty subjects (31 males and 29 females) were enrolled in six treatment groups and subsequently randomized to receive active or placebo treatment. There were four male and four female subjects in each HMS5552 group except the 50 mg HMS5552 group (five male and three female subjects). All demographic and baseline characteristics were comparable among the treatment groups (Table 1).
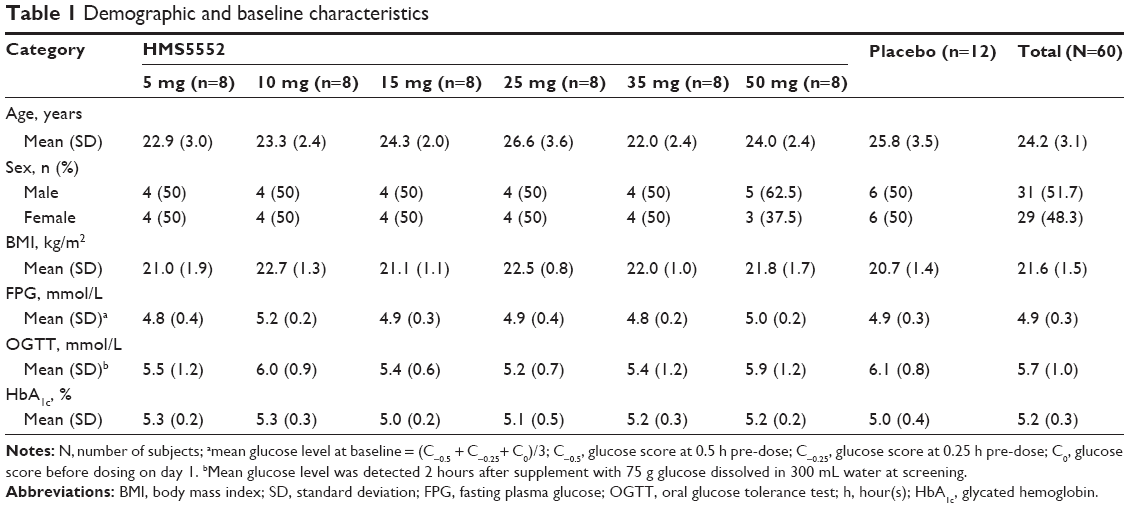 | Table 1 Demographic and baseline characteristics |
Safety and tolerability
Single oral doses of 5, 10, 15, 25, 35, and 50 mg of HMS5552 were safe and well-tolerated in this population of healthy volunteers. No subjects withdrew from the study due to AEs. AEs following HMS5552 or placebo administration are presented in Table 2. Of the 60 subjects, nine (15%) reported a total of 15 AEs following the administration of 5 (1/8), 10 (0/8), 15 (3/8), 25 (2/8), 35 (1/8), 50 mg (0/8) HMS5552, and placebo (2/12), respectively. The incidence of AEs was similar between HMS5552 and placebo (14.6% versus 16.7%, respectively), and all AEs were considered by the investigator to be mild in intensity. No serious AEs were reported, and six treatment-related AEs, including belching, dizziness, palpitation, cold sweat, and proteinuria, were reported in four subjects receiving either HMS5552 or placebo. Treatment-related AEs resolved spontaneously without any intervention. No apparent dose relationship could be observed for any AE.
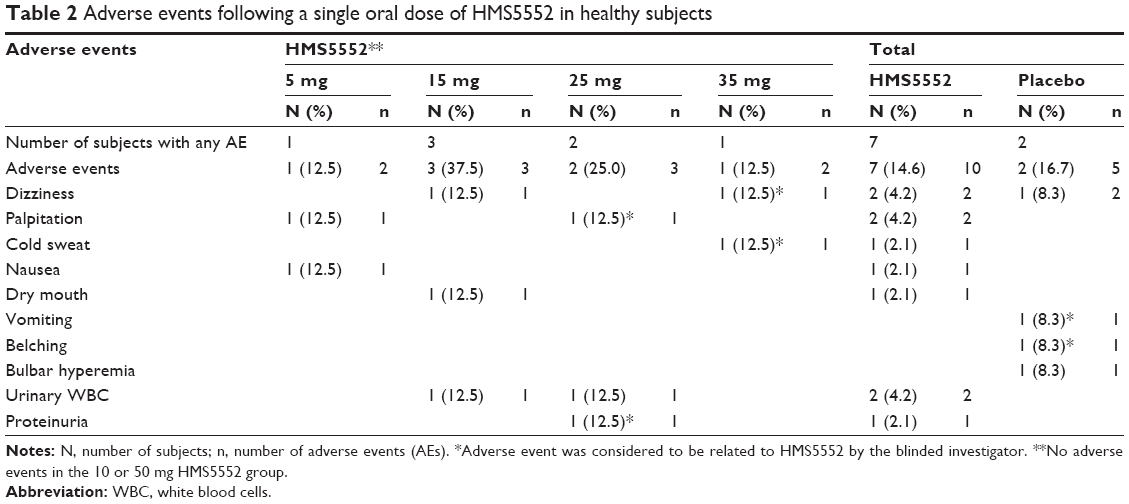 | Table 2 Adverse events following a single oral dose of HMS5552 in healthy subjects |
PK results
Mean plasma HMS5552 concentration versus time following a single oral administration was illustrated in Figure 1, and the derived PK parameters were shown in Table 3. The median Tmax of HMS5552 was achieved 1.25~2.50 hours after drug administration in all treatment groups. Cmax increased from 62.6 ng/mL at 5 mg dose to 582.0 ng/mL at 50 mg dose. The geometric mean t1/2 ranged from 4.48 to 7.51 hours, while CL/f was approximately 12 L/h across all doses (11.5~13.1 L/h). Renal excretion of HMS5552 appeared to be a secondary route of elimination. For all treatment groups, the fraction of HMS5552 in urine over the 72-hour interval was below 11% of the administered dose.
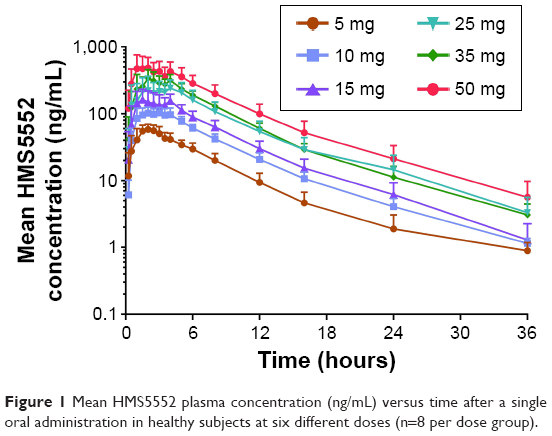 | Figure 1 Mean HMS5552 plasma concentration (ng/mL) versus time after a single oral administration in healthy subjects at six different doses (n=8 per dose group). |
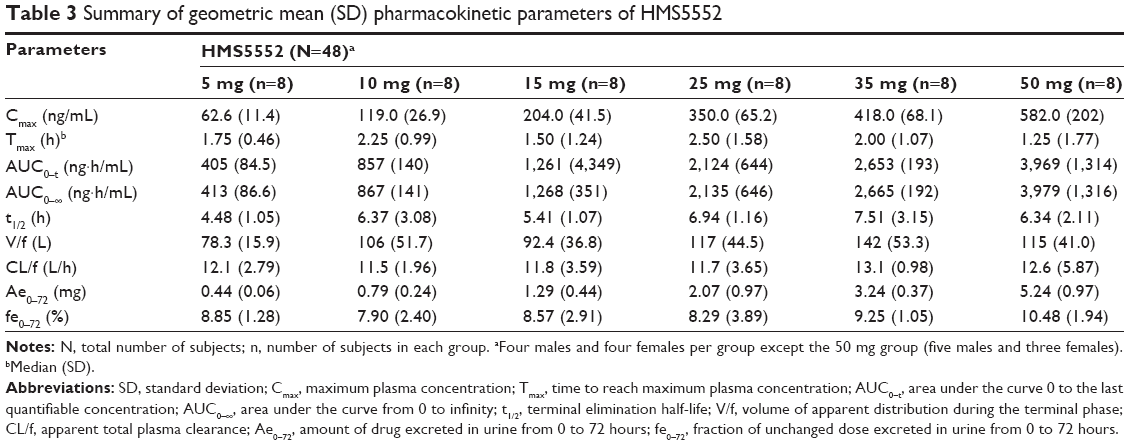 | Table 3 Summary of geometric mean (SD) pharmacokinetic parameters of HMS5552 |
Statistical analysis showed the slopes β for AUC0–t and Cmax were 0.932 (90% CI: 0.859–1.005, model: ln[AUC0–t] =0.932 * ln[Dose] −3.909) and 0.933 (90% CI: 0.865~1.001, model: ln[Cmax] =0.933 * ln[Dose] −2.169), respectively, indicating a dose-proportional increase of AUC0–t and Cmax in the dose range of 5~50 mg, which can be declared as 90% CIs of the slopes β were within the defined interval (0.8–1.25). No sex-related difference in the PK parameters was found except in Cmax in the 35 mg dose group.
Metabolites’ identification
There were eleven minor metabolites of HMS5552 detected in the human plasma and/or urine, which illustrated the pathways of oxidation, reduction, hydrolysis, and glucuronidation. They were assigned as M1~M11 according to metabolic pathways.
Based on the mass intensity, M10 and M11 were the main metabolites in human plasma and urine. Peaks of the parent and metabolites were normalized to yield the percentage of each compound relative to the combined total. In plasma, the only detectable metabolites M10 and M11 were each found at <3%, and M1 at ~1% (Figure 2A). In urine, M10 was approximately 14%, and M11 was 9% at 0~4 hour time period; this proportion remained relatively consistent at other time periods between 4 and 48 hours (Figure 2B). M1~M9 were detectable at much lower amounts in urine.
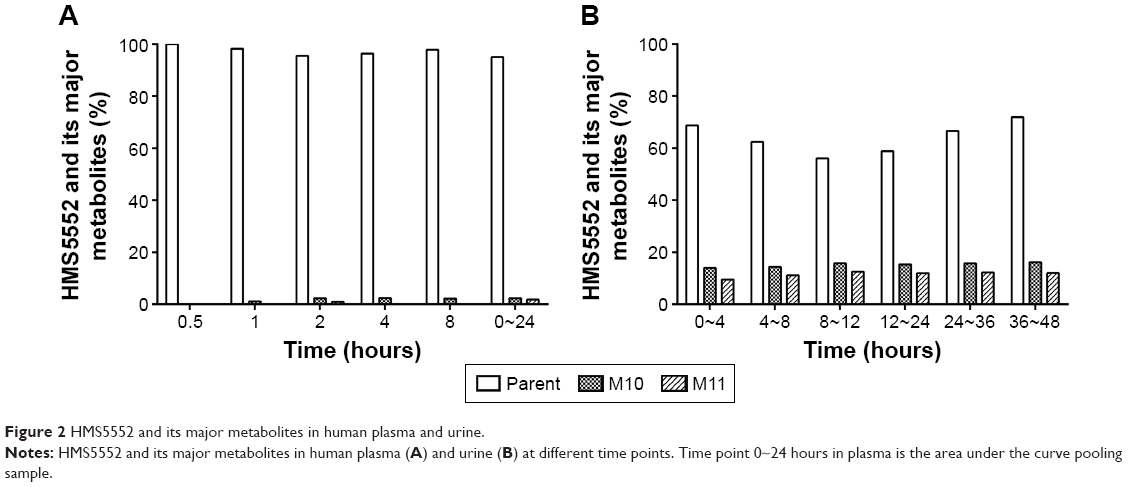 | Figure 2 HMS5552 and its major metabolites in human plasma and urine. |
PD results
Mean glucose concentration versus time was shown in Figure 3A. There was a dose-dependent reduction in FPG (0~4 hours). Maximum glucose lowering occurred in the 50 mg group (percent [%] maximum change from baseline was −31.49 as compared to −6.43, −11.32, −14.88, −18.39, −22.85, and −26.74 for the placebo, 5, 10, 15, 25, and 35 mg doses, respectively). The reductions in FPG demonstrated by maximum glucose change from baseline (%) and mean glucose AUEC0–4 change from baseline (%) were all dose-dependent and statistically significant (P<0.001).
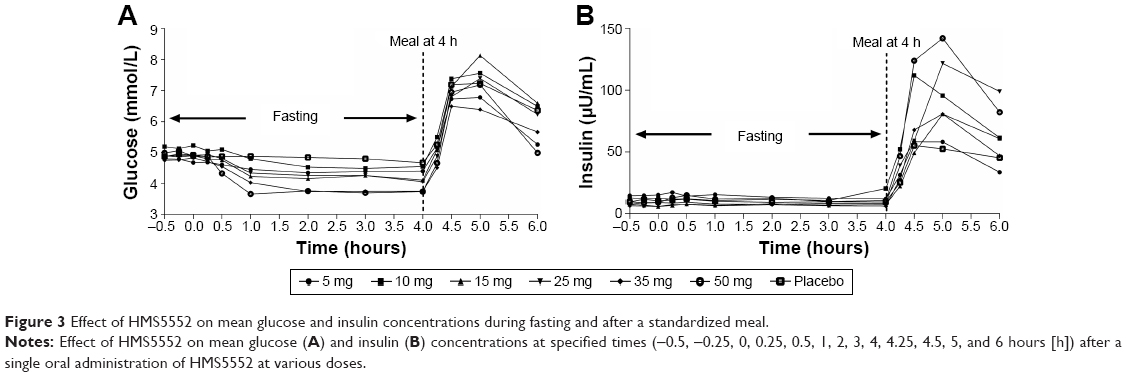 | Figure 3 Effect of HMS5552 on mean glucose and insulin concentrations during fasting and after a standardized meal. |
There were no significant changes in insulin levels versus dose in the fasting state. However, in the postprandial state, there appeared to be a clear dose-dependent relationship between insulin levels (maximum concentration and AUC4–6) and HMS5552 (Figure 3B). The 50 mg dose group achieved the maximal increase of insulin level (maximal concentration =152.278 μU/mL).
Discussion
In this study, HMS5552 was generally safe and well-tolerated over the evaluated doses ranging from 5 to 50 mg. The incidence and severity of AEs were comparable between placebo and active groups, and there were no dose-dependent increases in AEs observed. In addition, HMS5552 did not show any acute increases in systolic or diastolic blood pressure at any of the doses tested, nor acute perturbations in liver enzymes or lipids, particularly with regard to changes in triglycerides. Longer term T2DM patient studies with HMS5552 of up to 4 weeks dosing duration continue to support the safety findings obtained in this SAD study.15,16
The PK parameters for HMS5552’s plasma exposure (AUC0–t and Cmax) showed dose-proportionality, while the geometric mean t1/2 across all dose-cohorts was up to 7.51 hours. Additional data from multi-dose 1-week and 4-week trials of HMS5552 showed steady state t1/2 values of 8~10 hours.15,16 In contrast, the SAD study for AZD1656, a previous generation of GKA developed by AstraZeneca, showed that the geometric mean t1/2 was ~3 hours.17 MK-0941, another GKA developed by Merck, showed a similar duration of action (~4 hours).18 Besides, HMS5552 exhibited low renal elimination (<11%), indicating hepatic elimination might be the major route of excretion. Based on predictable and consistent PK parameters (AUC0–t and Cmax) across doses as demonstrated by dose-proportionality in this study, these results may provide useful information about dosage adjustments of this drug in subsequent clinical trials and clinical practice. The longer t1/2 also supports a twice daily regimen to improve compliance.
Of particular interest were HMS5552’s PD results showing dose-dependent decreases in FPG without concomitant increases in fasting insulin secretion (Figure 3, 0~4 hours). In contrast to the absence of insulin secretion in the fasting state, dose-dependent increases were observed in postprandial insulin secretion (Figure 3B, 4~6 hours). These findings are consistent with GKA’s overall mechanisms-of-action in decreasing hepatic glucose production and stimulating postprandial pancreatic GSIR,17,19,20 assuming a GKA that can maintain the sigmoidal glucose-dependency curve of the natural GK enzyme (nH =1.7).21–23 HMS5552’s Hill coefficient is very close to the natural value at the dose ranges tested in this study (nH ~1.6 at 50 mg with Cmax =1.26 μM), which is salient in demonstrating its glucose-dependent GSIR that could be clinically advantageous.
Conclusion
In summary, this first-in-human study of HMS5552 at doses from 5 to 50 mg in healthy subjects demonstrated a favorable safety profile with linear PK kinetics and dose-related glucose-lowering effects. The safety, efficacy, and β-cell sparing potential of HMS5552 are being investigated in multiple Phase I and Phase II clinical trials, including in monotherapy as well as in combination studies with metformin. These studies will provide a clearer basis for understanding the potential of HMS5552 to be a clinically differentiated T2DM treatment.
Acknowledgments
This work was supported by grants from the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (2014ZX09101002004), Shanghai Science and Technology Innovation Action Progress (14431908300, 1543907200), Program of Shanghai Subject Chief Scientist (13XD1528500), Shanghai Pudong Science and Technology Development Foundation (PKJ2014-506) and Shanghai Technology Program (PKJ2014-S06, 15XD1520500, and 15431907200).
The abstract of this paper was presented at the American Diabetes Association (ADA) 74th Scientific Sessions (June 13–17, 2014, San Francisco, California, USA): “Validating the Dual-Modes of Action of HMS5552, A Novel Pancreatic- and Hepatic-Targeting Glucokinase Activator” as a poster presentation. Primary findings of this study were also presented as part of a poster at the ADA 75th Scientific Sessions (June 5–9, 2015, Boston, USA): “A Novel Dual Pancreatic and Hepatic Acting Glucokinase Activator, HMS5552: Phase I studies in Healthy Subjects and T2DM Patients”.
Disclosure
John Choi, Guiyu Zhao, Tianxin Hu, Yongguo Li, Yi Zhang, and Li Chen are employees of Hua Medicine, Shanghai, People’s Republic of China. All other authors declare they have no conflict of interest.
References
Banegas JR, Lopez-Garcia E, Dallongeville J, et al. Achievement of treatment goals for primary prevention of cardiovascular disease in clinical practice across Europe: the EURIKA study. Eur Heart J. 2011;32(17):2143–2152. | ||
Kahn SE, Haffner SM, Heise MA, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355(23):2427–2443. | ||
Hinklin RJ, Boyd SA, Chicarelli MJ, et al. Identification of a new class of glucokinase activators through structure-based design. J Med Chem. 2013;56(19):7669–7678. | ||
Matschinsky FM. Assessing the potential of glucokinase activators in diabetes therapy. Nat Rev Drug Discov. 2009;8(5):399–416. | ||
Pfefferkorn JA. Strategies for the design of hepatoselective glucokinase activators to treat type 2 diabetes. Expert Opin Drug Discov. 2013;8(3):319–330. | ||
Matschinsky FM. GKAs for diabetes therapy: why no clinically useful drug after two decades of trying? Trends Pharmacol Sci. 2013;34(2):90–99. | ||
Theodorakis MJ, Carlson O, Michopoulos S, et al. Human duodenal enteroendocrine cells: source of both incretin peptides, GLP-1 and GIP. Am J Physiol Endocrinol Metab. 2006;290(3):E550–E559. | ||
Doliba NM, Fenner D, Zelent B, Bass J, Sarabu R, Matschinsky FM. Repair of diverse diabetic defects of beta-cells in man and mouse by pharmacological glucokinase activation. Diabetes Obes Metab. 2012;14 Suppl 3:109–119. | ||
Matschinsky FM, Magnuson MA. Glucokinase and Glycemic Diseases: From Basics to Novel Therapeutics. Frontiers in diabetes. Basel: Karger; 2004;16:65–74. | ||
Matschinsky FM, Zelent B, Doliba N, et al. Glucokinase activators for diabetes therapy: May 2010 status report. Diabetes Care. 2011;34 Suppl 2:S236–S243. | ||
Futamura M, Yao J, Li X, et al. Chronic treatment with a glucokinase activator delays the onset of hyperglycaemia and preserves beta cell mass in the Zucker diabetic fatty rat. Diabetologia. 2012;55(4):1071–1080. | ||
Wei P, Shi M, Barnum S, Cho H, Carlson T, Fraser JD. Effects of glucokinase activators GKA50 and LY2121260 on proliferation and apoptosis in pancreatic INS-1 beta cells. Diabetologia. 2009;52(10):2142–2150. | ||
Shirakawa J, Togashi Y, Sakamoto E, et al. Glucokinase activation ameliorates ER stress-induced apoptosis in pancreatic beta-cells. Diabetes. 2013;62(10):3448–3458. | ||
Grimsby J, Sarabu R, Corbett WL, et al. Allosteric activators of glucokinase: potential role in diabetes therapy. Science. 2003;301(5631):370–373. | ||
Zhu DL, Ding YH, Xiao DW, et al. Clinically differentiated glucokinase activator HMS5552: effective control of 24 hour glucose and improvement of β cell function in T2DM patients. Poster: 1167-P presented at: The 75th Annual Scientific Sessions of the American Diabetes Association; June 5–9, 2015; Boston, MA. | ||
Ding YH, Hu P, Zhu DL, et al. Clinical observed β-cell function improvement in Chinese T2D patients with 4 week treatment of HMS5552, a novel Glucokinase activator. Diabetes/Metabolism Research and Reviews: Supplement: The 19th Annual Meeting of the Chinese Diabetes Society; December 9–12, 2015; Suzhou, People’s Republic of China. | ||
Ericsson H, Roshammar D, Wollbratt M, et al. Tolerability, pharmacokinetics, and pharmacodynamics of the glucokinase activator AZD1656, after single ascending doses in healthy subjects during euglycemic clamp. Int J Pharmacol Ther. 2012;50(11):765–777. | ||
Meininger GE, Scott R, Alba M, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34(12):2560–2566. | ||
Bonadonna RC, Heise T, Arbet-Engels C, et al. Piragliatin (RO4389620), a novel glucokinase activator, lowers plasma glucose both in the postabsorptive state and after a glucose challenge in patients with type 2 diabetes mellitus: a mechanistic study. J Clin Endocrinol Metab. 2010;95(11):5028–5036. | ||
Morrow LA, Leonsson-Zachrisson M, Ericsson H, et al. Safety, pharmacokinetics and pharmacodynamics of multiple-ascending doses of the novel glucokinase activator AZD1656 in patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2012;14(12):1114–1122. | ||
Cuesta-Munoz AL, Huopio H, Otonkoski T, et al. Severe persistent hyperinsulinemic hypoglycemia due to a de novo glucokinase mutation. Diabetes. 2004;53(8):2164–2168. | ||
Larion M, Miller BG. 23-Residue C-terminal alpha-helix governs kinetic cooperativity in monomeric human glucokinase. Biochemistry. 2009;48(26):6157–6165. | ||
Molnes J, Teigen K, Aukrust I, et al. Binding of ATP at the active site of human pancreatic glucokinase – nucleotide-induced conformational changes with possible implications for its kinetic cooperativity. FEBS J. 2011;278(13):2372–2386. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.